

Abstract
Screening of “ drug-like” molecule from the molecular database produced through high throughput techniques and their large repositories requires robust classification. In our work, a set of heuristically chosen nine molecular descriptors including four from Lipinski's rule, were used as classification parameter for screening “drug-like” molecules. The robustness of classification was compared with four fundamental descriptors of Lipinski. Back propagation neural network based classifier was applied on a database of 60000 molecules for classification of, “ drug-like” and “non drug-like” molecules. Classification result using nine descriptors showed high classification accuracy of 96.1% in comparison to that using four Lipinski's descriptors which yielded an accuracy of 82.48%. Also a significant decrease of false positives resulted while using nine descriptors causing a sharp 18% increase of specificity of classification. From this study it appeared that Lipinski's descriptors which mainly deal with pharmacokinetic properties of molecules form the basis for identification of “drug-like” molecules that can be substantially improved by adding more descriptors representing pharmacodynamics properties of molecules.
Keywords: machine learning, small molecules, molecular descriptors, druglikeness, non druglikeness
Background
Discriminating “drug-like” molecules from non-drug like molecules is one of the prime focuses of current research in the field of computer aided drug design. Concept of drug likeness is important in both combinatorial chemistry and high throughput screening which deal with synthesis of libraries of molecules and their evaluation respectively [1]. Combinatorial chemistry has emerged as a new paradigm in the field of drug discovery as it provides a large library of compounds at a time. Moreover the number of commercially available compounds is also going on increasing. So the process of compound selection and prioritization is very crucial to reduce the time and computational cost of screening the libraries of compounds to get compounds feasible for synthesis and further testing [2]. In this scenario the concepts and methods to predict drug-likeness of enormous number of molecules available through miscellaneous sources in the libraries becomes important.
Drug-likeness can be deduced as a delicate balance among molecular properties affecting pharmaco-dynamics and pharmacokinetics of molecules which ultimately affects their absorption, distribution, metabolism, and excretion in human body like a drug. Molecular properties include molecular weight, electronic distribution, hydrophobicity, hydrogen bond donors/acceptors, solubility, viscosity, excess volume and other related properties. For computational studies these molecular properties have to be logically and quantitatively represented as molecular descriptors in such a way that their complete chemical information remains preserved [3]. Selection of suitable molecular descriptors for correctly predicting the druglikeness of a molecule is of prime importance for the screening the drug-like molecules.
Methods for drug-likeness prediction include from simple counting schemes like Lipinski's “rule of five” to machine learning approaches like artificial neural network and support vector machines [4]. Lipinski’s “rule of five” is a heuristic approach for predicting drug-likeness stating that molecules having molecular weight ≫500, log P ≫5, hydrogen bond donors ≫5 and hydrogen bond acceptors ≫10 have poor absorption or permeation [5]. This rule describes only the molecular properties related with pharmacokinetics of molecules which refers to the absorption, distribution, metabolism and excretion (ADME) of bioactive compounds in a higher organism. There is no consideration for pharmaco-dynamics aspect of molecules which deal with drug action on the body or on microorganisms and other parasites within or on the body. Moreover there are many violations of this rule among existing drugs and vice versa, and therefore, qualifying the “rule of five” does not guarantee that a molecule is “druglike” [6].
Other methods include ‘knowledge based method’ by Andrews et al., considering intrinsic binding energies and structural fragments i.e., functional groups. This method is based on intrinsic energies derived from 200 molecules [7]. Walter et al., proposed ’REOS (Rapid Elimination of Swill) approach‘ as a hybrid of simple counting method and functional group filtering method. Functional group filters used in this method identify reactive, toxic and undesirable moieties [8]. Initial filtering is based on seven properties including molecular weight, logP, hydrogen bond donors, hydrogen bond acceptors, formal charge, number of rotatable bonds and number of heavy atoms. REOS also employs a set of more than 200 rules based on presence of functional groups known to be problematic.
In addition, machine learning approaches are being used most successfully for drug-likeness prediction [6]. Sadowski and Kubinyi designed a feed forward neural network with 92 input nodes, five hidden nodes and one output node to predict drug-likeness [9]. They characterized the molecules using a set of 92 atom types originally devised for the purpose of predicting logP by Ghose and Krippen [10]. Their neural network classified 77% “drug-like” and 83% “non drug-like” molecules correctly. Two important aspects to be considered in machine learning based approaches are the choice of appropriate descriptors and the number of descriptors to be used at the time of creating the model. Representation of chemical structures by appropriate descriptors is the key to success in establishing useful relationships using machine learning approaches [11]. Evidently, larger the number of relevant descriptors more will be the efficiency for druglikeness classification using machine learning approach [12]. These facts suggest the requirement of improvement in classical approaches for predicting drug-likeness of molecules by adding more suitable descriptors as classifying parameters. In this context additional descriptors were deterministically selected in our work to account for pharmaco-dynamic properties of molecules. Use of these descriptors as input into back propagation network classifier appeared to be the possible cause of improvement in overall classification efficiency. Thus it is a tradeoff between requirement of additional descriptors and removal of redundant descriptors for better prediction of drug-likeness.
Methodology
Dataset
Data was downloaded from ZINC database [13]. Each molecule in ZINC database is represented by its three dimensional structure, SMILES representation and corresponding molecular descriptors. Descriptor values available in ZINC database are calculated using “Molinspiration MiTools” molecular processing toolkit and were used as such for our study [14]. For this study 30000 “drug-like” and 30000 “nondrug-like” molecules were used as working database. Within this database 20000 and 10000 molecules from both the groups constituted training and test set respectively
Selection of molecular descriptors
Two different data sets were prepared depending upon the choice of descriptors. In first data set molecules were represented by a vector of nine descriptors representing Molecular weight (MW), logP (LP), apolar desolvation energy (ADE), polar desolvation energy (PDE), number of H bond donors (HBD), number of H bond acceptors (HBA), total polar surface area (TPSA), charge (Ch) and number of rotatable bonds (RB). Second data set comprised of same molecules represented by four descriptors: molecular weight, log P, number of H-bond donors, and number of H-bond acceptors. These four descriptors are considered in Lipinski's “rule of five” and were used for the purpose of benchmarking. Descriptors considered in Lipinski's rule are directly related to pharmacokinetic properties (Absorption-Distribution- Metabolism-Excretion-Toxicity properties) of molecules. Other five descriptors selected i.e., apolar desolvation energy, polar desolvation energy, total polar surface area, charge and number of rotatable bonds are related to pharmacodynamic aspect of molecules. Thus the selected descriptors cover both the pharmacokinetic and pharmacodynamics aspects of drug action.
Preprocessing the data
Data was normalized to scale it down to the range between 0 and 1, using min max normalization method [15]. Normalized values can be represented by equation 1 in supplementary material.
Classification by back propagation Artificial Neural Network (ANN)
The classification decision was obtained with the help of a feed-forward back propagation network where normalized descriptor-vectors were used as input. Several neural network architectures were tried for training using data containing nine descriptors. The most efficient one was chosen as final network architecture. It consists of two hidden layers apart from input and output layers. While the input layer comprises of nine nodes, first and second hidden layers consist of seven and five nodes respectively. Output layer contains one node to represent two classes of our interest. Transfer function used in both hidden layers was tan sigmoid and that used in output layer was linear. Weights and biases of the neural network were optimized during training using gradient descent method. Mean square error was used as performance function. Similar neural network architecture with only exception of four input nodes was used for data set containing only Lipinski's descriptors. For training set target output was kept as 1 for “drug-like” molecules and -1 for “non druglike” molecules. For testing the trained neural network an error margin was taken as 0.10. Molecules giving output ≫0.90 were considered as “drug-like” and those giving output ≪-0.90 were classified as “non drug-like” molecules
Assessment of efficiency of classification
As the classification of small molecules into “drug-like” and “non drug-like” classes is a binary classification; sensitivity, specificity and accuracy are appropriate measures for assessing efficiency of classification [17]. While sensitivity is the proportion of true positives classified as positives, specificity can be defined as the proportion of true negatives classified as negatives. Accuracy is the proportion of true positives and true negatives classified as positives and negatives respectively. These measures were calculated for neural networks trained with nine descriptor data set and four descriptors data set separately.
Discussion
The study was aimed at comparing the efficiency of different descriptor sets in classifying small molecules into “drug-like” and “nondrug-like” classes, the result of which is summarized in table 2. There was consistent improvement in all the considered measures of performance of the network trained with all the nine descriptors in comparison to the network trained with Lipinski's four descriptors only. The improvements found in sensitivity, specificity and accuracy were 9%, 18.29% and 13.64% respectively, as visualized in histogram chart in figure 1. The most significant improvement was found in specificity as there was a decrease of ∼3.5 times in the number of false positives detected using nine descriptors data set in comparison to Lipinski's descriptors data set. The consistent improvement in efficiency measures of classification as shown in figure 1 indicates that there is further scope of improvement in building a protocol based on addition of more descriptors for distinguishing between “drug-like” and “non drug-like” molecules over that of Lipinski's “rule of five”. Our study also supports the studies done by Oprea (2000) and Frimurer (2000) which concluded that Lipinski's “rule of five” has limitations in distinguishing between “drug-like” and “non drug-like” molecules [1,18]. The basis behind these findings is that the distribution of descriptors considered in Lipinski's “rule of five”, does not differ significantly between the “drug-like” and “non drug-like” molecules most possibly because of the consideration of their pharmacokinetic properties only. Moreover, the contribution of a particular input descriptor in the final decision making stage can be evaluated by optimizing the weights of artificial neural network classifier linking inputs with outputs where very small value of weight will be assigned to the least contributing input [19]. It is well documented in literature that normalization of input data vector improves the efficiency of neural network classifier [15]. Moreover we considered the maximum error margin of 0.10. This makes our classifier very stringent in comparison to traditional neural network classifier where the molecules giving output ≪0 and ≫0, would have been classified into “non drug-like” and “drug-like” classes respectively. Thus our classifier had very stringent criteria for classification of molecules into their respective classes relaxing which a further improvement in efficiency can be obtained at the cost of degrading the reliability of decision. As discussed in the methodology section all the parameters related to neural network architecture were kept same for both the data sets with only exception in input layer. This reduced any chance of difference in efficiency of classifier due to any factor related to the classifier other then the descriptor set used in training and test sets.
Figure 1.
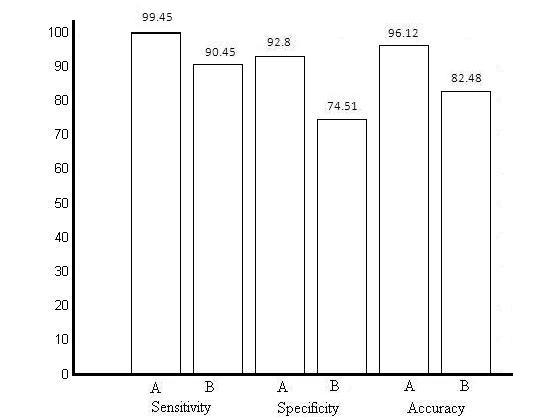
Measures of efficiency of classification. Bars named as ’A‘ represent values for nine descriptor data set and bars named as ’B‘ represent values for Lipinski's descriptor data set.
Conclusion
Our approach dealt with the problem of drug-likeness prediction in a different perspective. It showed that rule based approach for drug-likeness prediction has scope for improvement. Machine learning approach shows good efficiency for drug-likeness prediction. Appropriate choice and number of descriptors is of prime importance for application of machine learning approaches for druglikeness prediction. Another important aspect of this study was consideration of the reliability of the classifier that was achieved by keeping the permissible error margin of output as 0.1. Such stringent considerations made the classifier more reliable which most possibly helped to eliminate false positives which is crucial for screening “drug-like” molecules.
Supplementary material
Acknowledgments
We hereby acknowledge the helpful discussion we had with Manish Kumar, Indian Institute of Information Technology (IIIT), Allahabad, India. We are also grateful to Ministry of Human Resource and Development (MHRD) and Department of Science and Technology (DST) for their joint financial support to Bioinformatics Division of IIIT to continue this work.
Footnotes
Citation:Mishra et al, Bioinformation 3(9): 384-388 (2009)
References
- 1.Oprea TI. J Comput Aided Mol Des. 2000;14(3):251. doi: 10.1023/a:1008130001697. [DOI] [PubMed] [Google Scholar]
- 2.Kubinyi H. Perspect Drug Discov Design. 1998;9(11):225. [Google Scholar]
- 3. http://www.moleculardescriptors.eu/
- 4.Walters W Patrick, Murcko Mark A. Adv Drug Deliv Rev. 2002;54(3):255. doi: 10.1016/s0169-409x(02)00003-0. [DOI] [PubMed] [Google Scholar]
- 5.Lipinski CA. J Pharmacol Toxicol Methods. 2000;44(1):235. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 6.Kadam RU, Roy N. Indian J Pharm Sci. 2007;69(5):609. [Google Scholar]
- 7.Andrews PR, et al. J Med Chem. 1984;27(12):1648. doi: 10.1021/jm00378a021. [DOI] [PubMed] [Google Scholar]
- 8.Walters W Patrick, Numchuk Mark. Nat Rev Drug Discov. 2003;2(4):259. doi: 10.1038/nrd1063. [DOI] [PubMed] [Google Scholar]
- 9.Ghose AK, Crippen GM. J Comput Chem. 1986;7(4):565. [Google Scholar]
- 10.Sadowski J, Kubinyi H. J Med Chem. 1998;41(18):3325. doi: 10.1021/jm9706776. [DOI] [PubMed] [Google Scholar]
- 11.Gasteiger J, et al. J Phys Org Chem. 2003;16(4):232. [Google Scholar]
- 12.Li Q, et al. J Chem Inf Model. 2007;47(5):1776. doi: 10.1021/ci700107y. [DOI] [PubMed] [Google Scholar]
- 13.Irwin JJ, Shoichet BK. J Chem Inf Model. 2005;45(1):177. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. http://www.molinspiration.com/
- 15.Givehchi A, Schneider G. J Mol Model. 2004;10(3):204. doi: 10.1007/s00894-004-0186-9. [DOI] [PubMed] [Google Scholar]
- 16.Shalabi LA, et al. Journal of Computer Science. 2006;2(9):735. [Google Scholar]
- 17.Baldi P, et al. Bioinformatics. 2000;16(5):412. doi: 10.1093/bioinformatics/16.5.412. [DOI] [PubMed] [Google Scholar]
- 18.Frimurer TM, et al. J Chem Inf Comput Sci . 2000;40(6):1315. doi: 10.1021/ci0003810. [DOI] [PubMed] [Google Scholar]
- 19. http://www.makhfi.com/why_ann.htm.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.