

Abstract
The preparation of a series of monoquaternary pyridinium oximes bearing either a heterocyclic side chain or a functionalized aliphatic side chain and the corresponding in vitro evaluation for reactivation of paraoxon-inhibited electric eel acetylcholinesterase (EeAChE) and recombinant human acetylcholinesterase (rHuAChE) are reported. Several newly synthesized compounds efficiently reactivated inhibited EeAChE, but were poor reactivators of inhibited rHuAChE. Compounds bearing a thiophene ring in the side chain (20, 23, 26 and 29) showed better reactivation (24–37% for EeAChE and 5–9% for rHuAChE) compared to compounds with furan and isoxazole heterocycles (0–8% for EeAChE and 2–3% for rHuAChE) at 10−5 M. The N-pyridyl-CH2COOH analog 8 reactivated EeAChE (36%) and rHuAChE (15%) at 10−4 M with a kr value better than 2-pyridine aldoxime methiodide (2-PAM) for rHuAChE.
Keywords: organophosphorus, acetylcholinesterase, reactivators, monoquaternary pyridinium oximes, pralidoxime, 2-PAM
Reactivators of acetylcholinesterase (AChE; EC 3.1.1.7) are important therapeutic agents for the treatment of toxicity resulting from organophosphorus inhibitors (OPI) such as sarin, paraoxon, tabun, chlorpyrifos, cyclosarin, VX, and DFP. Some of these OPIs have been used as warfare agents (such as sarin, soman, tabun, VX) and are some of the most toxic man made compounds known.1 OPIs inhibit AChE via formation of a covalent bond at a serine hydroxyl group within the enzyme’s active site. Once inhibited, AChE is not able to hydrolyze acetylcholine and accumulates in the synaptic cleft leading to cholinergic over-stimulation, compromised respiration and sometimes death.1 AChE inhibited by OPI can be reactivated by 2-pyridine aldoxime methiodide (2-PAM, pralidoxime; 1)2 that nucleophilically attacks the phosphoryl moiety displacing it from the active site serine (Fig. 1). In addition to 2-PAM3–6, the bis-oximes trimedoxime (TMB-4, 2),7, 8 obidoxime (3)9, 10 and HI-6 (4)11, 12 show efficacy as reactivators in vivo and are widely used for the treatment of OPI poisoning.13, 14 Recently, several new pyridinium oxime reactivators were reported including bisquaternary pyridinium oximes connected with different linker chains viz. alkylene linker,15–18 but-2-ene linker,19–22 aromatic ring containing linkers23–25 and oxygenated linker chain.26–28 Monoquaternary pyridinium oximes include 2-PAM analogs with varying side chain29, 30 and 4-PAM analogs with modified oximes.31 Amongst these, but-2-ene linked bisquaternary 4,4′-bisoxime22 and 2-PAM analogs with a propyl side chain29 showed reactivation of paraoxon-inhibited AChE comparable to clinically known reactivators.
Figure 1.

Equation representing inhibition and reactivation of AChE by OPI and oximes. Selected examples of oximes are shown.
Although bis-quaternary oximes are considered more effective AChE reactivators, many countries use 2-PAM as the therapeutic agent.32,33 The efficacy of the oxime used is also dependent on the AChE type. Certain AChE types undergo rapid reactivation whereas others show little to no appreciable activity after oxime treatment. This and other limitations highlight that there has been no single broad-spectrum oxime reactivator identified to date.1,34 Therefore, the development and selection of new, more effective AChE oxime reactivators is very important. The focus of this work was to determine the effect of different heterocyclic and aliphatic side chains (capable of hydrogen bonding) on the ability of monoquaternary pyridinium oximes to reactivate two forms of AChE inhibited by paraoxon.
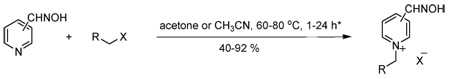
Monoquaternary pyridinium oximes 5–35 were prepared in 40–92% yield via reaction of 2-, 3- or 4-pyridine aldoximes with different substituted alkyl halides in acetonitrile or acetone at 60–80 °C for 1–24 h.35 Monoquaternary pyridinium oximes 5–16 were designed to contain an aliphatic side chain with a group capable of hydrogen bonding viz. carbamoyl, carboxyl and hydroxyl. Furthermore, highly electron rich heterocycles, viz. thiophene, furan and isoxazole were used as side chains 17–35 to explore interactions with cation-containing (cation-π bonding) or aromatic residues (π–π bonding) at or near the AChE active site. The oxime group was positioned at 2, 3 and 4 on the pyridinium ring to determine the best position for reactivation of AChE using the N-derivatized groups. All compounds were characterized by 1H NMR, 13C NMR, ESI-MS and IR prior to assay.35 Compounds 10, 11 and 13 have been reported as reactivators but not against EeAChE or rHuAChE.7,36 All compounds were assayed for their capacity to reactivate EeAChE and rHuAChE using a modified Ellman assay.37,38 2-PAM and paraoxon were chosen as a reference reactivator and as representative OPI.
Reactivation results are summarized in Table 1. Oximes with aliphatic side chains mostly showed concentration-dependent reactivation of AChE. Compounds containing carboxamide or carboxylic acid groups paired with the 2 or 4-oxime position (5, 7 and 8) gave 40–45% reactivation of EeAChE while compounds with hydroxyethyl side chain 11 and 13 showed 50 and 70% reactivation of EeAChE at 10−3 M comparable to 2-PAM 1. Compound 8 also showed promise as a reactivator of rHuAChE (15%) at 10−3 M. Compounds with aromatic side chains (thiophene, furan and isoxazole) displayed a reverse concentration-activity relationship. These compounds, with the exception of 19 and 35, showed ≤ 8% reactivation of EeAChE and no reactivation of rHuAChE at the highest concentration (10−3 M). Lower concentrations of oxime (10−4 to 10−5 M) 17–29 were better reactivators of both enzymes suggesting that these compounds may inhibit AChE at higher concentrations and explain, in part, the inability to reactivate inhibited AChE at 10−3 M.19,23
Table 1.
Synthesis and in vitro reactivation potency of monoquaternary pyridinium oximes 5–35 for paraoxon-inhibited EeAChE and rHuAChE
 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | R | Oxime position |
X | Reaction time (h) |
% Yield |
% Reactivation = [(Ar-Ai)/(Ao-Ai)] × 100 | |||||
| EeAChE (% R ± SEM) | rHuAChE (% R ± SEM) | ||||||||||
| 10−3 Ma | 10−4 Ma | 10−5 Ma | 10−3 Ma | 10−4 Ma | 10−5 Ma | ||||||
| 1 | H | 2 | I | - | - | 67 ± 1 | 67 ± 3 | 63 ± 3 | 42 ± 2 | 22 ± 2 | 7 ± 1 |
| 5 | CONH2 | 2 | I | 24 | 62 | 41 ± 3 | 9 ± 0 | 1 ± 0 | 2 ± 1 | 2 ± 0 | 1 ± 0 |
| 6 | CONH2 | 3 | I | 2 | 76 | 1 ± 1 | 0 | 0 | 0 | 1 ± 0 | 2 ± 0 |
| 7 | CONH2 | 4 | I | 2 | 84 | 44 ± 2 | 6 ± 1 | 0 | 0 | 1 ± 0 | 1 ± 0 |
| 8 | COOH | 2 | I | 24 | 61 | 45 ± 3 | 36 ± 3 | 34 ± 2 | 15 ± 1 | 15 ± 2 | 5 ± 1 |
| 9 | COOH | 3 | I | 2 | 71 | 1 ± 0 | 0 | 0 | 0 | 1 ± 0 | 1 ± 0 |
| 10 | COOH | 4 | I | 2 | 78 | 4 ± 0 | 0 | 0 | 0 | 1 ± 0 | 2 ± 0 |
| 11 | CH2OH | 2 | Br | 24 | 58 | 50 ± 4 | 37 ± 3 | 13 ± 1 | 2 ± 2 | 5 ± 0 | 2 ± 0 |
| 12 | CH2OH | 3 | Br | 2 | 60 | 8 ± 1 | 0 | 0 ± 0 | 0 | 1 ± 0 | 1 ± 0 |
| 13 | CH2OH | 4 | Br | 2 | 68 | 70 ± 2 | 2 ± 2 | 3 ± 1 | 6 ± 1 | 3 ± 0 | 2 ± 0 |
| 14 | CH2COOH | 2 | Br | 24 | 48 | 10 ±1 | 0 | 0 | 0 | 1 ± 0 | 1 ± 0 |
| 15 | CH2COOH | 3 | Br | 2 | 56 | 5 ± 0 | 0 | 0 | 0 | 1 ± 0 | 1 ± 0 |
| 16 | CH2COOH | 4 | Br | 2 | 68 | 15 ±1 | 0 | 0 | 0 | 2 ± 0 | 1 ± 0 |
| 17 | 4-methoxybenzen-1-yl | 2 | Cl | 12 | 70 | 8 ± 1 | 19 ± 2 | 18 ± 2 | 0 | 7 ± 1 | 3 ± 0 |
| 18 | 4-methoxybenzen-1-yl | 3 | Cl | 1 | 84 | 1 ± 1 | 0 | 0 | 0 | 3 ± 0 | 2 ± 0 |
| 19 | 4-methoxybenzen-1-yl | 4 | Cl | 1 | 92 | 19 ± 1 | 11 ± 1 | 3 ± 1 | 0 | 3 ± 1 | 2 ± 0 |
| 20 | thiophen-2-yl | 2 | Cl | 24 | 58 | 1 ± 0 | 24 ± 2 | 34 ± 2 | 0 | 9 ± 2 | 9 ± 1 |
| 21 | thiophen-2-yl | 3 | Cl | 2 | 65 | 0 | 3 ± 1 | 1 ± 0 | 0 | 4 ± 0 | 5 ± 0 |
| 22 | thiophen-2-yl | 4 | Cl | 2 | 78 | 3 ± 0 | 12 ± 2 | 12 ± 0 | 0 | 4 ± 1 | 5 ± 0 |
| 23 | 5-bromothiophen-2-yl | 2 | Cl | 12 | 74 | 0 | 29 ± 2 | 37 ± 2 | 0 | 7 ± 0 | 8 ± 1 |
| 24 | 5-bromothiophen-2-yl | 3 | Cl | 1 | 84 | 4 ± 1 | 7 ± 0 | 8 ± 1 | 0 | 5 ± 1 | 6 ± 0 |
| 25 | 5-bromothiophen-2-yl | 4 | Cl | 1 | 90 | 3 ± 1 | 14 ± 1 | 10 ± 1 | 0 | 5 ± 1 | 4 ± 0 |
| 26 | benzthiophen-3-yl | 2 | Cl | 24 | 67 | 0 | 28 ± 2 | 24 ± 3 | 0 | 7 ± 1 | 8 ± 1 |
| 27 | benzthiophen-3-yl | 3 | Cl | 2 | 79 | 0 | 5 ± 1 | 8 ± 1 | 0 | 2 ± 0 | 5 ± 1 |
| 28 | benzthiophen-3-yl | 4 | Cl | 2 | 85 | 0 | 0 | 1 ± 1 | 0 | 2 ± 0 | 2 ± 0 |
| 29 | thiophen-3-yl | 2 | Cl | 24 | 79 | 0 | 8 ± 1 | 28 ± 2 | 0 | 2 ± 0 | 5 ± 1 |
| 30 | furan-2-yl | 3 | Cl | 1 | 62 | 0 | 0 | 0 | 0 | 2 ± 0 | 2 ± 0 |
| 31 | furan-2-yl | 4 | Cl | 1 | 60 | 8 ± 1 | 14 ± 2 | 1 ± 1 | 0 | 3 ± 0 | 2 ± 0 |
| 32 | furan-3-yl | 3 | Cl | 1 | 46 | 0 | 0 | 0 | 0 | 3 ± 0 | 2 ± 0 |
| 33 | 3-methyl-isoxazol-5-yl | 2 | Br | 24 | 40 | 7 ± 1 | 25 ± 3 | 8 ± 1 | 0 | 7 ± 1 | 3 ± 0 |
| 34 | 3-methyl-isoxazol-5-yl | 3 | Br | 2 | 64 | 0 | 0 | 0 | 0 | 3 ± 1 | 2 ± 0 |
| 35 | 3-methyl-isoxazol-5-yl | 4 | Br | 2 | 67 | 20 ± 2 | 24 ± 2 | 6 ± 0 | 0 | 2 ± 1 | 2 ± 0 |
compounds 5–16 acetone, 60–70 °C, 2–24 h; 17–35 MeCN, 70–80 °C, 2–24 h;
reactivator concentration
Each reactivator varied with respect to the optimal concentration used, and four trends were identified from the data (Table 1). First, a correlation was found between percent reactivation and increasing concentration of oxime (5, 7, 8, 11–16). A second trend was found that inversely correlates increasing reactivation with decreasing concentration of oxime (20, 22–24, 27–29). Third, compounds 17, 21, 25, 26, 31, 33, 35 showed the best percent reactivation at mid-range concentrations of oxime.19 Last, analogs bearing the oxime in the 2-position were better reactivators than those with oximes in the 3- or 4-position. Of the compounds with heteroaromatic side chains, thiophene analogs 20, 23, 26 and 29 with the 2-oxime group reactivated paraoxon-inhibited EeAChE 34%, 37%, 24% and 28% at 10−5 M, respectively. These thiophene analogs also reactivated rHuAChE but only ~5% at 10−5 M. Oximes with a thiophen-2-yl side chain (e.g., 20 and 23) were slightly better reactivators than the corresponding thiophen-3-yl structures (26 and 29). The furan analogs 30–32 were poor reactivators but the 3-methylisoxazole analogs 33 and 35 showed 24–25% reactivation of EeAChE at 10−4 M. The 4-methoxybenzyl analogs 17–19 were poor reactivators of paraoxon-inhibited EeAChE and rHuAChE although the 2- and 4-positional oximes 17 and 19 reactivated AChE up to 19% of its activity.
Owing to their greater activity, compounds 8, 13, 20, 23 and 26 were selected for analysis of the oxime-mediated reactivation rate constants (kr; characterizes the dissociation of the enzyme and the phosphorylated-oxime). 39 The kr was calculated using the equation from the linear portion of activity curves (0–15 min).40
As expected from the preliminary data (Table 1), these oximes showed 8 to 21-fold greater kr values for reactivation of paraoxon-inhibited EeAChE than rHuAChE. Compound 8 showed the highest kr values among the analogs tested against for both EeAChE and rHuAChE, and 8 was superior to 2-PAM for rHuAChE reactivation. Also interesting was the trend of kr values 8 > 20 > 23 > 26 ≥ 13 that was the same for both EeAChE and rHuAChE (Table 2).
Table 2.
Reactivation rate constant (kr) for 8, 13, 20, 23 and 26
| Entry | kr [min−1] | |
|---|---|---|
| EeAChE | rHuAChE | |
| 2-PAM (1) | 0.1766 ± 0.0134 | 0.0041 ± 0.0003 |
| 8 | 0.0406 ± 0.0021 | 0.0048 ± 0.0004 |
| 13 | 0.0108 ± 0.0002 | 0.0010 ± 0.0002 |
| 20 | 0.0339 ± 0.0026 | 0.0016 ± 0.0001 |
| 23 | 0.0189 ± 0.0004 | 0.0012 ± 0.0001 |
| 26 | 0.0138 ± 0.0008 | 0.0010 ± 0.0001 |
Paraoxon-inhibited EeAChE undergoes greater than 60% reactivation with 2-PAM from 10−3M to 10−5M (Table 1). Conversely, 2-PAM reactivates paraoxon-inhibited rHuAChE to a far lesser extent at 10−3M (42%) and is barely reactivatable at 10−5M (7%). This suggests that rHuAChE will eventually become unable to reactivate at all following inhibition by paraoxon. This may be due to one or more non-reactivation processes including denaturation or aging. The amino acid sequences of rHuAChE41 and EeAChE42 are 89% homologous and do not reveal any obvious differences to account for the 8 to 21 fold greater rate of reactivation observed for EeAChE. However, species-specific differences in reactivation rates following inhibition by a variety of OP agents are known43,44 and any data comparisons should consider these differences.
In conclusion, a new series of monoquaternary pyridinium oximes with heteroaromatic side chains have been reported of which thiophene-containing analogs 20, 23 and 26 were the better reactivators. Compound 8, bearing an acetic acid side chain, was found to be the most effective reactivator of the 31 novel compounds tested with a higher kr value than pralidoxime (1) for paraoxon-inhibited rHuAChE.
Acknowledgements
The research in this study was supported by NIH UO1-ES016102 (CMT) and SBIR grants to ATERIS Technologies LLC (R43 ES016392 U44 NS058229). Support from the Core Laboratory for Neuromolecular Production (NIH P30-NS055022) and the Center for Structural and Functional Neuroscience (NIH P20-RR015583) is appreciated.
Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the U.S. Army or the Department of Defense.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Bajgar J. Adv. Clin. Chem. 2004;38:151. doi: 10.1016/s0065-2423(04)38006-6. [DOI] [PubMed] [Google Scholar]
- 2.Wilson IB, Ginsburg S. Biochim. Biophys. Acta. 1955;18:168. doi: 10.1016/0006-3002(55)90040-8. [DOI] [PubMed] [Google Scholar]
- 3.Thiermann H, Szinicz L, Eyer F, Worek F, Eyer P, Felgenhauer N, Zilker T. Toxicol. Lett. 1999;107:233. doi: 10.1016/s0378-4274(99)00052-1. [DOI] [PubMed] [Google Scholar]
- 4.Johnson DD, Stewart WC. Can. J. Physiol. Pharm. 1970;48:625. doi: 10.1139/y70-092. [DOI] [PubMed] [Google Scholar]
- 5.Harris LW, Stitcher DL. Drug Chem. Toxicol. 1983;6:235. doi: 10.3109/01480548309017814. [DOI] [PubMed] [Google Scholar]
- 6.Sidell FR, Groff WA. Toxicol. Appl. Pharmacol. 1974;27:241. doi: 10.1016/0041-008x(74)90195-1. [DOI] [PubMed] [Google Scholar]
- 7.Poziomek E, Hackley BJ, Steinberg G. J. Org. Chem. 1958;23:714. [Google Scholar]
- 8.Hackley BE, Jr, Poziomek EJ, Steinberg GM. 3,077,476. US patent. 1963
- 9.Luttringhaus A, Hagedorn I. Arzneimittelforschung. 1964;14:1. [PubMed] [Google Scholar]
- 10.Heilbronn E, Tolagen B. Biochem. Pharmacol. 1965;14:73. doi: 10.1016/0006-2952(65)90059-6. [DOI] [PubMed] [Google Scholar]
- 11.Hagedorn I, Gündel WH, Schoene K. Arzneimittelforschung. 1969;19:603. [PubMed] [Google Scholar]
- 12.Inns RH, Leadbeater L. J. Pharm. Pharmacol. 1983;35:427. doi: 10.1111/j.2042-7158.1983.tb04316.x. [DOI] [PubMed] [Google Scholar]
- 13.Hobbiger FW, Sadler PW. Nature. 1958;182:1672. doi: 10.1038/1821672a0. [DOI] [PubMed] [Google Scholar]
- 14.Worek F, Diepold C, Eyer P. Arch. Toxicol. 1999;73:7. doi: 10.1007/s002040050580. [DOI] [PubMed] [Google Scholar]
- 15.Pang YP, Kollmeyer TM, Hong F, Lee JC, Hammond PI, Haugabouk SP, Brimijoin S. Chem. Biol. 2003;10:491. doi: 10.1016/s1074-5521(03)00126-1. [DOI] [PubMed] [Google Scholar]
- 16.Kuca K, Bielavsky J, Cabal J, Bielavska M. Tetrahedron Lett. 2003;44:3123. [Google Scholar]
- 17.Musilek K, Holas O, Kuca K, Jun D, Dohnal V, Dolezal M. Bioorg. Med. Chem. Lett. 2006;16:5673. doi: 10.1016/j.bmcl.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 18.Srinivas Rao C, Venkateswarlu V, Achaiah G. Bioorg. Med. Chem. Lett. 2006;16:2134. doi: 10.1016/j.bmcl.2006.01.065. [DOI] [PubMed] [Google Scholar]
- 19.Musilek K, Kuca K, Jun D, Dohnal V, Dolezal M. Bioorg. Med. Chem. Lett. 2006;16:622. doi: 10.1016/j.bmcl.2005.10.059. [DOI] [PubMed] [Google Scholar]
- 20.Musilek K, Jun D, Cabal J, Kassa J, Gunn-Moore F, Kuca K. J. Med. Chem. 2007;50:5514. doi: 10.1021/jm070653r. [DOI] [PubMed] [Google Scholar]
- 21.Musilek K, Holas O, Kuca K, Jun D, Dohnal V, Opletalova V, Dolezal M. J. Enz. Inhib. Med. Chem. 2008;23:70. doi: 10.1080/14756360701383981. [DOI] [PubMed] [Google Scholar]
- 22.Musilek K, Holas O, Kuca K, Jun D, Dohnal V, Opletalova V, Dolezal M. Bioorg. Med. Chem. Lett. 2007;17:3172. doi: 10.1016/j.bmcl.2007.03.025. [DOI] [PubMed] [Google Scholar]
- 23.Musilek K, Holas O, Kuca K, Jun D, Dohnal V, Dolezal M. J. Enz. Inhib. Med. Chem. 2007;22:425. doi: 10.1080/14756360601164960. [DOI] [PubMed] [Google Scholar]
- 24.Acharya J, Gupta AK, Dubey DK, Raza SK. Eur. J. Med. Chem. 2009;44:1335. doi: 10.1016/j.ejmech.2008.02.029. [DOI] [PubMed] [Google Scholar]
- 25.Acharya J, Gupta AK, Mazumder A, Dubey DK. Eur. J. Med. Chem. 2009;44:1326. doi: 10.1016/j.ejmech.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 26.Kim TH, Kuca K, Jun D, Jung YS. Bioorg. Med. Chem. Lett. 2005;15:2914. doi: 10.1016/j.bmcl.2005.03.060. [DOI] [PubMed] [Google Scholar]
- 27.Oh KA, Yang GY, Jun D, Kuca K, Jung YS. Bioorg. Med. Chem. Lett. 2006;16:4852. doi: 10.1016/j.bmcl.2006.06.063. [DOI] [PubMed] [Google Scholar]
- 28.Yang GY, Oh KA, Park NJ, Jung YS. Bioorg. Med. Chem. 2007;15:7704. doi: 10.1016/j.bmc.2007.08.056. [DOI] [PubMed] [Google Scholar]
- 29.Musilek K, Kucera J, Jun D, Dohnal V, Opletalova V, Kuca K. Bioorg. Med. Chem. 2008;16:8218. doi: 10.1016/j.bmc.2008.07.036. [DOI] [PubMed] [Google Scholar]
- 30.Ohta H, Ohmori T, Suzuki S, Ikegaya H, Sakurada K, Takatori T. Pharmaceut. Res. 2006;23:2827. doi: 10.1007/s11095-006-9123-1. [DOI] [PubMed] [Google Scholar]
- 31.Picha J, Kuca K, Kivala M, Kohout M, Cabal J, Liska F. J. Enz. Inhib. Med. Chem. 2005;20:233. doi: 10.1080/14756360400021858. [DOI] [PubMed] [Google Scholar]
- 32.Bajgar J, Fusek J, Kuca K, Bartosova L, Jun D. Mini-Rev. Med. Chem. 2007;7:461. doi: 10.2174/138955707780619581. [DOI] [PubMed] [Google Scholar]
- 33.Luo C, Tong M, Chilukuri N, Brecht K, Maxwell DM, Saxena A. Biochemistry. 2007;46:11771. doi: 10.1021/bi701002f. [DOI] [PubMed] [Google Scholar]
- 34.Kuca K, Cabal J, Musilek K, Jun D, Bajgar J. J. Appl. Toxicol. 2005;25:491. doi: 10.1002/jat.1084. [DOI] [PubMed] [Google Scholar]
- 35.Typical procedure for synthesis of monoquaternary pyridinium salts 5–35: Hydroxyiminomethylpyridine (1 mmol) and the alkylating agent (1.5 mmol) in solvent (20 mL) were stirred at 60–80 °C for 1–24 h. The mixture was cooled to rt, the precipitate collected, washed with acetone (3 × 20 mL), dried under vacuum, and characterized by 1H NMR, 13C NMR, ESI-MS and IR. Representative compounds as follows: 1-Carboxymethyl-2-hydroxyimino methylpyridinium iodide (8): light green solid; yield: 61%; mp 138–140 °C; 1H NMR (400 MHz, CD3OD): δ 8.80 (d, J = 5.2 Hz, 1H), 8.65 (s, 1H), 8.58-8.42 (m, 2H), 8.01 (t, J = 8.0 Hz, 1H), 4.41 (s, 2H); 13C NMR (100 MHz, CD3OD): δ 169.11, 147.01, 146.53, 145.36, 141.25, 127.52, 125.99, 46.46; IR (Neat): νmax 3059, 3098, 3048, 2965, 2880, 2743, 1692, 1628, 1508, 1480, 1288, 1160, 1018 cm−1; ESI-MS: m/z 181.06 [M]+ (calcd for [C8H9N2O3]+ 181.06); 1-(2-Hydroxy)-ethyl-2-hydroxyimino methyl pyridinium bromide (11): brown solid; yield: 58%; mp 184–186 °C; 1H NMR (400 MHz, D2O): δ 8.64 (d, J = 6.4 Hz, 1H), 8.57 (s, 1H), 8.37 (t, J = 8.0 Hz, 1H), 8.26 (d, J = 8.4 Hz, 1H), 7.87 (t, J = 6.8 Hz, 1H), 4.71 (t, J = 4.4 Hz, 2H), 3.89 (t, J = 4.4 Hz, 2H); 13C NMR (100 MHz, D2O): δ 147.20, 146.38, 145.85, 142.49, 127.78, 127.03, 60.33, 60.07; IR (Neat): νmax 3377, 3072, 2991, 2865, 2734, 2620, 1627, 1590, 1504, 1430, 1313, 1152, 1072, 1000 cm−1; ESI-MS: m/z 167.08 [M]+ (calcd for [C8H11N2O2]+ 167.08); 1-(Thiophen-2-yl)-methyl-2-hydroxyiminomethyl-pyridinium chloride (20): off white solid; yield: 58%; mp 150–152 °C; 1H NMR (400 MHz, D2O): δ 8.73 (d, J = 6.4 Hz, 1H), 8.63 (s, 1H), 8.38 (t, J = 8.0 Hz, 1H), 8.20 (d, J = 8.0 Hz, 1H), 7.86 (t, J = 6.4 Hz, 1H), 7.40 (d, J = 5.2 Hz, 1H), 7.09 (d, J = 3.6 Hz, 1H), 6.94 (t, J = 4.0 Hz, 1H), 5.98 (s, 2H); 13C NMR (100 MHz, D2O): δ 146.15, 145.23, 142.28, 139.01, 133.52, 130.28, 129.20, 128.25, 127.94, 127.50, 56.61; IR (Neat): νmax 3069, 3013, 2945, 2831, 2735, 1711, 1628, 1577, 1514, 1474, 1321, 1253, 1020 cm−1; ESI-MS: m/z 219.21 [M]+ (calcd for [C11H11N2OS]+ 219.06)
- 36.Joshi HC, Ramachandran PK, Haksar CN. Curr. Sci. 1980;49:213. [Google Scholar]
-
37.In vitro reactivation screening - An AChE stock solution (0.2 mg/mL in PBS7.2) was treated with ethanol (0.1% v/v) and paraoxon (100 µM in ethanol, 0.1% v/v) as control and experiment vessels. After 20–50 min, 90% AChE inhibition was achieved and halted with a 32-fold dilution (PBS). A 16 µL aliquot from control and experiment vessels was diluted to 20 µL with PBS, and incubated for 30 min as activity control and inhibition control. The initial activity (A0) or inhibition (Ai) was analyzed by adding DTNB (final concentration of 0.3 mM, 200 µL total volume) and ATChI (final concentration of 1 mM, 200 µL total volume). Oxime reactivator (4 µL, 1 ~ 0.01 mM final conc) was added to a 16 µL aliquot and after 30 min incubation, the reactivated activity was determined by Ellman assay as Ar. The % reactivation for each reactivator was determined with n ≥ 3:
- 38.Ellman GL, Courtney KD, Andres VJ, Feather-Stone RM. Biochem. Pharmacol. 1961;7:88. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- 39.Determination of reactivation rate constant (kr) for selected compounds - Similar experiments as in vitro reactivation screening were accomplished with different reactivation time (0–15 min). By plotting ln(reactivation %-age) vs. reactivation time (t), kr is presented as the negative slope of the plot. ln(reactivation %-age) = −kr • t. Each experiment was repeated with n ≥ 3. Plots with R2 > 0.9 were chosen for kr calculations
- 40.Thompson CM, Ryu S, Berkman CE. J. Am. Chem. Soc. 1992;114:10710. [Google Scholar]
- 41.Kryger G, Harel M, Giles K, Toker L, Velan B, Lazar A, Kronman C, Barak D, Ariel N, Shafferman A, Silman I, Sussman JL. Acta Crystallogr.,Sect.D. 2000;56:1385. doi: 10.1107/s0907444900010659. [DOI] [PubMed] [Google Scholar]
- 42.Bourne Y, Grassi J, Bougis PE, Marchot P. J. Biol. Chem. 1999;274:30370. doi: 10.1074/jbc.274.43.30370. [DOI] [PubMed] [Google Scholar]
- 43.Kuca K, Cabal J, Kassa J. Journal of Enzyme Inhibition and Medicinal Chemistry. 2005;20:227. doi: 10.1080/14756360500043208. [DOI] [PubMed] [Google Scholar]
- 44.Worek F, Reiter G, Eyer P, Szinicz L. Archives of Toxicology. 2002;76:523. doi: 10.1007/s00204-002-0375-1. [DOI] [PubMed] [Google Scholar]