

Abstract
Expression of Migration inducting gene-7 (Mig-7) is limited to tumor cells and to date not found in normal tissues. Multiple tumor microenvironment factors, such as epidermal and hepatocyte growth factors, in concert with αvβ5 integrin ligation induce Mig-7 mRNA expression. Gain or loss of Mig-7 protein studies demonstrate that Mig-7 promotes invasion of colon and endometrial carcinoma cells. These data led us to hypothesize that targeting Mig-7 through various methods could decrease invasion, enhance monocyte cell killing of tumor cells and inhibit disease progression. To begin testing this hypothesis an in vitro chemoinvasion assay of endometrial carcinoma cells treated with Mig-7 specific or control antibodies was used. Mig-7 antibody significantly reduced invasion by >60% compared to controls. In another approach to test this hypothesis, an in vitro analysis of peptide-stimulated human peripheral blood monocyte cells (MC) and their killing of MCF-7 breast carcinoma cells was used. Mig-7 peptide treatment increased MC tumor necrosis factor expression and killing of MCF-7 cells 30-fold over no peptide stimulation and 3-fold over MUC-1 or control peptide treatments. Furthermore, stably expressing Mig-7-specific short hairpin RNA resulted in significantly reduced Mig-7 protein levels and early primary tumor growth in a xenograft nude mouse model. Reduced phosphorylation of ERK1/2, Akt, and S6 kinase as well as decreased membrane-type 1 matrix metalloproteinase activity were mechanisms through which Mig-7 protein caused these effects. Based on these collective data, Mig-7 expression could be a potential candidate for future targeted cancer therapies.
Keywords: immunomodulation, shRNA targeting
Introduction
Discovery and targeting of tumor cell-specific gene expressions could lead to more effective cancer treatments with less toxic side effects. Furthermore, targeting tumor cell proteins that facilitate their invasion, during which tumor cells are resistant to current therapies (1), could provide additional efficacy and less recurrence of disease. We have discovered one such human protein, Migration inducting gene-7 (Mig-7), that is regulated by tumor microenvironment factors (2;3;4;5).
Mig-7 protein is cysteine-rich and is primarily localized to the membrane fraction of carcinoma cells. Mig-7 expression, a result of receptor tyrosine kinase (RTK) activation, also requires αvβ5 integrin ligation (1,2) that acts in concert with RTK-activation causing tumor cell invasion and dissemination in vivo (6;7). Antisense to Mig-7, but not sense, oligonucleotide treatment inhibits carcinoma cell scattering (2). In previous studies to date, 87% of tumors from breast, endometrial, colon, lung, ovary, stomach, kidney, thyroid, cervix, small intestine, and prostate (n >200 patients), blood from untreated cancer patients, and metastatic sites possess cells expressing Mig-7 mRNA. Notable from these studies is that Mig-7 mRNA is not detectable in 25 different normal tissues (n=6 each tissue) or in blood from normal subjects (2;4).
Consistent with Mig-7 expression causing invasion, its cDNA is 99% homologous to expressed sequence tags isolated from early invasive stage placenta (2). During placental development, trophoblast cells from the implanted blastocyst invade through the endometrium and one third of the myometrium. These plastic cells can also mimic endothelial cells to remodel the maternal spiral arteries; a process that provides sufficient blood flow for fetal growth and development. Thus, the only normal cells found to date that express Mig-7 are trophoblast cells (3) that behave like aggressive tumor cells (8;9;10).
HT29 colon carcinoma cell Mig-7 expression induces invasion and vessel-like structure formation in three dimensional (3D) cultures (3). In addition, Mig-7 expression in these cells reduces their adhesion to laminin and increases production of laminin 5 γ2 chain promigratory fragments known to promote invasion and vessel-like structure formation by aggressive melanoma cells (11). Furthermore, knockdown of Mig-7 by stable shRNA expression in RL95 endometrial carcinoma cells causes reduced invasion in 3D cultures (2).
In vivo, tumor cells can line lumens of irregular vessels in multiple tumor types, including melanoma, ovarian carcinoma, Ewing sarcoma, and hepatocarcinoma (12;13;14;15). In three dimensional cultures, aggressive melanoma cells have been found to form these vessel-like structures, a process termed vasculogenic mimicry (16). In our nude mouse model of metastasis, both RL95 and HEC1A endometrial carcinoma cells that express Mig-7 localize to vessel-like structures formed by these invasive tumor cells (3). Adhesion assays to various components of the extracellular matrix suggest that a mechanism for Mig-7 in vessel formation by tumor cells is due, at least in part, to significantly lower adhesion to laminins, probably through facilitating cleavage of laminin 5 γ2 chain (3). Membrane tethered metalloproteinase, MT1-MMP, cleaves laminin 5 and is required for invasion, metastases and tumor growth in extracellular matrix (ECM) (17;18;19). These studies led us to hypothesize that targeting Mig-7 by multiple methods would decrease invasion, enhance immune cell killing of tumor cells and inhibit disease progression.
As an extension of previous Mig-7 cancer specificity studies, the present work shows that Mig-7 expression was detected in tumor cells of breast carcinoma and a subset of precancerous breast samples but not in cells of normal breast tissue samples. Several experimental systems were used to demonstrate, as proof of concept, that Mig-7 can be targeted. First, treatment with peptides specific to Mig-7 increased monocyte expression of tumor necrosis factor (TNF) and killing of breast carcinoma cells in vitro. Second, targeting Mig-7 with an antibody to its first nine amino acids reproducibly and significantly inhibited endometrial carcinoma cell invasion in vitro. Third, in vivo studies showed that shRNA decreased Mig-7 expression significantly impaired early tumor growth in an endometrial carcinoma cell xenograft nude mouse model. Active states of MT1-MMP (also known as MMP-14), ERK1/2, Akt, and S6 kinase were all reduced with Mig-7 targeting.
Methods
Cell cultures and transfections
Methods for constructing expression vectors, FLAGMig-7 and shRNA, as well as transfecting, selecting and culturing HT29 colon carcinoma, HEC1A, RL95 endometrial carcinoma (2;3;20), and MCF-7 breast carcinoma (21) cell lines were previously described. Mig-7 sequence (Accession DQ080207) of the previously unpublished shRNA construct insert, 4-2 antisense-loop-sense, is TCATTCACCTGCTATAGACTTCAAGAGAGTCTATAGCAGG-TGAATGA (bp 1303 to 1321). Under Institutional Review Board (IRB) approval, human monocyte cells (MC) from breast adenocarcinoma patients were isolated and cultured at 2 × 106 cells/ml in AIM-VR serum-free lymphocyte medium (Gibco, Invitrogen, Carlsbad, CA) as previously described (21).
Modified Boyden chamber invasion assay
Chemoinvasion assays were performed as previously described (22). Briefly, transwell filters (Costar, Corning, NY, 8 μm) were blocked in 1% bovine serum albumin (BSA)-DMEM/F12 for 30 minutes and rinsed with PBS. Matrigel (BD Biosciences, San Jose, CA) was diluted in ice cold PBS to 1 mg/mL to coat the lower side of each transwell insert. After incubating at 37° C for one hour, inserts were washed with PBS containing Ca2+ and Mg2+. HEC1A cells were detached using trypsin without EDTA, neutralized with soybean trypsin inhibitor, centrifuged for 5 minutes at 1000 rpm (4° C), and washed one time in DMEM/F12 media. Cell count and viability were determined using trypan blue exclusion and a hemocytometer. HEC1A cells were preincubated with 10 μg/mL affinity-purified anti-Mig-7 peptide (first nine amino acids) rabbit polyclonal antibody (3;4) or control normal rabbit IgG antibody (23) for 15 minutes in a 37° C, humidified incubator. For HEC1A cells, media containing 20 ng/ml of the chemoattractant, hepatocyte growth factor (HGF, R&D Systems Inc., Minneapolis, MN), was added to bottom wells. Media without HGF containing 50,000 cells was added to each top well. Cells were allowed to invade for 24 (HT29) or 72 (HEC1A) hours at 37° C in 5% CO2, 95% air humidified incubator. Then filters were rinsed with PBS and fixed in Hema3 fixative (Fisher Scientific, Inc., Pittsburgh, PA) for at least 30 minutes. Non-invaded cells in the upper chamber were removed with a cotton swab. Filters were dried and stained with Hema3 (Fisher Diagnostics, Middletown, VA) according to manufacturer's instructions. Filters were mounted on slides with gridded coverslips to count invaded cells using a microscope (Electron Microscopy Science, Hatfield, PA) at 400× magnification with a count of 10 squares (0.6 × 0.6 mm each) per filter from each treatment. Total numbers of invaded HT29 cells were counted and for HEC1A percent invasion was calculated by extrapolating the average cell count for the entire filter surface area and dividing by initial cell number. No cells were observed in bottom wells that could have invaded through the Matrigel. All treatments were performed in triplicate and experiments were repeated three times.
Apoptosis Assay
Apoptosis was assessed using the Vybrant Apoptosis Assay Kit #4 (Molecular Probes, Eugene, OR). For RL95 shRNA expressing cell lines, 1×106 cells were plated in triplicate on 6-well ultra-low attachment plates (Corning, Inc., Corning, NY) for 18 hours prior to analysis. For HEC1A cells, 1×106 cells were plated in 24-well plates and treated with Mig-7 antibody or rabbit IgG (10 μg/ml each) for 72 hours prior to analysis. Cells were trypsinized and centrifuged to pellet followed by washing once in PBS. After suspending in 250 μl of PBS, cells were transferred to 96-well plates. One μl of a 1:4 dilution of both YO-PRO-1 (100 μM) and propidium iodide (PI) (1 mg/ml) was added to cells and incubated on ice for 30 min. Cells left unstained, or stained with YO-PRO-1 or PI alone, were also included for controls. Listmode data were collected using a FACSCalibur (Becton Dickinson, Bedford, MA) flow cytometer and Winlist analysis software (Verity Software, Topsham, ME). Single-stained samples were used to perform compensation. Cells were gated to exclude large cell clumps and very small debris. YO-PRO-1 positive staining indicated apoptotic cells. YO-PRO-1 and PI double staining indicated dead cells. All cell lines or treatments were analyzed in triplicate and the experiments were performed twice with similar results.
Proliferation assay
Cell proliferation was assessed by using PI nuclear staining dye and flow cytometry as previously described (24). HEC1A cells were counted and 5×105 cells were plated in a 24-well plate and treated with IgG or Mig-7 antibodies as indicated above. For RL95 shRNA expressing cell lines, 5×105 cells were plated and grown for two days prior to collection. All cells were fixed in 1 ml of 70% ethanol overnight. After fixation, cells were centrifuged (470 × g, 5 min., 4° C) and the pellets washed once in 1X staining buffer (Dulbecco's PBS containing 2% FBS and 0.01% NaN3). Cells were then treated with 1 mg/ml RNase A (Sigma, St. Louis, MO) in PBS at 37° C for 30 min. After removing the RNase A solution, 300 μl of staining buffer containing 20 μg PI was added to resuspended cells and incubated 30 min. at room temperature. PI was not added to one sample as a negative control. After 30 min., PI staining was analyzed using the FACSCalibur (Becton Dickinson, Bedford, MA). Levels of PI staining correlated to the different cell cycle phases, and the numbers of cells in the G2/M phase (highest PI expression) were compared as an indicator of proliferation. Cells were gated to exclude large cell clumps and small debris. All cell lines or treatments were analyzed in triplicate and the experiments were performed twice with similar results.
α2-Macroglobulin capture assay
This assay was performed as previously described (25). Briefly, indicated cell lines were plated at confluency and treated as indicated in a 6-well plate on 1 mg/ml Matrigel for 18-20 hours before the assay. Cells were then removed by scraping and each sample was split into two wells of a 24-well plate. Purified human α2-macroglobulin (MP Biomedicals, Solon, OH) was added to one of the two wells for each sample at a concentration of 1 mg/ml and incubated at room temperature for 15 min. Cells without α2-macroglobulin served as control. Following incubation, lysis buffer [2% SDS, 60 mM Tris pH 6.8, 10% glycerol and 2X protease inhibitors (Complete, Roche, Indianapolis, IN)] was added, at a 1:2 final dilution, and immunoblots performed as detailed below. Assays were performed in triplicate for each cell line and treatment. Lysates from cells without α2 macroglobulin treatment were pooled for immunoblot analyses. Individual density background levels for each lane were subtracted from the band of interest density level then divided by respective tubulin band density level for densitometry calculations.
Protein phosphorylation analyses
For protein phosphorylation analyses, confluent RL95 shRNA-expressing cells were plated on a 1:10 dilution of Matrigel (10.5 mg/ml) for 19 hours prior to harvest. Confluent cells were trypinized, pelleted at 4° C at 1000 rpm, 4 min. and washed three times in media, followed by three washes in cold TBS. Multiprotein phosphorylation analysis using antibodies and Luminex Bead-based immunoassays was performed by AssayGate, Inc. (Ijamsville, MD). Briefly, indicated proteins in each cell lysate were determined quantitatively and simultaneously with a Bio-Plex 200 Bead Reader System. Antibodies were labeled with differing concentrations of two fluorophores to generate distinct bead sets. Each bead set was coated with capture antibody specific for each indicated total or phospho-protein. Captured analyte was detected using a biotinylated detection antibody and streptavidin-phycoerythrin (S-PE). Analyses were performed using a dual laser, flow-based, sorting and detection platform. One laser was bead-specific and determined a given antibody bound to protein. The other laser determined the magnitude of PE-derived signal, which is in direct proportion to the amount of protein bound. Protein concentrations of samples were determined by 5-parameter logistic regression algorithm with analysis of the median fluorescence intensity readings of an 8-point protein standard curve. Once a regression equation was derived, the fluorescence intensity values of the standards were treated as unknowns and the concentration of each standard was calculated. A ratio of the calculated value to the expected value of this standard was determined. A ratio between 70 and 130% for each of the standards indicated a good fit. Precision was evaluated by the coefficient of variation which equals the standard deviation divided by the mean. All samples indicated a good level of precision.
Antibodies to phosphorylation sites analyzed included Ser473 for Akt, Thr202 /Tyr204 and Thr185 /Tyr187 for extracellular signal-regulated kinase 1/2 (ERK1/2), Thr246 for proline-rich Akt substrate of 40 kDa (PRAS40), Thr421/Ser424 for S6 kinase, and Tyr1135 /Tyr1136 for insulin-like growth factor-1 receptor (IGF-1R). Antibodies to each protein were also used to detect total protein levels. Samples were tested in triplicate.
Immunoblotting
Cell lysates and immunoblots were performed as previously described (2;3) with the following modifications. Cultured cells were lysed in 2% SDS, 60 mM Tris, 10% glycerol containing 2X protease inhibitors (Complete, Roche, Indianapolis, IN) and quantitated using RC/DC Protein Assay (Bio-Rad, Hercules, CA). Lysates were boiled for 5 min. in the presence of 100 mM dithiothreitol and 0.01% bromophenol blue. Equal amounts of protein were loaded onto a 12% polyacrylamide gel and run at constant 200 V for 30-40 min. Gels were semi-dry transferred to polyvinylidene fluoride membranes and blocked in TBS-Tween (0.05%) containing 5% dry milk for one hour at room temperature. Endogenous Mig-7 protein was detected using previously described affinity-purified Mig-7 antibody (3) at 0.16 μg/ml in TBS-T. After extensive washings, horseradish peroxidase (HRP)-labeled secondary anti-rabbit IgG antibody (Zymed Laboratories, San Francisco, CA) was used to detect the Mig-7 antibody at 0.038 μg/ml in TBS-T. Mouse anti-β-tubulin (clone AA2, Upstate, Inc., Lake Placid, NY) was used at 0.2 μg/ml in TBS-T followed by extensive washing and incubation with HRP-labeled secondary anti-mouse IgG antibody (Cell Signalling, Danvers, MA). After stringent washings, Chemiluminescence Reagent (Amersham, Piscataway, NJ) allowed detection of HRP-labeled antibodies when exposed to film.
For α2-macroglobulin assay blots, lysates were not reduced or boiled before electrophoresis and transferring to nitrocellulose membrane. MT1-MMP (MMP-14) was detected using a goat anti-human MMP-14 antibody (R&D Systems, Inc.) at 0.1 μg/ml and an HRP-labeled secondary anti-goat IgG antibody (Zymed Laboratories, San Francisco, CA) at 0.038 μg/ml.
Xenograft nude mouse studies
Nude mouse studies were performed under Institutional Animal Care and Use Committee approval as previously described with modifications (3;4). Briefly, RL95 parental cells and cells stably transfected with shRNA expression vectors containing Mig-7 specific sequences 1-3, 3-1, or 4-2 G418 selected, pooled colonies were described previously (2;3). Cells were lifted off the plate using 1X trypsin-EDTA (Gibco, Invitrogen), followed by inhibition of trypsin using Defined Trypsin Inhibitor (Cascade Biologics, Portland, OR). Viable cells were counted using trypan blue exclusion and a hemocytometer. For each injection, shRNA 1-3, 3-1, or 4-2 expressing RL95 cell lines were suspended at 5×105 each in serum-containing media with 10 ng/ml EGF (Gibco, Invitrogen) and 1:2 diluted Matrigel (10.3 mg/ml, BD Biosciences) and injected subcutaneously into the dorsal neck region of nu/nu athymic mice (National Cancer Institute, Bethesda, MD). Negative controls were mice injected with Matrigel alone (no cells) and shRNA 3-1 RL95 cells that express Mig-7 at parental levels. Five animals were injected per cell line. Tumor size was measured with a caliper every 2-3 days and volume was calculated using (length × width2)/2 as previously described (26). Mice were euthanized after 4 weeks.
Immunohistochemistry
Breast core punch biopsies (10% formalin fixed, paraffin embedded, 5 μm) on tissue array slides were obtained from Cybrdi, Inc (Frederick, MD). Detection of Mig-7 protein was performed using Mig-7-specific affinity-purified antibody as previously described (3;4). Briefly, after deparaffinization and rehydration, antigen retrieval was performed for 2 seconds on ice in a microwave oven. Slides were washed two times in Dulbecco phosphate buffered saline (D-PBS) and then permeabilized with 0.01% digitonin (Sigma-Aldrich, St. Louis, MO) in PBS at room temperature for 30 minutes. After washing two times in PBS, slides were blocked in 10% horse serum (Gibco, Invitrogen) in D-PBS for 30 minutes at room temperature. Affinity-purified, polyclonal, rabbit anti-peptide (first nine amino acids of Mig-7) primary antibody (0.32 μg/μl), was diluted 1:50 and incubated on the tissue sections overnight at 4 °C. Slides were washed two times in PBS then incubated for 20 minutes in 3% H2O2 in methanol. After washing two times in PBS, slides were incubated for 30 minutes with secondary antibody, goat anti-rabbit IgG coupled to horseradish peroxidase (HRP), in PBS containing 0.5% BSA (Fisher Scientific). Slides were washed two times in PBS and then developed using 3,3′-diaminobenzidine (DAB) substrate (Vector Laboratories, Burlingame, CA) until brown specific staining was detected by microscopy (less than 3 minutes). After washing in water for 5 minutes, slides were counterstained in Hematoxylin QS (Vector Laboratories). Slides were dehydrated in 75%, 95%, and absolute ethanol, air dried, and coverslips mounted with DPX (BDH Laboratory Supplies, Poole, UK). Secondary antibody alone or normal rabbit IgG instead of Mig-7 antibody served as controls.
Images were taken on a Nikon Microphot microscope with a Retiga 2000R digital CCD camera.
RNA isolation and RT-PCR
Total RNA from normal breast tissue and from breast carcinoma cell lines were purchased from Ambion, Inc. (Austin, TX). Isolation of total RNA from MCF-7 breast carcinoma cells, DNasing, and RT-PCR was performed as previously described (2;3;4).
Peptides
Control (MAA SRC SGL YIV RND TSG; YIV RND TSG LSG SQW VDS; LSG SQW VDS PLK SPC QVW) and Mig-7 (RVH MRA CSA GSA YLK QMK; GSA YLK QMK FCR MAA SLD; FCR MAA SLD KVK KTD RGE RG) peptides (Accession DQ080207) were synthesized by Biosource, Inc. Previously characterized MUC-1 peptide (GNN APA HGV NNA PDN RPA P) (21) was synthesized by American Peptide Co., Inc. Peptides, 95% pure, were evaluated by mass spectrometry and solubilized in media.
Stimulation of human monocyte cells and MCF-7 killing assays
Stimulations were performed as previously described (21). Briefly, IL-2 (Cetus, Inc., Emeryville, CA) was added to isolated MC (see cell cultures) twice per week at 100 IU/ml on days 0 and 4. Cells were stimulated with 1 μg/ml of indicated peptides on days 0 and 7. MCF-7 cells (5 × 103 per well) were plated into 96-well tissue culture plates. Peptide-stimulated MC/effectors were added to each well in two effector cell to target cell ratios (E:T) 10:1 and 5:1. Cell lysis was evaluated on day 8 of peptide stimulation using a tetrazolium salt XTT assay (Roche, Inc.) as previously described (27). Treatments were in replicates of 6 and each experiment was performed at least twice.
Formation of formazan in XTT assays indicates viable cells. Formazan formed by target (MCF-7 cells) alone, MC/effector MC alone, or background (no cells) was determined as the mean of six wells each. The percent specific lysis (%SL) was calculated as previously described (21):
ELISA Cytokine Assay
TNF-α concentration in media from each peptide treated monocyte cell culture were determined using the human BD OptEIA™ cytokine assay that is a solid phase sandwich Enzyme-Linked Immunosorbent Assay (ELISA) (BD Biosciences), according to manufacturer's instructions. Briefly, stimulated monocyte culture supernatant was added to TNF-α antibody coated wells and incubated for two hr. After washing, captured TNF was detected with a streptavidin-HRP conjugate mixed with a biotinylated anti-human TNF-α antibody. After washing, 3,3′,5,5′-tetramethylbenzidine substrate was added and the reaction was stopped after development. Amounts of TNF-α were determined by measuring absorbance at 450 nm and comparison with a standard curve. The background of empty wells was subtracted before statistical analyses. Experiments were performed in replicates of six for each peptide treated monocyte experiment.
Statistical Analysis
Statistical significance of the in vitro cytotoxicity assays and of cytokine assays were determined by the Mann-Whitney Rank Sum test. Data from HEC1A invasion assays and nude mouse xenograft assays were statistically analyzed by One-Way ANOVA and Tukey-Kramer post test and considered significant at p ≤ 0.05. Data from HT29 invasion assays, signaling phosphorylation studies, and α2-macroglobulin capture by MT1-MMP densitometry analyses were evaluated by a paired t-test and considered significant at p < 0.05.
Results
Antibody to Mig-7 treatment of HEC1A cells results in decreased invasion in vitro
A method to quantitatively assess tumor cell invasion is the transwell chemoinvasion assay (22). The effect of amino terminus FLAG-tagged Mig-7 (FLAGMig-7) overexpression in stably transfected HT29 colon carcinoma cells was quantitatively analyzed compared to vector alone transfected cells. We chose this cell line because it does not endogenously express Mig-7 and it stably overexpresses FLAGMig-7 at the protein level (2;3;20). Overexpression in HT29 cells significantly increased the total number of invaded cells by 8-fold (p = 0.002) over control (Figure 1a).
Figure 1.
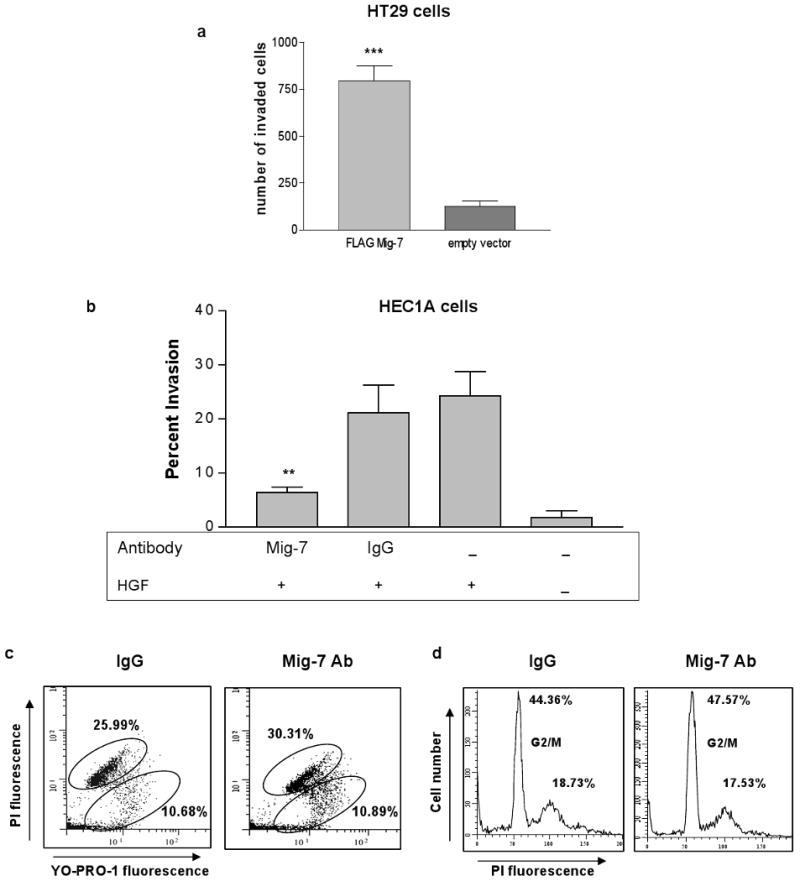
Mig-7 specific antibody decreases HEC1A endometrial carcinoma cell chemoinvasion. (a) Overexpression of amino terminus FLAG-tagged Mig-7 significantly increased Matrigel invasion of HT29 cells compared to vector alone stably transfected cells. Bars represent SEM. Results are after 24 hours of invasion performed in quadruplicate. (b) Mig-7 affinity-purified antibody, but not IgG rabbit isotype antibody, significantly decreased chemoinvasion of HEC1A cells into Matrigel toward HGF (p<0.01). Ab = antibody, + HGF = bottom wells contained 20 ng/ml HGF. Results shown are after 72 hours of invasion and are representative of three independent experiments performed in triplicate. Bars represent SEM. (c) Representative flow cytometry histograms of control normal rabbit IgG and Mig-7 antibody-treated HEC1A cells stained with YO-PRO-1 and propidium iodide (PI). YO-PRO-1 single positive cells (lower oval) are apoptotic, and double positive cells (upper oval) are dead. Mean percentages of three samples are shown. (d) Representative flow cytometry histograms of control IgG and Mig-7 antibody-treated HEC1A cells stained with PI. Cells in G2/M phase are indicated on histograms. Mean percentages of three samples are shown. Flow cytometry experiments were conducted twice with similar results.
HGF, the growth factor used to isolate Mig-7 (2), acts as a chemoattractant of invasive HEC1A cells in transwell in vitro assays (28). Therefore, we used this cell line, previously shown to express Mig-7 (2;3), treated with affinity-purified antibody to Mig-7 peptide representing its first nine amino acids to determine if this antibody could inhibit cell invasion.
Mig-7 antibody treatment of HEC1A cells significantly decreased the average percentage of invaded cells counted as described in Methods. Chemoinvasion toward HGF by HEC1A cells was significantly (p = 0.0046) inhibited by 70% when treated with Mig-7 antibody and compared to normal rabbit IgG antibody treated cells (Figure 1b). Furthermore, treatment with normal rabbit IgG antibody did not significantly inhibit HEC1A cell chemoinvasion when compared to no antibody treatment. No HGF chemoattractant reduced invasion of HEC1A cells by >90% (Figure 1b). Flow cytometric analysis with the apoptotic dye YO-PRO-1 and propidium iodide (PI) showed no significant increase in apoptosis due to Mig-7 antibody compared to normal rabbit IgG antibody treatment (Figure 1c). In addition, flow cytometric cell cycle analysis showed no significant decrease in cell proliferation of Mig-7 antibody-treated cells compared to untreated and control-treated cells (Figure 1d).
Antibody or expression of shRNA specific to Mig-7 decreases activity of MT1-MMP
MT1-MMP (17;18) activity is required for tumor cell invasion and Mig-7 expression significantly increases this activity (2;3). Mig-7 protein is primarily membrane-localized and is cysteine-rich (1). A free thiol group on one of the many cysteine residues in membrane-localized Mig-7 could activate MT1-MMP without cleavage via the “cysteine switch” (29;30). Therefore, we used the α2-macroglobulin capture assay to test for MT1-MMP activation. In addition, this test was used instead of zymography because the SDS in gel zymography activates the “cysteine switch” (31).
HEC1A cells treated with Mig-7 antibody showed a significant 70% decrease in the levels of activated MT1-MMP, as indicated by the upper, α2 macroglobulin captured band (∼190 kD), compared to cells treated with IgG control. The lower uncaptured, unactivated MT1-MMP band was 62 kD as indicated. Levels of activated MT1-MMP were determined by densitometry normalized with tubulin levels for each sample (Figure 2a).
Figure 2.
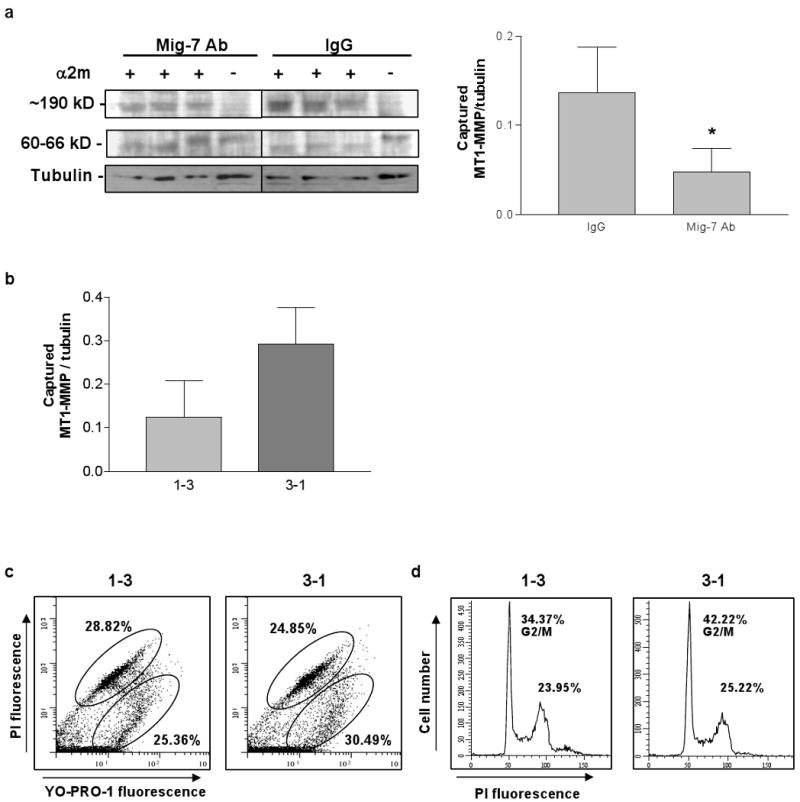
Antibody or expression of shRNA specific to Mig-7 decreases activity of MT1-MMP. (a) Representative immunoblot and densitometry results for α2-macroglobulin capture assay of active MT1-MMP in lysates from equal numbers of HEC1A cells treated with normal rabbit IgG or with Mig-7 antibody. All samples were run on the same gel. Mig-7 antibody treatment significantly decreased MT1-MMP activity by 70% compared to IgG treatment (p=0.0496). Densitometry data are for the upper, captured MT1-MMP band in each lane normalized to its respective tubulin band. Bars represent SEM. (b) Densitometry analysis of the upper, captured MT1-MMP band from α2-macroglobulin capture assay in lysates from RL95 cells stably transfected with shRNA 1-3 (decreased Mig-7 expression) and 3-1 (control, endogenous levels of Mig-7 expression). Results are normalized to β-tubulin. RL95 cells expressing shRNA 1-3 showed a 57% decrease in MT1-MMP activity compared to 3-1 control cells. Bars represent SEM. (c) Representative flow cytometry histograms of RL95 shRNA 1-3 and 3-1 stably transfected cells stained with YO-PRO-1 and propidium iodide (PI). YO-PRO-1 single positive cells (lower oval) were apoptotic, and double positive cells (upper oval) were dead. Mean percentages of three samples are shown. (d) Representative flow cytometry histograms of RL95 shRNA 1-3 and 3-1 stably transfected cells stained with propidium iodide for cell cycle analysis. Cells in G2/M phase are indicated on histograms. Mean percentages of three samples are shown. Flow cytometry experiments were conducted twice with similar results.
Given that Mig-7 antibody treatment reduced levels of active MT1-MMP, we next tested whether stable knockdown of Mig-7 expression in RL95 cells could inhibit MT1-MMP activation. In this study, we used previously characterized RL95 endometrial carcinoma cells stably expressing shRNA constructs 1-3 and 3-1. In that study, the 1-3 construct expression significantly inhibits Mig-7 protein levels by at least 50% compared to the 3-1 control construct expression that does not significantly reduce the Mig-7 protein levels (3). In Figure 2b, α2-macroglobulin capture assays revealed that 1-3 shRNA RL95 cells decreased levels of active MT1-MMP by 57% compared to control (2). Analysis of these cell lines by flow cytometry for apoptosis and proliferation showed no significant differences between 1-3 and control cell lines (Figure 2c,d).
Expression of shRNA specific to Mig-7 decreases phosphorylation of ERK1/2, Akt, and S6 kinase
In order to further define potential mechanisms by which Mig-7 exerts its effects on invasion, a multiplex protein phosphorylation analysis was utilized to determine changes in protein activation. Phosphorylation status of proline-rich Akt substrate of 40 kD (PRAS40), ribosomal protein S6 kinase, extracellular signal-regulated kinase1/2 (ERK1/2), insulin-like growth factor-1 receptor (IGF-1R) and Akt (also known as protein kinase B) were analyzed as described in Methods. In cell lysates from RL95 cells with decreased Mig-7 protein levels due to stable shRNA 1-3 expression (2) phosphorylation of AKT Ser473, ERK1/2 Thr202 /Tyr204, Thr185 /Tyr187, and S6 kinase Thr421/Ser424 were significantly reduced by 10, 40, and 30%, respectively, as compared to control (Figure 3a). No significant differences in phosphorylation between these two RL95 cell lines were detected for PRAS40 or for IGF-1R (Figure 3b).
Figure 3.

Expression of shRNA specific to Mig-7 decreases S6 kinase, ERK1/2 and Akt phosphorylation in RL95 cells. (a) Normalized fluorescence intensity from analysis of phosphorylated ERK1/2, Akt and S6 kinase in RL95 shRNA 1-3 (knockdown) and 3-1 (control) stably transfected cells. Cells were plated and prepared as given in Methods. 1-3 shRNA expressing cells in which levels of Mig-7 are significantly reduced by >50% (2) showed significant phosphorylation decreases in ERK1/2 by 40% (p = 0.002), in Akt by 10% (p = 0.0449), and in S6 kinase by 30% (p = 0.0155) compared to control RL95 cells that express levels of Mig-7 similar to the RL95 parental cell line. (b) Normalized fluorescence intensities from analysis of phosphorylated PRAS40 and IGF-1R. All phosphorylation fluorescence intensities were normalized to total respective protein fluorescence intensities. These experiments were performed in triplicate.
Stimulation of human peripheral blood monocytes (MC) with peptides specific to Mig-7 increases TNF expression and killing of MCF-7 breast carcinoma cells in vitro
Cancer immunotherapies include ex vivo stimulation of cancer patients' immune cells with tumor antigens. We hypothesized that Mig-7 peptides could stimulate breast cancer patients' MC to increase their killing of breast carcinoma cells in vitro. To test this hypothesis, we utilized our previously described method of MC isolated from two different breast cancer patients and the MCF-7 breast carcinoma cell line (32;21). Because Mig-7 expression induces invasion (3), we wanted a cell line that lacks invasive properties to test if Mig-7 could still be used as a target in such a model cell line. The MCF-7 cell line is lowly invasive due to deficiency of MT1-MMP and MMP-2 (33). In addition, MCF-7 cells express αvβ5 integrin (34;35) that is required for Mig-7 expression (2). Further, our use of MCF-7 cells in the current study was warranted by the fact that this experimental system is optimized with this cell line (21;32), and that MCF-7 cells expressed Mig-7 mRNA and protein (Figure 4a).
Figure 4.
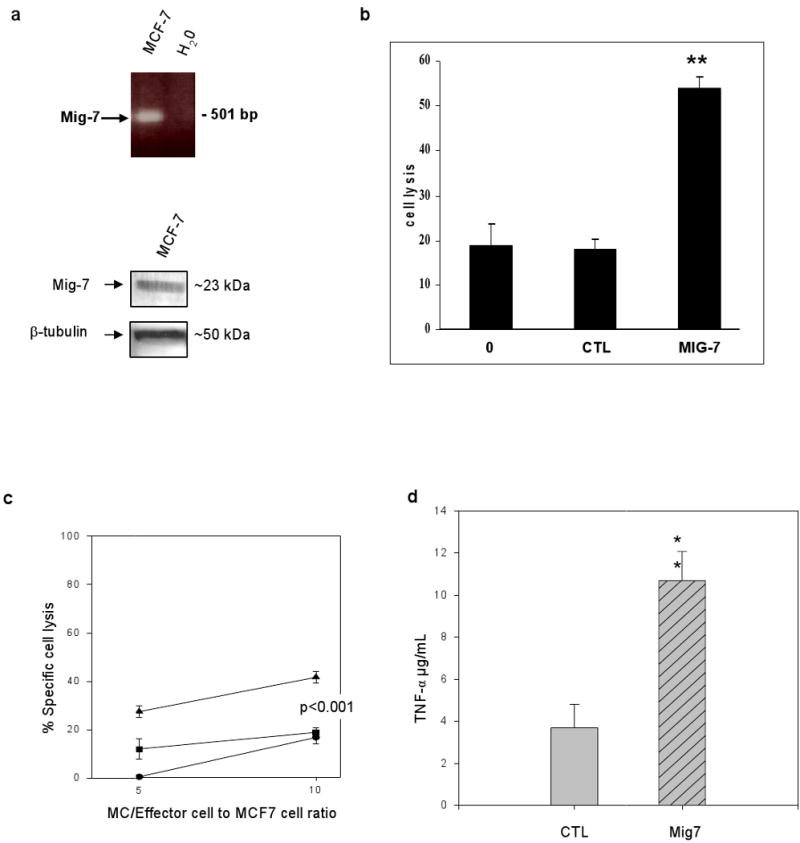
Mig-7 peptides enhance human monocyte killing of MCF-7 breast carcinoma cells. (a) MCF-7 cells expressed Mig-7 mRNA by RT-PCR and protein by immunoblot analyses. Experiments were repeated twice. (b) Human MC cells stimulated with IL-2 and either MUC-1 peptide (0), pooled control peptides (CTL), or pooled Mig-7 peptides (sequences given in Methods). MC/effector cell lysis of MCF-7 cells as described in Methods. Note that Mig-7 peptides significantly (p < 0.001) enhanced MC/effector killing of MCF-7 carcinoma cells. Experiments have been repeated 3 times in replicates of 6 for each treatment group. MCs were isolated from two different individuals. (c) MC after stimulations to effector cells ratio to MCF-7 cells and respective percentages of MCF-7 cell specific lysis.  represents no peptide,
represents no peptide,  represents MUC-1 peptide and
represents MUC-1 peptide and  represents MUC-1 plus Mig-7 peptides treatment groups. Bars indicate ± SEM. Experiments were repeated twice with replicates of six for each treatment group. (d) TNF-α production by human isolated MC after indicated peptide stimulation. TNF-α production was measured by ELISA assay as described in Methods after MC were cultured for 8 days with control peptides or with Mig-7 peptides. Assays were performed in replicates of six in three independent experiments with identical results.
represents MUC-1 plus Mig-7 peptides treatment groups. Bars indicate ± SEM. Experiments were repeated twice with replicates of six for each treatment group. (d) TNF-α production by human isolated MC after indicated peptide stimulation. TNF-α production was measured by ELISA assay as described in Methods after MC were cultured for 8 days with control peptides or with Mig-7 peptides. Assays were performed in replicates of six in three independent experiments with identical results.
Mig-7 peptides representing the +1 frameshifted protein sequence or control peptides representing the sequence in the non-coding reading frame, i.e. the frame that did not produce protein (20), were used to stimulate MC in vitro. In addition, these peptides included overlapping sequences (see Methods) to prevent the possible omission of an epitope. None of the peptides were significantly homologous to any other sequence than the Mig-7 banked sequence in databases available through the National Center for Biotechnology Information. MUC-1 peptide served as an internal control because of its previous use and optimization in this assay. These previous studies of stimulating MC cells with MUC-1 peptide and IL-2 support the presentation of peptide by antigen-presenting cells that stimulate cytotoxic T lymphocytes in this heterogeneous cell population (21;32).
Stimulation with Mig-7 peptides significantly enhanced MC/effector killing of MCF-7 cells by 3-fold over MUC-1 peptide alone (0) or control (CTL) peptides. There was no significant difference between control peptides and MUC-1 peptide alone treatment groups (Figure 4b). Figure 4c demonstrates that a ratio of 10:1 MC/effector:MCF-7 cells resulted in no difference between no peptide and MUC-1-stimulated MC/effector killing of MCF-7 cells. In contrast, at this ratio Mig-7 with MUC-1 peptides significantly increased MC/effector killing of MCF-7 cells >2-fold over no peptide or MUC-1-stimulated cells. Also at this higher MC/effector cell ratio, IL-2, that enhances cytotoxic activity of T and natural killer cells (36;21), treatment alone of MC/effectors increased their level of killing to that detected for cells stimulated with IL-2 plus MUC-1 peptide. At a 5:1 ratio, Mig-7 with MUC-1 peptides stimulation of MC significantly enhanced their killing of MCF-7 cells 1.9-fold over MUC-1-peptide stimulation and at least 30-fold over no peptide stimulation (Figure 4c). Mig-7 peptide stimulation also significantly increased levels of MC/effector-produced TNF-α >3-fold over control peptide stimulated cells as determined by ELISA (Figure 4d).
Mig-7 expression is specific to breast carcinoma and not to normal breast
To date, Mig-7 expression is detected in multiple types of tumor cells, but not in normal cells (2;3;4). However, normal breast cells were absent in our previous studies. Here, we extend our analyses of Mig-7 specificity with analyses of cells in breast tissues of pre-cancerous states and carcinomas compared to normal breast tissues. First, relative RT-PCR was used as previously described (2;3;4) to analyze commercially available total RNA from three breast carcinoma cell lines, T47D, MDA-MB453, DU4475, and from normal breast tissue from three subjects with no previous history of cancer. No amplification product specific to Mig-7 was detected in normal breast tissue RNA samples. However, all three breast carcinoma cell lines expressed Mig-7 mRNA (Figure 5a).
Figure 5.
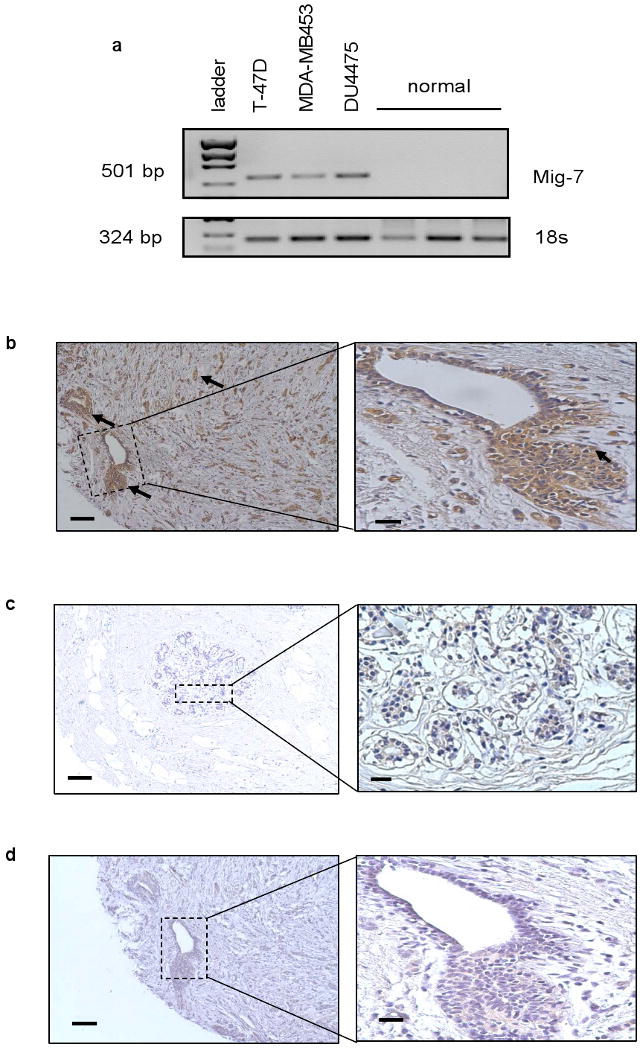
Mig-7 expression is specific to breast carcinoma tissue and cell lines. (a) Representative Mig-7 specific relative RT-PCR of three breast carcinoma cell lines (T47D, MDA-MB453, DU4475) and commercially available RNA from normal breast tissue of three different human subjects without previous histories of cancer. (b-d) Representative images of Mig-7 antibody IHC on breast tissue array from Cybrdi, Inc. as described in Methods. Core samples from (b) breast carcinoma. Arrows point to positive Mig-7 staining. (c) Representative normal breast tissue IHC with Mig-7 antibody, and (d) Representative image of control normal rabbit IgG instead of Mig-7 as primary antibody IHC of breast carcinoma tissue section (serial section to that shown in b). Hematoxylin was used to counterstain. Note a lack of specific staining in d compared to b. Images were taken at 100× magnification with inserts at 400× magnification. Scale bars indicate 100 μm for 100× images and 20 μm for inserts.
Immunohistochemistry was performed on 70 human breast tissue arrayed core samples of normal, adenosis, papillomatosis, and carcinoma. Our results showed that primarily carcinoma tissues (Figure 5b) stained positive for Mig-7, whereas normal breast tissues lacked specific Mig-7 staining (Figure 5c). No staining was detected with normal rabbit IgG antibody in breast carcinoma tissue serial section to that in Figure 5b (Figure 5d). A summary of the overall pathology report of Mig-7 staining for all 70 samples is as follows: normal: 0 positive, 3 negative; breast adenosis: 5 positive, 23 negative; breast papillomatosis: 0 positive, 3 negative; breast carcinoma: 19 positive, 17 negative. Pearson Chi-square analysis with 3 degrees of freedom to test goodness of Mig-7 fit for detection of breast carcinoma was statistically significant (p = 0.008).
Stable knockdown of Mig-7 expression reduces early primary tumor growth in vivo
Mig-7 induces HT29 colon carcinoma cell invasion in three-dimensional cultures (3) and MT1-MMP proteolysis enables tumor cells to circumvent three-dimensional extracellular matrix growth restraint (19). These data led us to test the effect of stable Mig-7 knockdown, through shRNA expression, on primary tumor growth in vivo.
Three RL95 endometrial carcinoma cell lines stably transfected with different Mig-7 shRNA containing expression plasmids, 1-3, 4-2, and negative control 3-1 were each injected subcutaneously into nude mice and tumor growth measured as described in Methods. We confirmed our previous results (3) that cells expressing shRNA 1-3 have reduced Mig-7 protein levels compared to parental and 3-1 expressing cells by Western blot (Figure 6a). 1-3 expressing cells showed an approximate 50-60% decreased Mig-7 protein levels compared to 3-1 expressing cells that in previous studies show levels of Mig-7 protein similar to parental RL95 cell line (2). In addition, expression of a previously unpublished Mig-7 shRNA construct, 4-2, reduced Mig-7 protein levels by amounts similar to 1-3 expressing cells (Figure 6a).
Figure 6.
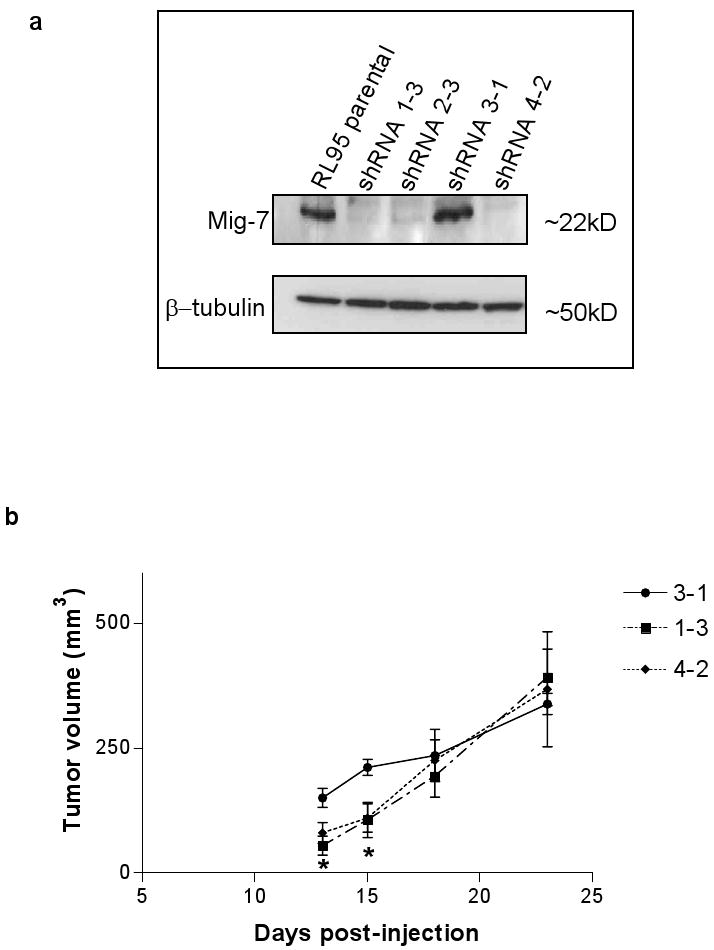
Stable knockdown of Mig-7 expression in RL95 cells decreases early primary tumor growth in nude mice. (a) Representative immunoblot of equal amounts of protein lysates from RL95 parental and Mig-7-specific shRNA stably transfected pooled clones 1-3, 3-1, and 4-2 cell lines. The top panel is after probing with Mig-7 antibody. Blot was reprobed with tubulin antibody to confirm equal loading and transfer. Statistical analyses of 1-3, 3-1 and parental Mig-7 knockdown and sequences of specific shRNAs were shown previously (2) and 4-2 levels of Mig7 protein were equivalent to those found in 1-3 shRNA knockdown of Mig-7 protein. See Methods for 4-2 shRNA sequence. (b) Representative graph showing tumor volumes (mm3) measured 13, 15, 18, and 23 days after injection of Matrigel containing RL95 cell lines stably transfected with shRNA constructs 1-3, 4-2, or 3-1 (control) into nude mice (five animals per cell line, as described in Methods). Note that cells expressing shRNAs 1-3 or 4-2, that significantly knockdown Mig-7 protein levels, showed significant 60% and 40-50% (p<0.05) decreased tumor volume, respectively, at days 13 and 15 of tumor growth. Bars represent SEM. Anova with Tukey's Multiple Comparison Test was used for statistical analyses. This experiment was performed twice with similar results.
Upon measuring (see Methods) primary tumors of all five mice for each cell line, RL95 cells expressing shRNA 1-3 and 4-2 showed significant (p < 0.05) decreased tumor volume, 60% and 40-50%, respectively, at 13 and 15 days after injection compared to mice injected with control cells expressing shRNA 3-1 (Figure 6b). While tumor volume trended lower for shRNA expressing cells at days 18 and 23 this was not significantly different than control cell line growth (Figure 6b). Mice injected with Matrigel alone showed no tumor formation across the entire time period (data not shown).
Discussion
Targeting Mig-7 protein with an antibody to its amino terminus nine amino acids significantly decreased HEC1A endometrial carcinoma cell chemoinvasion and MT1-MMP activation. Compared to controls, stimulating breast cancer patients' isolated MC with Mig-7 specific peptides increased their killing of MCF-7 breast carcinoma cells as well as MC production of TNF-α in vitro. In addition, specific shRNA knockdown of Mig-7 protein levels in RL95 cells decreased MT1-MMP, ERK1/2, Akt and S6 kinase active states in vitro as well as early tumor growth in vivo. These results are consistent with our previous reports that show Mig-7 contributes to the invasive phenotype of carcinoma cells (2;3).
Immunohistochemical and RT-PCR data in the present study further demonstrate the specificity of Mig-7 expression to breast carcinoma tissue and not to normal breast tissues. These data are not surprising because even with 40 cycles of PCR Mig-7 mRNA is not detected in 25 different normal human tissues (n=6 each tissue) or in blood from normal subjects (2;4). This specificity of Mig-7 expression may be due to multiple tumor microenvironment factors such as ligation of αvβ5 integrin and multiple growth factors leading to Mig-7 transcription (2;3) as well as multiple events required for Mig-7 translation (20). The percentage of total breast carcinoma tissues staining for Mig-7 was 53%. This result may be due to the fact that only a small core of each tumor tissue is represented on these tissue microarrays and the invasive front of carcinoma cells may not be represented in all of these core samples. Our previous data, however, show that 98% of cDNA from breast carcinoma tissue samples (n=50) contain moderate to high levels of Mig-7 (4). Mig-7 is also expressed in a subset of human breast adenosis samples with hyperplasia, which is considered “pre-cancerous” tissue (37). A similar subset of pre-cancerous endometrial lesions are positive for Mig-7 (4). These data further support our previous findings (2;4) that Mig-7 expression is specific to tumor cells cells and not found in normal adult cells.
Mig-7 antibody significantly inhibited invasion of endometrial carcinoma cells in vitro, suggesting that treatment with Mig-7 antibody in vivo may inhibit invasion. Previous studies show that antibody to CXCR4, a chemokine receptor, inhibits invasion of carcinoma cells (38). Further studies are needed to determine the effects of Mig-7 specific antibodies on tumor progression in vivo.
MT1-MMP activity causes activation of MMP-2 (gelatinase A) (39), cleavage of laminin-5 (17), increased invasion (40) and is required for tumor cell vasculogenic mimicry (11). Thus, because Mig-7 expression significantly increases invasion, decreases adhesion to laminins, increases laminin 5 γ2 domain 3 fragmentation and appears to play a role in vessel-like structure formation by endometrial carcinoma cells (3), we hypothesized that Mig-7 expression facilitates MT1-MMP activation. Data from the present studies show that inhibiting Mig-7 function by antibody treatment or expression by Mig-7 shRNA leads to decreased levels of activated MT1-MMP, suggesting a mechanism by which Mig-7 promotes the above cellular events. MT1-MMP can be activated through the “cysteine switch” mechanism (29;30) and Mig-7 is highly cysteine-rich (2;20). Additional studies beyond the scope of this work are needed to determine if one or more Mig-7 cysteine residues play a role in MT1-MMP activation.
Consistent with Mig-7 facilitating activation of MT1-MMP and invasion are data showing that Mig-7 expression leads to increased levels of phosphorylated ERK1/2, Akt, and S6 kinase. MT1-MMP catalytic activity or binding to tissue inhibitor of metalloproteinases-2 (TIMP-2) without proteolytic activity induces ERK1/2 phosphorylation (41;42). Other signaling molecules analyzed in the present study, IGF-1R and PRAS40, which are known to play roles in tumor progression (6;43), showed no decrease in phosphorylation due to reduced Mig-7 protein levels which further supports specific effects of Mig-7 expression on ERK1/2, Akt, and S6 kinase signaling molecules.
ERK1/2 and Akt phosphorylation signals invasion downstream of RTK activation and αv integrin ligation (44;45;46). RTK activation by EGF or HGF in concert with αvβ5 ligation also induce Mig-7 expression (2;3). Similarly, S6 kinase activation downstream of HGF, ERK1/2 and Akt signaling is important for tumor cell migration (47). Not surprisingly, activation of ERK1/2 and Akt downstream of RTK signaling facilitates MT1-MMP activation (48;49;50). Taken together, these data suggest that Mig-7 is an intermediate signaling protein downstream of RTK activation and αvβ5 integrin ligation (2), and upstream of MT1-MMP, ERK1/2, Akt as well as S6 kinase.
Enhanced killing of non invasive, MT1-MMP negative, Mig-7 expressing MCF-7 breast carcinoma cells by monocytes stimulated with IL-2 and Mig-7-specific peptides suggests that Mig-7 is a tumor cell antigen. In addition, these experiments show that Mig-7 peptides combined with MUC-1 peptides are superior in stimulating MC to kill MCF-7 cells over MUC-1 peptide or control peptides. Furthermore, Mig-7 peptide stimulation of monocytes enhanced production of TNF-α, a cytokine known to cause tumor cell death (51). This is probably the mechanism of cell killing in our assay because MCF-7 cells are responsive to TNF-α (52). These data suggest that Mig-7 peptides could be used ex vivo to stimulate MC/effector cells and test their tumor cell killing in vivo.
Inhibition of Mig-7 expression by stable Mig-7-specific shRNA expression significantly reduced early tumor growth in vivo. With two different Mig-7 knockdown RL95 cell lines (1-3 and 4-2), significant 40-60% decreased tumor growth compared to control was measured at days 13 and 15 after injection. This result is likely due to decreased invasion and decreased MT1-MMP proteolytic activity from reduced Mig-7 expression. In support of this conclusion, MCF-7 cells expressing MT1-MMP with a mutation E240A rendering it proteolytically inactive produces tumors 50% the size of wild type MT1-MMP (42). In our previous studies, HT29 colon carcinoma cells overexpressing FLAG-tagged Mig-7 or RL95 parental cells strictly invade Matrigel ECM during the first ten days of three dimensional culture (3). This probably also occurred with the current in vivo studies when RL95 cell lines were combined with Matrigel and injected subcutaneously. We propose that the RL95 3-1 cell line, expressing parental levels of Mig-7 protein, efficiently invades the Matrigel while knockdown cell lines (1-3 and 4-2) poorly invade the Matrigel in vivo as we have previously observed in Matrigel three dimensional cultures (3). This lack of invasion could inhibit proliferation due to ECM constraint at least at these earlier time points.
MT1-MMP activity was reduced in 1-3 expressing RL95 cells as compared to 3-1 cells that express Mig-7 protein at parental cell line levels. In another report, three different cell lines, SCC-1, Panc-1 and HT-1080, MT1-MMP confers these cell lines with a significant growth advantage in three dimensional cultures in vitro and in vivo (19). This advantage requires MT1-MMP activity and proteolysis to release the constraints of the surrounding ECM (19). Primary tumor sizes in the present study were not significantly different at days 18 and 23 post injection. Consistent with these data Mig-7 expression caused no difference in proliferation or apoptosis in these cell lines in vitro when plated on Matrigel. One explanation for these later in vivo time points is that MT1-MMP binds to TIMP-2 that induces MAPK and ERK1/2 activation as well as cell growth without proteolysis in vivo (42). Alternatively, Matrigel may not be stable at these later time points and therefore its constraint on proliferation may be absent. We propose that this may have occurred resulting in more rapid growth in the knockdown cell line tumors without invasion at these later time points.
Based on these data and because Mig-7 facilitates invasion in 3D cultures (2), it is likely that reduction of Mig-7 expression in RL95 cells slows early primary tumor growth by inhibiting local tumor cell invasion in Matrigel. Different models of tumor growth and metastases such as an orthotopic model may provide additional information on how Mig-7 expression impacts disease progression. Future studies beyond the scope of this present work are required to determine if Mig-7 plays a role in MT1-MMP tyrosine573 phosphorylation that is required for this growth advantage in ECM (53) or if ERK1/2 phosphorylation is affected by Mig-7 activation of a non proteolytic MT1-MMP (42) in the presence or absence of TIMP-2 at physiological levels.
Acknowledgments
This research was funded by NIH grant CA93925 (to J.S.L.), the Laura W. Bush Institute for Women's Health (to S.E.W. & J.S.L.) and Ladies Auxiliary Dept Texas Veterans of Foreign Wars (to S.E.W.).
Abbreviations
- Mig-7
Migration inducting gene 7
- shRNA
short hairpin RNA
- RTK
receptor tyrosine kinase
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- IGF-1R
insulin-like growth factor-1 receptor
- PRAS40
proline-rich Akt substrate of 40 kDa
- ERK1/2
extracellular signal-regulated kinase 1/2
- MC
monocyte cells
- BSA
bovine serum albumin
- HGF
hepatocyte growth factor
- IL-2
interleukin-2
- TNF
tumor necrosis factor, IFN-γ, interferon-gamma
- ELISA
enzyme-linked immunosorbent assay
- TBS
tris-buffered saline
- HRP
horseradish peroxidase
- DAB
3,3′-diaminobenzidine
- PI
propidium iodide
References
- 1.Condeelis J, Singer RH, Segall JE. The great escape: When cancer cells hijack the genes for chemotaxis and motility. Annu Rev Cell Dev Biol. 2005;21:695–718. doi: 10.1146/annurev.cellbio.21.122303.120306. [DOI] [PubMed] [Google Scholar]
- 2.Crouch S, Spidel CS, Lindsey JS. HGF and ligation of αvβ5 integrin induce a novel, cancer cell-specific gene expression required for cell scattering. Exp Cell Res. 2004;292:274–287. doi: 10.1016/j.yexcr.2003.09.016. [DOI] [PubMed] [Google Scholar]
- 3.Petty AP, Garman KL, Winn VD, Spidel CM, Lindsey JS. Overexpression of carcinoma and embryonic cytotrophoblast cell-specific Mig-7 induces invasion and vessel-like structure formation. Am J Pathol. 2007;170:1763–1780. doi: 10.2353/ajpath.2007.060969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Phillips TM, Lindsey JS. Carcinoma cell-specific Mig-7: A new potential marker for circulating and migrating cancer cells. Oncol Rep. 2005;13:37–44. [PubMed] [Google Scholar]
- 5.Robertson GP. Mig-7 linked to vasculogenic mimicry. Am J Pathol. 2007;170:1454–1456. doi: 10.2353/ajpath.2007.070127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brooks PC, Klemke RL, Schon S, Lewis JM, Schwartz MA, Cheresh DA. Insulin-like growth factor receptor cooperates with integrin alpha v beta 5 to promote tumor cell dissemination in vivo. J Clin Invest. 1997;99:1390–1398. doi: 10.1172/JCI119298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klemke RL, Yebra M, Bayna EM, Cheresh DA. Receptor tyrosine kinase signaling required for integrin alpha v beta 5-directed cell motility but not adhesion on vitronectin. J Cell Biol. 1994;127:859–866. doi: 10.1083/jcb.127.3.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beard J. Embryological aspects and etiology of carcinoma. The Lancet. 1902;1:1758–1761. [Google Scholar]
- 9.Red-Horse K, Zhou Y, Genbacev O, et al. Trophoblast differentiation during embryo implantation and formation of the maternal-fetal interface. J Clin Invest. 2004;114:744–754. doi: 10.1172/JCI22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soundararajan R, Rao AJ. Trophoblast ‘pseudo-tumorigenesis’: Significance and contributory factors. Reprod Biol Endocrinol. 2004;2:15. doi: 10.1186/1477-7827-2-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seftor REB, Seftor EA, Koshikawa N, et al. Cooperative interactions of laminin 5 γ2 chain, matrix metalloproteinase-2, and membrane type-1 matrix/metalloproteinase are required for mimicry of embryonic vasculogenesis by aggressive melanoma. Cancer Res. 2001;61:6322–6327. [PubMed] [Google Scholar]
- 12.Maniotis AJ, Folberg R, Hess A, et al. Vascular channel formation by human melanoma cells in vivo and in vitro: Vasculogenic mimicry. Am J Pathol. 1999;155:739–752. doi: 10.1016/S0002-9440(10)65173-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sood AK, Seftor EA, Fletcher MS, et al. Molecular determinants of ovarian cancer plasticity. Am J Pathol. 2001;158:1279–1288. doi: 10.1016/S0002-9440(10)64079-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun B, Zhang S, Zhang D, et al. Vasculogenic mimicry is associated with high tumor grade, invasion and metastasis, and short survival in patients with hepatocellular carcinoma. Oncol Rep. 2006;16:693–698. [PubMed] [Google Scholar]
- 15.van der Schaft DWJ, Hillen F, Pauwels P, et al. Tumor cell plasticity in Ewing sarcoma, an alternative circulatory system stimulated by hypoxia. Cancer Res. 2005;65:11520–11528. doi: 10.1158/0008-5472.CAN-05-2468. [DOI] [PubMed] [Google Scholar]
- 16.Hendrix MJC, Seftor EA, Kirschmann DA, Quaranta V, Seftor REB. Remodeling of the microenvironment by aggressive melanoma tumor cells. Ann NY Acad Sci. 2003;995:151–161. doi: 10.1111/j.1749-6632.2003.tb03218.x. [DOI] [PubMed] [Google Scholar]
- 17.Koshikawa N, Minegishi T, Sharabi A, Quaranta V, Seiki M. Membrane-type matrix metalloproteinase-1 (MT1-MMP) is a processing enzyme for human laminin {gamma}2 chain. J Biol Chem. 2005;280:88–93. doi: 10.1074/jbc.M411824200. [DOI] [PubMed] [Google Scholar]
- 18.Koshikawa N, Giannelli G, Cirulli V, Miyazaki K, Quaranta V. Role of cell surface metalloprotease MT1-MMP in epithelial cell migration over laminin-5. J Cell Biol. 2000;148:615–624. doi: 10.1083/jcb.148.3.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hotary KB, Allen ED, Brooks PC, Datta NS, Long MW, Weiss SJ. Membrane type I matrix metalloproteinase usurps tumor growth control imposed by the three-dimensional extracellular matrix. Cell. 2003;114:33–45. doi: 10.1016/s0092-8674(03)00513-0. [DOI] [PubMed] [Google Scholar]
- 20.Petty AP, Dick CL, Lindsey JS. Translation of an atypical human cDNA requires fidelity of a purine-pyrimidine repeat region and recoding. GENE. 2008;414:49–59. doi: 10.1016/j.gene.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 21.Wright SE, Kilinski L, Talib S, et al. Cytotoxic T lymphocytes from humans with adenocarcinomas stimulated by native MUC1 mucin and a mucin peptide mutated at a glycosylation site. J Immunother. 2000;23:2–10. doi: 10.1097/00002371-200001000-00002. [DOI] [PubMed] [Google Scholar]
- 22.Hendrix MJC, Seftor EA, Seftor REB. Comparison of tumor cell invasion assays: human amnion versus reconstituted basement membrane barriers. Invasion Metastasis. 1989;9:278–297. [PubMed] [Google Scholar]
- 23.Yang AD, Camp ER, Fan F, et al. Vascular endothelial growth factor receptor-1 activation mediates epithelial to mesenchymal transition in human pancreatic carcinoma cells. Cancer Res. 2006;66:46–51. doi: 10.1158/0008-5472.CAN-05-3086. [DOI] [PubMed] [Google Scholar]
- 24.Noguchi P. Use of flow cytometry for DNA analysis. In: Coligan J, Kruisbeek A, Margulies D, Shevach E, Strober W, editors. Current Protocols in Immunology. New York: Wiley-Interscience; 1991. pp. 5.7.1–5.7.4. [Google Scholar]
- 25.Windsor LJ, Bodden MK, Birkedal-Hansen B, Engler JA, Birkedal-Hansen H. Mutational analysis of residues in and around the active site of human fibroblast-type collagenase. J Biol Chem. 1994;269:26201–26207. [PubMed] [Google Scholar]
- 26.Williams NS, Burgett AWG, Atkins AS, Wang X, Harran PG, McKnight SL. Therapeutic anticancer efficacy of a synthetic diazonamide analog in the absence of overt toxicity. PNAS. 2007;104:2074–2079. doi: 10.1073/pnas.0611340104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roehm NW, Rodgers GH, Hatfield SM, Glasebrook AL. An improved colorimetric assay for cell proliferation and viability utilizing the tetrazolium salt XTT. J Immunol Methods. 1991;142:257–265. doi: 10.1016/0022-1759(91)90114-u. [DOI] [PubMed] [Google Scholar]
- 28.Bae-Jump V, Segreti EM, Vandermolen D, Kauma S. Hepatocyte growth factor (HGF) induces invasion of endometrial carcinoma cell lines in vitro. Gynecol Oncol. 1999;73:265–272. doi: 10.1006/gyno.1999.5353. [DOI] [PubMed] [Google Scholar]
- 29.Bescond A, Augier T, Chareyre C, Garcon D, Hornebeck W, Charpiot P. Influence of homocysteine on matrix metalloproteinase-2: Activation and activity. Biochem Biophys Res Commun. 1999;263:498–503. doi: 10.1006/bbrc.1999.1391. [DOI] [PubMed] [Google Scholar]
- 30.Wart HEV, Birkedal-Hansen H. The cysteine switch: A principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. PNAS. 1990;87:5578–5582. doi: 10.1073/pnas.87.14.5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Birkedal-Hansen H, Taylor RE. Detergent-activation of latent collagenase and resolution of its component molecules. Biochem Biophys Res Commun. 1982;107:1173–1178. doi: 10.1016/s0006-291x(82)80120-4. [DOI] [PubMed] [Google Scholar]
- 32.Wright SE, Rewers-Felkins KA, Quinlin IS, et al. Adoptive immunotherapy of mucin1 expressing adenocarcinomas with mucin1 stimulated human peripheral blood mononuclear cells. Int J Mol Med. 2002;9:401–404. [PubMed] [Google Scholar]
- 33.Rozanov DV, Deryugina EI, Ratnikov BI, et al. Mutation analysis of membrane type-1 matrix metalloproteinase (MT1-MMP). The role of the cytoplasmic tail Cys574, the active site Glu240, and furin cleavage motifs in oligomerization, processing, and self-proteolysis of MT1-MMP expressed in breast carcinoma cells. J Biol Chem. 2001;276:25705–25714. doi: 10.1074/jbc.M007921200. [DOI] [PubMed] [Google Scholar]
- 34.Doerr ME, Jones JI. The roles of integrins and extracellular matrix proteins in the insulin-like growth factor I-stimulated chemotaxis of human breast cancer cells. J Biol Chem. 1996;271:2443–2447. doi: 10.1074/jbc.271.5.2443. [DOI] [PubMed] [Google Scholar]
- 35.Silvestri I, Cattani IL, Franco P, et al. Engaged urokinase receptors enhance tumor breast cell migration and invasion by upregulating αvβ5 vitronectin receptor cell surface expression. Int J Cancer. 2002;102:562–571. doi: 10.1002/ijc.10744. [DOI] [PubMed] [Google Scholar]
- 36.Nelson BH, Willerford DM. Biology of the interleukin-2 receptor. Adv Immunol. 1998;70:1–81. doi: 10.1016/s0065-2776(08)60386-7. [DOI] [PubMed] [Google Scholar]
- 37.Celis JE, Moreira JMA, Gromova I, et al. Characterization of breast precancerous lesions and myoepithelial hyperplasia in sclerosing adenosis with apocrine metaplasia. Mol Oncol. 2007;1:97–119. doi: 10.1016/j.molonc.2007.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Müller A, Homey B, Soto H, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 39.Lehti K, Lohi J, Valtanen H, Keski-Oja J. Proteolytic processing of membrane-type-1 matrix metalloproteinase is associated with gelatinase A activation at the cell surface. Biochem J. 1998;334:345–353. doi: 10.1042/bj3340345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sato H, Takino T, Okada Y, et al. A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature. 1994;370:61–65. doi: 10.1038/370061a0. [DOI] [PubMed] [Google Scholar]
- 41.Gingras D, Bousquet-Gagnon N, Langlois S, Lachambre MP, Annabi B, Béliveau R. Activation of the extracellular signal-regulated protein kinase (ERK) cascade by membrane-type-1 matrix metalloproteinase (MT1-MMP) FEBS Letters. 2001;507:231–236. doi: 10.1016/s0014-5793(01)02985-4. [DOI] [PubMed] [Google Scholar]
- 42.D'Alessio S, Ferrari G, Cinnante K, et al. Tissue inhibitor of metalloproteinases-2 binding to membrane-type 1 matrix metalloproteinase induces MAPK activation and cell growth by a non-proteolytic mechanism. J Biol Chem. 2008;283:87–99. doi: 10.1074/jbc.M705492200. [DOI] [PubMed] [Google Scholar]
- 43.Madhunapantula SV, Sharma A, Robertson GP. PRAS40 deregulates apoptosis in malignant melanoma. Cancer Res. 2007;67:3626–3636. doi: 10.1158/0008-5472.CAN-06-4234. [DOI] [PubMed] [Google Scholar]
- 44.Hollier BG, Kricker JA, Van Lonkhuyzen DR, Leavesley DI, Upton Z. Substrate-bound insulin-like growth factor (IGF)-I-IGF binding protein-vitronectin-stimulated breast cell migration is enhanced by coactivation of the phosphatidylinositide 3-kinase/AKT pathway by {alpha}v-integrins and the IGF-I receptor. Endocrinology. 2008;149:1075–1090. doi: 10.1210/en.2007-0740. [DOI] [PubMed] [Google Scholar]
- 45.Matsuo M, Sakurai H, Ueno Y, Ohtani O, Saiki I. Activation of MEK/ERK and PI3K/Akt pathways by fibronectin requires integrin alphav-mediated ADAM activity in hepatocellular carcinoma: A novel functional target for gefitinib. Cancer Sci. 2006;97:155–162. doi: 10.1111/j.1349-7006.2006.00152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou HY, Pon YL, Wong AST. Synergistic effects of epidermal growth factor and hepatocyte growth factor on human ovarian cancer cell invasion and migration: Role of extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase. Endocrinology. 2007;148:5195–5208. doi: 10.1210/en.2007-0361. [DOI] [PubMed] [Google Scholar]
- 47.Wong AST, Roskelley CD, Pelech S, Miller D, Leung PCK, Auersperg N. Progressive changes in Met-dependent signaling in a human ovarian surface epithelial model of malignant transformation. Exp Cell Res. 2004;299:248–256. doi: 10.1016/j.yexcr.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 48.Lehti K, Allen E, Birkedal-Hansen H, et al. An MT1-MMP-PDGF receptor-{beta} axis regulates mural cell investment of the microvasculature. Genes Dev. 2005;19:979–991. doi: 10.1101/gad.1294605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Takino T, Miyamori H, Watanabe Y, Yoshioka K, Seiki M, Sato H. Membrane type 1 matrix metalloproteinase regulates collagen-dependent mitogen-activated protein/extracellular signal-related kinase activation and cell migration. Cancer Res. 2004;64:1044–1049. doi: 10.1158/0008-5472.can-03-1843. [DOI] [PubMed] [Google Scholar]
- 50.Tsubaki M, Matsuoka H, Yamamoto C, et al. The protein kinase C inhibitor, H7, inhibits tumor cell invasion and metastasis in mouse melanoma via suppression of ERK1/2. Clin Exp Metastasis. 2007;24:431–438. doi: 10.1007/s10585-007-9080-z. [DOI] [PubMed] [Google Scholar]
- 51.Bonta IL, Ben Efraim S. Interactions between inflammatory mediators in expression of antitumor cytostatic activity of macrophages. Immunol Lett. 1990;25:295–301. doi: 10.1016/0165-2478(90)90199-z. [DOI] [PubMed] [Google Scholar]
- 52.Xiang J, Moyana T, Qi Y. Genetic engineering of a recombinant fusion possessing anti-tumor F(ab′)2 and tumor necrosis factor. J Biotechnol. 1997;53:3–12. doi: 10.1016/s0168-1656(96)01639-2. [DOI] [PubMed] [Google Scholar]
- 53.Nyalendo C, Beaulieu E, Sartelet H, et al. Impaired tyrosine phosphorylation of membrane type 1-matrix metalloproteinase reduces tumor cell proliferation in three-dimensional matrices and abrogates tumor growth in mice. Carcinogenesis. 2008;29:1655–1664. doi: 10.1093/carcin/bgn159. [DOI] [PubMed] [Google Scholar]