

Abstract
Obesity is an energy balance disorder in which intake is greater than expenditure, with most excess calories stored as triglyceride (TG). We previously reported that mice lacking β-isoform of protein kinase C (PKCβ), a diacylglycerol- and phospholipid-dependent kinase, exhibit marked reduction in the whole body TG content, including white adipose tissue (WAT) mass. To investigate the role of this signaling kinase in metabolic adaptations to severe dietary stress, we studied the impact of a high fat diet (HFD) on PKCβ expression and the effect of PKCβ deficiency on profound weight gain. We now report that HFD selectively increased PKCβ expression in obesity-prone C57BL/6J mice, specifically in WAT; the expression levels were little or unchanged in the liver, muscle, kidney, and heart. Basal PKCβ expression was also found to be elevated in WAT of obese ob/ob mice. Remarkably, mice lacking PKCβ were resistant to HFD-induced obesity showing significantly reduced WAT and slightly higher core body temperatures. Unlike lean lipodystrophic mouse models, these mice did not have fatty livers or exhibited insulin resistance. Moreover, PKCβ−/− mice exhibited changes in lipid metabolism gene expression and such alterations were accompanied by significant changes in serum adipokines. These observations suggest that PKCβ deficiency induced a unique metabolic state congruous with obesity resistance, thus raising the possibility that dysregulation of PKCβ expression could contribute to dietary fat-induced obesity and related disorders.
Keywords: high-fat diet, TG metabolism, signal transduction, fatty acid oxidation, transcriptional induction
Introduction
There is a worldwide epidemic of obesity and type 2 diabetes1, 2 that parallels the “westernization” of diet, which is characterized by high-fat and higher caloric intake3. Since 80% of the patients with type 2 diabetes are obese, and obesity is associated with insulin resistance4, attention has been focused on the identification of molecular targets for fat storage, and the characterization of links between obesity and insulin resistance. One mechanism that has been proposed as a cause of obesity-induced insulin resistance is an elevation in intracellular fatty acid metabolites, such as fatty acyl coenzyme A and diacylglycerol. This results in tissue-specific activation of specific isoforms of protein kinase C (PKC) that lead to the impairment of insulin signaling and activity5–10. Alterations in the expression levels and/or activities of several lipid-dependent PKC isoforms have been associated with insulin resistance in type 2 diabetic patients, animal models of diabetes, and cellular models11,12. In mammals, the PKC family is quite heterogeneous, comprising 11 isoforms divided into three major subsets: conventional (α, βI, βII, and γ), novel (δ, ε, λ, μ, and θ), and atypical isoforms (λ and ζ)13,14. Each isoform is encoded by a separate gene, with the exception of the β1 and βII isoforms, which are splice variants. In fact, among the ubiquitously expressed family of serine/threonine kinases, PKCβ is the only subtype that is expressed through two splice-variants15. The biological significance of this heterogeneity has not been clarified, but it appears that the members of this enzyme family are activated in specific intracellular compartments in different ways, depending on the distribution of membrane lipid metabolites, and play distinct roles in the control of major cellular functions14,16.
Previous studies have firmly established that diabetic conditions activate diacylglycerol-PKC signaling10,12. Diabetes-induced PKC activity does not appear to be isoform generalized, rather restricted to a few “diabetic-related” isoforms in a tissue-specific manner. PKCβ is one of those isoforms and has been most directly linked to important aspects of hyperglycemia in vivo and in vitro. Insulin is known to stimulate PKCβ activity in order to promote glucose disposal17. PKCβ is also shown to inhibit several components of the insulin signaling cascade, and the downstream metabolic enzymes including glycogen synthase18. There is also evidence that PKCβ may act upstream of other serine and threonine kinases. For example, PKCβ activation can enhance the ability of kinases, such as Jun amino-terminal kinase and inhibitor of kB kinase, to phosphorylate insulin receptor substrate-1 at Ser307, a key regulatory site located near the domain that interacts with the insulin receptor19,20. On the other hand, PKCβ is also known to act upstream of mitogen/extracellular-regulated kinase to promote Ser612 phosphorylation of insulin receptor substrate-1, which modulates phosphatidylinositol-3-kinase activation21–23. Moreover, variations in the promoter region of the human PKCβ gene are associated with alterations in insulin sensitivity24, 25. Finally, in vivo inhibition of PKCβ reverses diabetes-induced vascular dysfunction, thereby identifying a definitive role for PKCβ in the pathogenesis of diabetic complications26.
Metabolic pathways leading to adiposity from consumption of dietary fat are incompletely understood. Due to link between lipids and PKC activation, dietary fat has also been examined as a modulator of PKC activity in several tissue types from a range of animal species27–30. Providing various types, and levels, of fat in the diet of animals has been shown to produce alterations in PKCβ translocation and activation in a variety of tissues. For example, increased translocation of cardiac PKCβII has been shown to accompany mild cardiac hypertrophy in rats that were fed a diet rich in saturated fat31. The extent to which PKCβ exerts control on visceral adiposity, TG storage, and insulin sensitivity is not clear. The importance of PKCβ in lipid metabolism was recently unraveled using the PKCβ deficient (PKCβ−/−) mice, where we showed that the loss of PKCβ function is associated with a marked reduction in adipose mass, significantly reduced TG stores in insulin-sensitive tissues, increased metabolic rate and oxygen consumption, and reduced body weight despite increased food intake32. The marked decrease of TG levels in PKCβ−/− mice suggested alterations in lipid uptake or metabolism of PKCβ mice. To understand further the physiological role of PKCβ, and its potential to regulate systemic TG homeostasis, we evaluated whether PKCβ expression and its distribution is influenced by high fat feeding and also assessed the impact of PKCβ deficiency on obesity and its accompanying metabolic disorders under conditions of severe dietary stress. We now report tissue- and isoform-specific upregulation of adipose PKCβ expression in two obesity mouse models. Mice with a targeted disruption of the PKCβ are resistant to the dietary fat-induced obesity and the development of insulin resistance. Moreover, PKCβ−/− mice exhibited changes in lipid metabolism gene expression and such alterations were accompanied by significant alterations in serum adipokines. Our data raise the possibility that dysregulation of PKCβ expression can be associated with susceptibility to high fat diet (HFD)-induced obesity.
Materials and Methods
Animals and Diet
Production of PKCβ-deficient (PKCβ−/−) mice in C57BL/6J background and genotypic determination were performed as described previously (35). Six weeks old PKCβ−/− and WT controls were fed ad libitum for 12 weeks continuously either a HFD (D12492; Research Diets, New Brunswick, NJ) in which 60% of the total calories were derived from fat (soybean oil and lard) or a standard diet containing 15% kcal from fat (7912 rodent chow; Harland Tekland) and were fed ad libitum. PKCβ−/− and WT mice were housed under controlled temperature (23°C) and lighting (12 h of light, 07:00-19:00; 12 h of dark, 19:00-07:00) with free access to water and standard mouse chow diet (6% fat by calories, Harlan Tekland, Madison, WI). All procedures on mice followed the guidelines established by the Ohio State University College of Medicine Animal Care Committee. Unless indicated, all experiments were performed on male animals starved for ~16 h.
Histological Analysis of Tissues
Liver, WAT, and BAT were isolated from WT and PKCβ−/− mice and fixed in 4% paraformaldehyde in phosphate-buffered saline and processed for paraffin sections and stained by hematoxylin and eosin. Cross-sectional areas of adipocytes were measured using Image J software by counting at least 300 individual cells from random fields in histological sections. Non-perfused livers and WAT were digested in chloroform-methanol to determine hepatic TG levels. Lipid layers were separated using H2SO4 and the TG concentrations were determined with a TG assay kit according to the manufacturer’s instructions. Livers were fixed either in 4% paraformaldehyde-phosphate buffered saline and stained by hematoxylin and eosin, or snapped frozen in 5-methylbutane. Cryostat sections of livers were fixed with 4% paraformaldehyde-phosphate buffered saline for 1 h and cryoprotected with 20% sucrose prior to staining with 4% oil red O for 2 h. Liver tissues were fixed with 10% formalin in phosphate buffered saline for 2 h then stained with 4% oil red O solution for 2 h.
Glucose Tolerance Test (GTT) and Insulin Tolerance Test (ITT)
Both GTT and ITT were performed on fasted (16 h) mice. Mice were weighed and then injected intraperitoneally with either glucose (1.5 mg/kg of body weight) or insulin (Humulin; Lilly; 0.8 units/kg of body weight). Blood samples were obtained via retro-orbital bleeds at base line and at indicated time intervals after glucose or insulin injection and analyzed for glucose and insulin levels. Overnight fasted mice that have been on a HFD were intraperitoneally injected with glucose. Tail vein blood was collected at 0, 30, 60, and 120 min following glucose injection and blood glucose level was measured with a Beckman Glucose Analyzer 2. Plasma insulin levels were measured by radioimmuneassay using kit from LINCO Research (St. Charles, MO). Insulin tolerance was assessed after overnight fast by administration of human insulin and blood glucose monitoring. Hypoglycemia was assessed using a SureStep Ultra (Lifescan) glucometer.
Blood, Serum, and Tissue Analyses
Plasma TG and cholesterol concentrations were determined by colorimetric kit assays (Roche Diagnostics). Serum glucose was measured using a One Touch SureStep glucometer from LifeScan (Johnson & Johnson). Immunoreactive insulin was measured using a sensitive insulin RIA kit from Linco Inc. Serum leptin and adiponectin levels were measured by ELISA assay kits (LINCO, Inc.) as mentioned in the manufacturer’s protocol. NEFAs were measured by NEFA C from Wako. Serum TG was measured using the Serum TG determination kit from Sigma-Aldrich. Tissue TG contents were determined with a TG 320A kit (Sigma) as described. For qualitative analysis of tissue lipids, lipids were extracted from tissues and separated on ALSILG Silica Gel TLC plates (Whatman) using hexane:ethyl ether:acetic acid (83:16:1).
Tissue Extraction
WAT were fractionated as described earlier33. Epididymal fat pads were homogenized and centrifuged at 1,000 g, and supernatants were collected and subjected to another centrifugation at 100,000 g for 1 h at 4°C. The supernatants were collected as soluble fractions and referred to as the cytosol fraction. The pellets were dissolved in with ice-cold buffer in a volume equal to that of soluble fractions and were termed the membrane fraction.
Western blotting
Tissue samples were snap frozen, pulverized, and dispersed inlysis buffer32,34. Sample were loaded onto 8% acrylamide gel and blotted. Anti-PKCβ antibody was obtained from Santa Cruz Biotechnology, CA. Immunoreactive proteins visualized by enhanced chemiluminescence (ECL plus; Amersham, Arlington Heights, IL) and quantified by densitometry using Molecular Analyst software (Bio-Rad).
Gene expression analysis
Gene expression analysis was performed as described previously32,34. To ensure the validity of the SYBR-green-based mRNA quantifications, most of the mRNAs were quantified using the 32P-labeled dCTP. This alternative method and the SYBR green method gave similar results. All data are expressed as the fold induction relative to each control value.
Statistical Analysis
Statistical comparison of results was performed using the unpaired Student’s t test.
Results
Dietary fat induces PKCβ expression in an isoform- and tissue-specific manner
It is well demonstrated that C57BL/6J mice develop severe obesity accompanied by hyperglycemia when fed a HFD35. Therefore, these mice are among the several strains of mice classified as sensitive to diet-induced obesity. To address the relationship between dietary fat and PKCβ expression, we examined effects of HFD feeding on expression levels of PKC isoforms in different tissues. After 12 weeks of feeding, PKCβ mRNA levels were significantly increased selectively in white adipose tissue (WAT) (4-fold; P<0.001) and brown adipose tissue (BAT) (3-fold; P<0.05) compared with those on a low-fat diet; expression levels were little or unchanged in the liver, muscle, kidney or heart (Fig. 1A & 1B). The expression levels of other PKC isoforms were not altered significantly, except slight increases in adipose PKCα and PKCθ expression on a HFD. To further verify that PKCβ was overexpressed upon HFD feeding, PKCβ protein levels were examined in the WAT. Indeed, expression level of PKCβ protein was found to be increased in the WAT of HFD-fed mice (Fig. 1D).
Fig. 1.
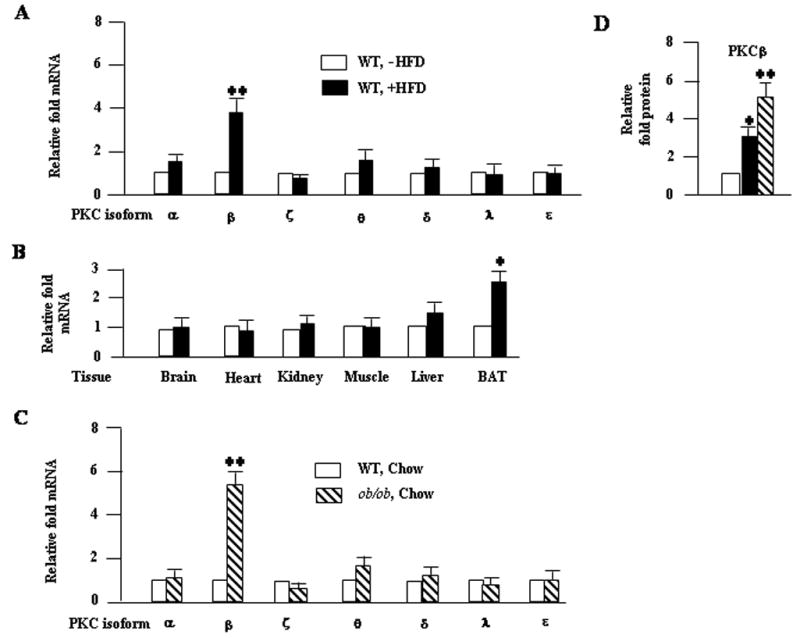
Induction of adipose PKCβ expression by HFD in C57BL/6J mice and ob/ob mice fed chow diet exhibited elevated adipose PKCβ expression. (A) Relative mRNA levels of different PKC isoforms in WAT of starved (16 h) C57BL/6J WT mice fed either a normal chow or a HFD for 12 weeks. (B) Relative PKCβ mRNA levels in different tissues of HFD-fed WT mice. (C) Expression levels of different PKC isoforms mRNA in the WAT of starved (16 h) ob/ob mice fed a chow diet and control lean animals on the same diet. Total RNA was isolated and used for assessment of levels of different PKC isoforms. β-Actin was used as a control in all experiments. (D) Abundance of PKCβ was determined by Western blot analysis using anti-PKCβ serum (Santa Cruz Biotechnology, CA). Densitometric analysis of PKCβ band is shown. Results are means±S.D. Asterick denote statistically significant differences between genotypes. *, P<0.05; **, P<0.01.
Elevated basal adipose PKCβ expression in ob/ob mice
The obese ob/ob mouse is an excellent model of obesity induced by insulin resistance, overeating and overweight36. To evaluate the regulation of PKCβ by obesity and hyperinsulinemia in vivo, we measured PKCβ expression in different tissues of 18 weeks old C57BL/6J ob/ob mice and their lean counterparts C57BL/6J WT mice. As shown in Figure 1C, basal PKCβ mRNA expression was significantly increased in ob/ob mice WAT by 531±56% compared with C57Bl/6J WT mice (P<0.001), while no significant differences were observed in other tissues (Fig. 1C). Moreover, expression levels of other PKC isoforms were little or unchanged in ob/ob WAT.
PKCβ deficiency ameliorated diet-induced obesity
Diet-induced changes in adipose PKCβ expression raised the possibility that expression of this kinase might be regulated physiologically to enhance dietary fat storage. To address the relationship between PKCβ expression and susceptibility to obesity, we compared responses of two strains of mice under conditions of severe dietary stress. Both PKCβ−/− and WT mice were fed HFD for 12 weeks, beginning at six weeks of age. Body weights and food intake were monitored weekly, and at the end we examined weights of WAT, BAT, and various metabolic parameters. A significant increase in WT mice body weights were evident after only three weeks on a HFD, and this trend continued throughout the dietary protocol (Fig. 2A). However, PKCβ−/− mice fed a HFD gained less weight and exhibited an obese-resistant phenotype (Figs. 2A & 2B). While body weights of PKCβ−/− mice were less than WT mice, PKCβ−/− mice appeared to be mildly hyperphagic and consistently consumed a greater caloric load (Fig. 2A). When energy intake was calculated relative to increased body weight (feed efficiency) over the 12 week period, PKCβ−/− mice consistently exhibited a lower feed efficiency (i.e. required more energy intake relative to body weight gained) compared to WT mice (2.1±0.29 WT versus 1.27±0.12 body weight gain/gram of HFD/week PKCβ−/−; P<0.001) (Fig. 2A). It is thus clear that the decreased body weight was not a result of decreased food intake.
Fig. 2.
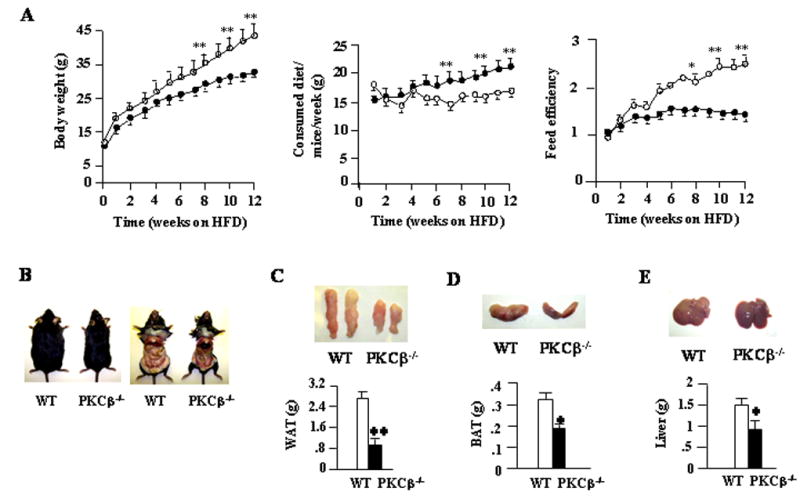
PKCβ−/− mice are resistant to HFD-induced obesity. (A) Body weights, food intakes and feed efficiencies in WT and PKCβ−/− mice fed a HFD. At 6 weeks of age, mice were fed a HFD for a period of 12 weeks and were weighed weekly. Values represent means±S.D. of WT and PKCβ−/− mice (n=7). (B) Gross representative images of WT and PKCβ−/− mice and the abdominal view of the fat pads under the skin following 12 weeks of HFD. (C, D & E) Pictures and weights of livers, WAT and BAT for the same group (n=6). Values represent means±S.D. of WT and PKCβ−/− mice (n=6). *, P<0.05; **, P<0.001.
To determine if changes in weights were associated with differences in adiposity, we compared changes in adipose fat pads in WT and PKCβ−/− mice on a HFD. The weights of WAT (2.42±0.27 WT versus 0.85±0.12 gram PKCβ−/−; P<0.001) (Fig. 2C) and BAT (0.32±0.07 WT versus 0.19±0.001 gram PKCβ−/−; P<0.05) (Fig. 2D) in PKCβ−/− mice were dramatically reduced when compared with WT mice, indicating that these mice are strongly protected from diet-induced obesity. The weights of PKCβ−/− livers were also lower than those of WT (1.48±0.17 WT versus 0.9±0.1 g PKCβ−/−; P<0.001) (Fig. 2E), whereas various other tissues were of similar weight. There was a slight change in the liver color of PKCβ−/− mice, however, no obvious morphological differences were observed in other tissues. Histological examination of these livers revealed increased numbers and sizes of intracellular vacuoles, an indication of fatty liver, in WT mice compared with PKCβ−/− mice (Fig. 3A). Oil red O staining of liver sections verified deposition of increasing quantities of lipids in WT liver (Fig. 3A). Histological analysis of WAT revealed that adipocytes from PKCβ−/− mice were significantly smaller than those from WT mice (Fig. 3A). The thickness of adipose tissue beneath the dermis in PKCβ−/− mice was also clearly reduced compared with WT mice (Fig. 3B). As expected, based on histological analysis, chemical analysis of livers and muscles showed elevated TG contents in WT mice compared with PKCβ−/− mice, with a greater than 7-fold and 3fold difference, respectively (Figures 3C–3E). Elevated hepatic TG in WT mice is reflected by slight color change in the livers of these animals, even though their cholesterol levels remained same (Fig. 3F).
Fig. 3.
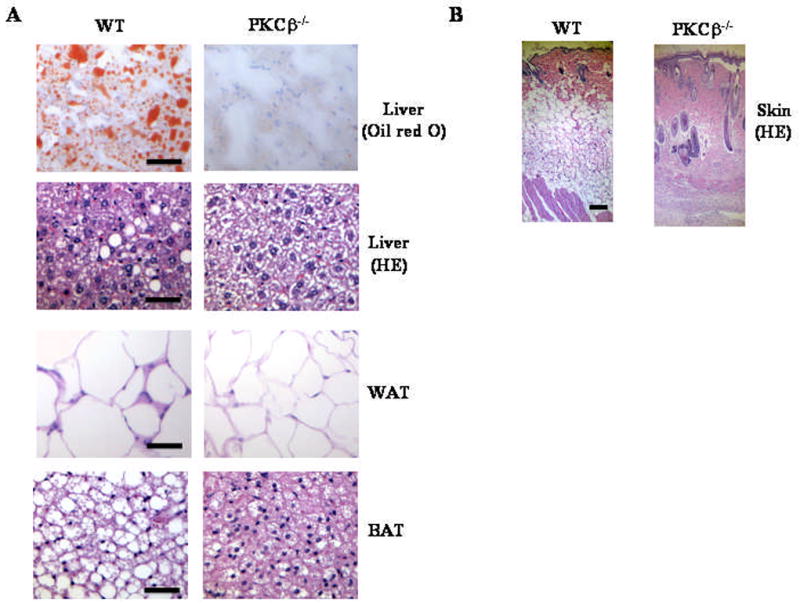
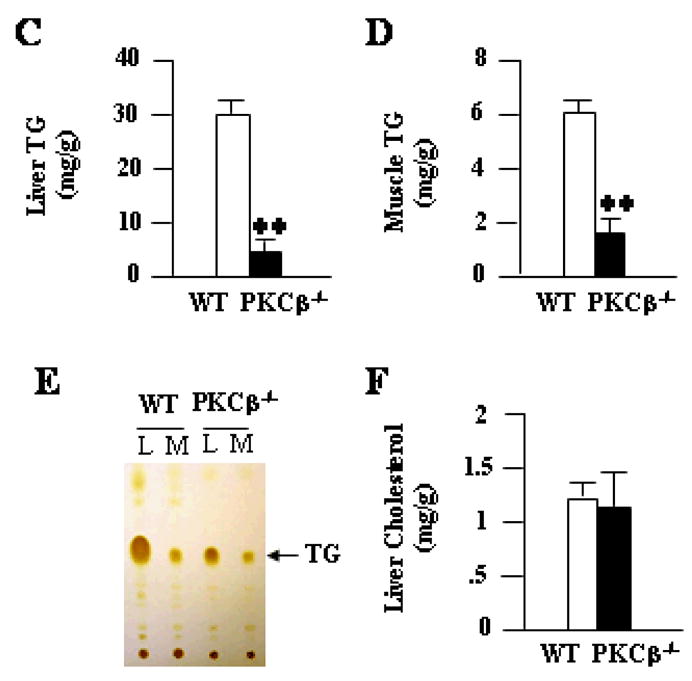
Histological and biochemical changes in liver, adipose, and skin from WT and PKCβ−/− mice. (A & B) Histological sections of livers (20x), WAT (20x), BAT (20x), and skin (4x) were prepared and stained with hematoxylin and eosin or oil red O. Bar=50 μm for liver, WAT and BAT, whereas Bar=100 μm for skin. Results are representative of n=5 in each group. Each value represent means±S.D. (C, D & E) Relative amounts of lipid contents in the liver and muscle of WT and PKCβ−/− mice following HFD feeding for 12 weeks (fasted 16 h). Each value represent means±S.D. of n=5–10 mice per group. TLC of total lipid extracts from livers (L) and muscles (M) of WT and PKCβ−/− mice is also shown. Each lane represents lipids from liver and muscle of three mice. *, P<0.001. (F) Liver cholesterol contents of WT and PKCβ−/− mice. Each value represent means±S.D.
Consistent with adipose tissue mass, PKCβ−/− mice fed HFD for 12 weeks exhibited significantly lower levels of serum leptin (29.1±2.5 WT versus 0.9±0.3 mEq/1 PKCPβ−/−; P<.001) (Fig. 4A). In contrast, plasma adiponectin levels were slightly elevated, but never reached a significant level, in PKCβ−/− mice compared with WT mice (32.9±4.3 WT versus 38.9±6.4 ng/ml PKCβ−/−; P=NS) (Fig. 4A). Consequently, food intake, which is regulated by plasma levels of leptin, was >20% higher in PKCβ−/− mice than in WT mice (Fig. 3A). Leptin values observed in PKCβ−/− mice were significantly lower than what would be expected based on reduction in WAT. Decreased leptin values in PKCβ−/− mice were more than could be explained by reduction in body weight alone. In view of these results, leptin and adiponectin mRNA levels were assessed in WAT and BAT. Leptin mRNA was 10-fold lower in PKCβ−/− mice on a HFD (Fig. 4B), suggesting that decreased expression of this hormone in PKCβ−/− mice further contributed to lower circulating levels. Surprisingly, adiponectin mRNA is not significantly different between WT and PKCβ−/− mice, despite 3-fold decrease of WAT in PKCβ−/− mice.
Fig. 4.
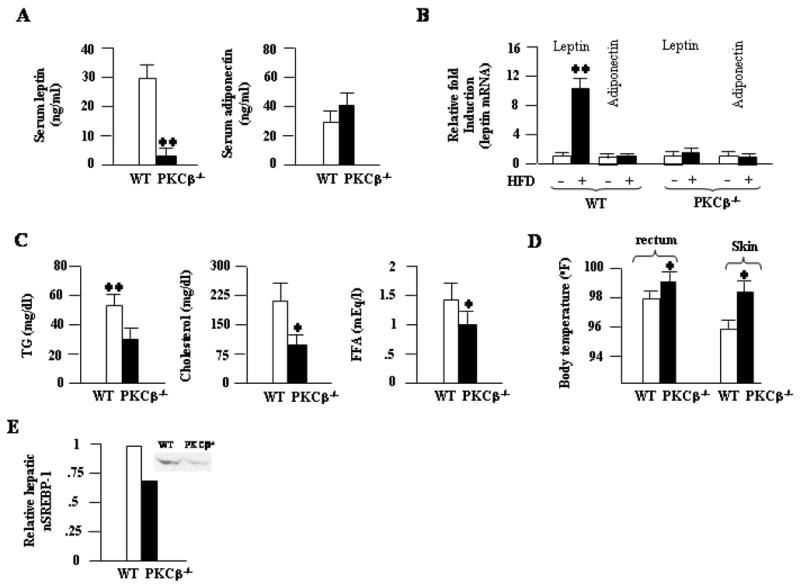
Comparison of metabolic profiles of WT and PKCβ−/− mice fed a HFD for 12 weeks. (A) Serum leptin and adiponectin levels; (B) Relative leptin and adiponectin mRNA levels in the WAT of WT and PKCβ−/− mice fed either a chow diet or a HFD. (C) plasma TG, cholesterol, and FFA levels; (D) Comparison of body temperatures. Each value represent means±S.D. of n=5–10 mice per group. *, P<0.05; **, P<0.001. (E) Comparison of hepatic mature SREBP-1 levels in WT (given an arbitrary value of 1) and PKCβ−/− mice as revealed by Western blotting using anti-SREBP-1 (K-10) from Santa Cruz Biotechnology. A representative autoradiograph is shown in the inset. Autoradiographs were quantitated densitometrically and the results are normalized to the levels of hepatic mature SREBP-2. Values shown are averages of two different experiments.
Explanations for the surprising phenotype include deficiency in intestinal food absorption and/or abnormalities in lipid clearance, metabolism, or storage. To distinguish among these possibilities, we examined circulating TG, cholesterol, and free fatty acid (FFA) levels. We hypothesized that lipid levels would be increased if lipid storage or clearance were altered. Total circulating cholesterol, TG, and FFA levels were much lower in PKCβ−/− mice compare with WT mice (Fig. 4C). Fasting FFA levels were not statistically different, but there was a tendency for elevation of these levels in PKCβ−/− mice (results not shown). Mean rectal temperature (97.8±0.3 WT versus 98.9±0.4°C PKCPβ−/−; P<.01) or skin temperature (95.9±0.4 WT versus 98.1±0.5°C PKCPβ−/−; P<.01) was slightly higher in PKCβ−/− mice relative to those of control mice (Fig. 4D). Increased body temperatures in PKCβ−/− mice compared to WT mice point toward increased thermogenesis in the mutant mice. In short, these results demonstrate that PKCβ deficiency imparts resistance to diet-induced obesity and liver steatosis.
Altered expression of genes involved in lipolysis, fatty acid synthesis and oxidation in HFD-fed PKCβ−/− mice
Having shown that HFD-induced adiposity is ameliorated in mice lacking PKCβ, we next examined changes in the expression profiles of genes involved in lipid uptake, storage, and metabolism. The most striking changes were seen in genes involved in lipogenesis and fatty acid β-oxidation in the liver. The expression levels of acetyl-CoA carboxylase-2 (ACC-2), a negative regulator of carnitinepalmitoyltransferase-1 activity and fatty acid oxidation, were significantly reduced in PKCβ−/− mice, indicating an increase in β-oxidation. Reduced hepatic expression levels of genes encoding fatty acid synthase, sterol response element-binding protein (SREBP)-1a, and SREBP-1c point to a decrease in the TG biosynthesis (Table 1). Consistent with mRNA levels, western blot analysis revealed reduced amount of mature SREBP-1 in the livers of PKCβ−/− mice compared with WT mice (Fig. 4E).
Table 1.
Expression of various mRNAs in WAT, BAT, and liver of mice fed HFD for 11 weeks. The fold change is calculated as the ratio of knockout/control expression and is an average of two experiments. ND, not determined.
| Gene | Relative-fold change |
||
|---|---|---|---|
| WAT | BAT | Liver | |
| SREB-1a | 0.57 | 0.53 | 0.61 |
| SREBP-1c | 0.51 | 0.6 | 0.4 |
| SREBP-2 | 1.7 | 1.4 | 1.0 |
| C/EBPα | 1.0 | 1.0 | ND |
| LRP-1 | 0.9 | 1.0 | 1.0 |
| FAS | 0.3 | 0.53 | 0.7 |
| Perilipin | 0.2 | 0.85 | ND |
| ACC-2 | ND | 0.9 | 0.47 |
| HSL | 1.5 | ND | 1.0 |
| ATGL | 1.0 | 1.0 | ND |
| CD36 | 1.0 | ND | ND |
| SCD-1 | 1.0 | 1.0 | 1.0 |
| UCP-1 | ND | 1.0 | ND |
| UCP-2 | 1.3 | 1.3 | 1.0 |
| UCP-3 | 0.34 | 0.37 | 0.5 |
To begin to understand changes in adipose tissue resulting from PKCβ deficiency, differentially regulated genes were also characterized. We focused on enzymes and proteins that reside at the lipid droplet surface that are thought to play a pivotal role in the lipolysis and/or lipid droplet formation. Hormone sensitive lipase mRNA was elevated in the adipose of PKCβ−/− mice compared with WT adipose tissue, whereas mRNA level for adipose triglyceride lipase did not change in PKCβ−/− mice. The mRNA for perilipin A, a lipid droplet protein intimately involved in lipolysis that may serve as a scaffold for droplet-associated regulatory proteins, was reduced in the PKCβ−/− adipose tissue. In contrast, multiple genes involved in lipid homeostasis (C/EBPα, LDL receptor related protein-1, and CD36) are not changed in the PKCβ−/− mice compared with WT mice (Table 1). We also examined expression of genes encoding uncoupling proteins (UCP), since it appears that PKCβ−/− mice expend more energy in thermogenesis than control mice. Expression of UCP-2 in WAT was slightly elevated in PKCβ−/− WAT and BAT, but not in the liver, whereas expression of UCP-3 was significantly reduced in all three tissues examined.
PKCβ−/− mice exhibited increased insulin sensitivity
Diet-induced obesity is frequently associated with insulin resistance, so we next investigated whether depletion of PKCβ would affect insulin sensitivity. Plasma glucose and insulin levels were significantly higher in fasted WT mice compared with those in PKCβ−/− mice after 12 weeks of HFD feeding, indicating resistance to the emergence of insulin resistance (Fig. 5A). Glucose tolerance tests indicated that PKCβ−/− mice on a HFD were more efficient in clearing an intraperitoneally injected bolus of glucose than WT mice on the same diet (Fig. 5B). Protection from diet-induced insulin resistance in PKCβ−/− mice was also confirmed by an insulin tolerance test. Insulin sensitivity, as measured by the degree of reduction in plasma glucose after insulin administration, reflected that, on a HFD, PKCβ−/− mice were more efficient at insulin-mediated suppression of plasma glucose than WT mice (Fig. 5B). These results demonstrate that the absence of PKCβ clearly hindered the development of HFD-induced insulin resistance in HFD-induced obesity and suggest that supporting mechanisms may be linked to reduced TG contents.
Fig. 5.
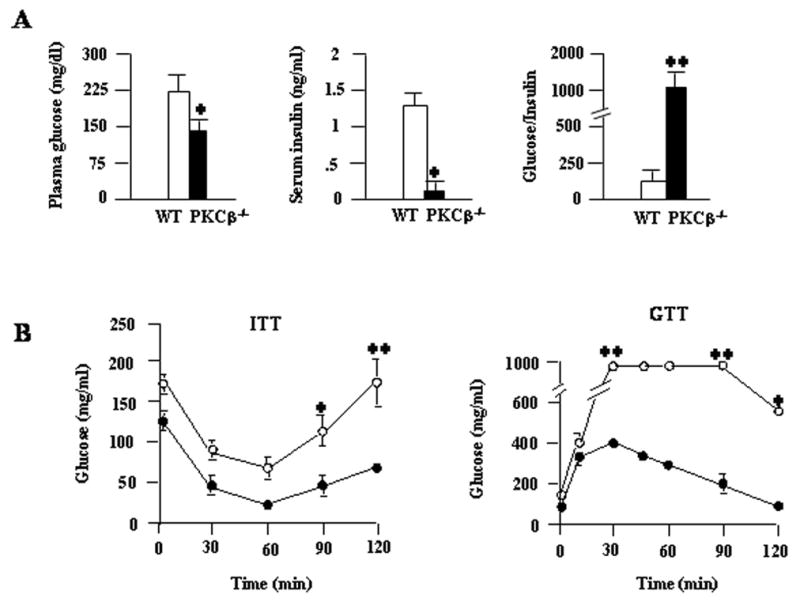
Increased in vivo insulin sensitivity in the PKCβ−/− mice compared to WT mice following 12 weeks of a HFD. (A) Plasma glucose and plasma insulin were measured after 12 weeks on a HFD in WT and PKCβ−/− mice. Data are means±S.D. (n=8). (*, P<0.05). (B) Changes in glucose levels in insulin tolerance tests (ITT) (left panel) and glucose tolerance tests (GTT) (right panel). For ITT, mice fasted for ~16 h were injected with insulin (0.75 units/kg of body weight), blood samples were obtained at the indicated times, and glucose levels were monitored. For GTT, mice fasted for ~16 h were injected with a bolus of glucose, and blood samples were obtained at the indicated time and analyzed for glucose levels. Data are means±S.D. (n=6). *, P<0.05; **, P<0.001.
We finally investigated the relationship of adipose PKCβ expression with any of the parameters characteristic for obesity and insulin resistance. Dietary fat may increase PKCβ levels directly through an effect on lipid metabolic machinery or indirectly by changing other metabolic parameters. Dietary studies were performed using another group of WT mice, and plasma insulin and adipose PKCβ expression levels were determined after HFD feeding for varying periods (0, 2, 4, or 8 weeks). As shown in Fig. 6A, the increase in PKCβ expression was observed with HFD in a time-dependent manner, which paralleled an increase in plasma insulin levels. To confirm the association between adipose PKCβ expression and insulin levels, we further investigated the effect of insulin on PKCβ expression in primary adipocytes from C57BL/6J mice. The freshly isolated adipocytes were plated in serum containing medium, and after 16 h cells were switched to media containing 10 nM insulin and harvested after varying intervals. Treatment of these cells with insulin induced PKCβ expression in a time- and dose-dependent manner (Fig. 6B), thus supporting involvement of hyperinsulinemia in HFD-dependent induction of adipose PKCβ expression.
Fig. 6.
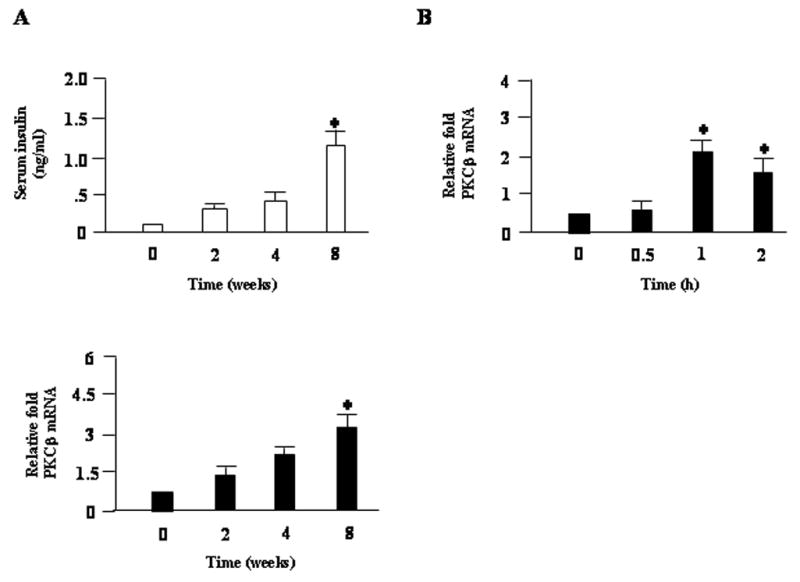
Plasma insulin and adipose PKCβ expression in WT mice fed a HFD for the indicated periods. (A) In order to test the efficacy of a HFD treatments, plasma insulin levels and adipose PKCβ expression were measured in WT mice fed a normal chow or a HFD for 0, 2, 4, and 8 weeks. (B) Time-dependent changes in PKCβ expression by insulin in primary adipocytes from WT mice. Freshly isolated primary adipocytes were treated with 10 nM insulin for the indicated time. Results are plotted relative to the control value set at 1. For all panels, data are means±S.D. (n=4). *, P<0.05.
Discussion
This study demonstrates, for the first time, nutritional regulation of the PKCβ gene in vivo. In particular, we report prominent upregulation of PKCβ expression in the fat tissues of two obese animal models, ob/ob and HFD fed mice, suggesting tissue- and isoform-specific differences in the mechanisms regulating PKC isoforms expression in response to dietary fat. Differential regulation of individual PKC isoforms has earlier been observed under various physiological and pathological conditions14,16. An important question is whether the delayed change in PKCβ gene expression is a result of exposure to HFD or the resulting insulin resistance. It is conceivable that diet-induced insulin resistance may contribute to the induction of adipose PKCβ gene expression. As a consequence of increased insulin resistance, insulin levels are elevated in these mice, which in turn can induce PKCβ expression. In support of this idea, we observed an increase in plasma insulin levels in HFD fed WT mice that paralleled closely with increased adipose tissue PKCβ expression. Furthermore, hyperinsulinemic ob/ob mice exhibited elevated basal PKCβ expression in the WAT. Lastly, induction of adipose PKCβ expression can be reproduced ex vivo by treating adipose tissue organ cultures with insulin, the classic hormone of the fed state (Fig. 6 & ref. 37). Given that insulin stimulates SREBP-1c expression and activity38,39, the mechanism of induction may, at least in part, include multiple SRE-1 sites present in the PKCβ promoter40,41. We therefore propose that increased PKCβ expression observed in HFD fed mice is secondary to increased caloric consumption and the consequent insulin resistance, rather than to the HFD itself. The nature of the signaling pathway that triggers differential PKCβ expression in a tissue-specific manner remains to be characterized. Adipose selective PKCβ induction may be related to tissue-specific promoter events as transgenic mice overexpressing SREBP-1c in the liver42 or adipose tissue43 exhibited different phenotypes, possibly due to tissue-specific action of SREBP-1c resulting in differential gene expression. Additionally, HFD-dependent development of hepatic insulin resistance could prevent insulin-mediated changes in gene expression. Future studies to identify the precise mechanism(s) underlying this effect will be of interest.
The adipose-selective induction of PKCβ expression, in addition to being a consequence of HFD-induced hyperinsulinemia, may also have direct effects on the development of obese phenotype in WT mice. In accordance with this possibility, we report that PKCβ−/−mice are resistant to HFD-induced obesity, as reflected by lower body weight, smaller visceral fat pads, decreased fat accumulation in the liver, and better insulin sensitivity. The PKCβ−/−phenotype is distinct from lipodystrophic mice, a class of “lean” mice, which have defects in white adipocyte differentiation or function43–45. Lipodystrophic mice lack normal adipocytes and accumulate fat in non-adipose tissues, resulting in fatty livers, elevated circulating TG levels, and insulin resistance. In contrast, PKCβ−/−mice exhibit decreased lipid storage in adipose tissue, but do not exhibit increased circulating TG levels or hepatic steatosis. In fact, reduced accumulation of fat in the liver is characteristic of PKCβ−/−mice and is consistent with reduced expression of lipogenic genes, such as FAS, SREBP-1c, and SREBP-1a in the liver. PKCβ−/−mice also differs from other classes of lean mice resulting from alterations in CNS-mediated gene targets. For example, leptin transgenic mice46, or knockout mice in melanin concentrating hormone47 or the muscarinic M3 receptor48,49, exhibit increased metabolic rates, but rarely exhibit compensatory increase in food intake. Interestingly, PKCβ−/− mice exhibit some similarity to a class of lean mice resulting from a reduction in adipose tissue storage capacity, or by enhanced lipolysis, which can be induced by the disruption of a number of genes50–53. Reduced energy storage consistently results in increased energy expenditure due to repartitioning of energy substrates up to a point where increased food intake can not compensate.
Possible scenarios can be envisioned that may account for mechanism(s) by which PKCβ deficiency imparts resistance to HFD-induced obesity. Increased oxygen consumption and differential thermogenesis may contribute to protection in PKCβ−/−mice (Bansode et al., 2008). Body temperatures of PKCβ−/−mice were slightly higher than WT mice, suggesting that protection from obesity in PKCβ−/−mice most likely involves increased energy expenditure. It is consistent with previous studies that have shown that diet-induced thermogenesis is higher in lean humans compared to obese individuals53. Potential mechanisms regulating this process may involve reduced hepatic malonyl-CoA levels in PKCβ−/−mice. Recent reports have indicated that reduced malonyl-CoA levels are associated with increased fat oxidation, because reduction of ACC-2 expression in PKCβ−/−mice would down-regulate mitochondrial malonyl-CoA, thus releasing its inhibition of CPT-1 activity, and thereby stimulating β-oxidation54,55. Decreased plasma insulin levels may be responsible for reduced hepatic ACC-2 expression in PKCβ−/− mice56. Moreover, altered expression of genes involved in fatty acid synthesis in the liver may further contribute to changes in the PKCβ−/− mice. The lean phenotype could be due to reduced lipogenesis, because we have observed significantly reduced expression of SREBP-1c and its target gene encoding fatty acid synthase. It appears that the combined effect of increased fatty acid oxidation and decreased lipid synthesis in the livers of PKCβ−/− mice may in part account for resistance of the PKCβ−/−mice to acquisition of a fatty liver.
Our results also uncovered an important regulatory role for PKCβ in altering adipose lipid metabolism, including misregulated lipolysis, storage, and lipogenesis. Our analysis indicates elevated adipose hormone-sensitive lipase mRNA in PKCβ−/−mice. Also, promoting the increased lipolysis in the PKCβ−/−mice adipose is decreased perilipin A expression. Furthermore, FAS expression was decreased in the PKCβ−/− adipose tissue relative to WT mice tissue. In addition, UCP-3 expression is decreased in both WAT and BAT of PKCβ−/−mice. In rodents and humans administration of fatty acids increased muscle UCP-3 expression57,58. However, UCP-3 is also regulated by several factors, including thyroid hormone, β3-adrenergic agonists and leptin, as well as fat feeding in rodents. One or more of these factors may be responsible for the down-regulation of UCP-3 in PKCβ−/−mice. Our results may reconcile studies that have shown increased UCP-3 mRNA in type 2 diabetic patients, characterized by higher TG levels58. Our observation that PKCβ−/−mice exhibit decreased UCP-3 expression and reduced fat mass is consistent with human association studies. We thus favor the interpretation that protection from obesity with the PKCβ deficiency is mechanistically linked to reduced energy storage as a consequence of increased oxidation of energy that would otherwise be stored. It remains to be seen if high levels of PKCβ may be a component of the obesity risk profile.
It is also likely that contributions from other sites of PKCβ action may regulate energy expenditure. In particular, PKCβ is also expressed in the brain (Mehta, K.D. et al, Unpublished results), and it may influence PKCβ-dependent signaling events mediated by either insulin and/or leptin at this site. While it has been suggested that insulin-mediated signaling in the hypothalamus controls sympathetic outflow and indirectly stimulates energy expenditure, it is not firmly established that the metabolic effects of insulin in the hypothalamus are dependent upon the activity of PKCβ. A recent report has linked PKCβ to β-adrenergic signaling in adipocytes59. Likewise, occurrence of adaptive changes in lipid metabolism in liver that does not induce this gene in response to HFD indicates that importance of cross-talk between adipose and liver, and has been documented repeatedly60–63. Adipokines are known to act via endocrine, paracrine, autocrine, and/or juxatacrine modes of action to modulate fat depot size and body fat redistribution and ultimately influence the obesity and liver damage. Both leptin and adiponectin have each been demonstrated to increase rates of fatty acid oxidation and decrease body’s lipid contents63,64. An increase in body fat causes an increase in circulating leptin levels, which will normally decrease the energy intake and increase energy expenditure, yet in the WT mice, the ratio of leptin to body weight and fat mass was much higher than that in the PKCβ−/−mice. Thus, unlike PKCβ−/−mice, WT mice develop leptin resistance with HFD, mimicking humans with diet-induced obesity. There are at least two potential mechanisms by which PKCβ may influence leptin sensitivity in mice. First, it may positively regulate leptin transcription and PKCβ deficiency reduces leptin expression. Our results demonstrating that the leptin mRNA expression is markedly reduced in PKCβ−/−mice implicates PKCβ-dependent mechanisms in the transcription control of leptin promoter. Modulation of transactivation potential of Sp1 and HIF-1α through phosphorylation by multiple kinases, including PKC and p42/44MAPK strongly supports such a possibility65. Alternatively, PKCβ may be necessary for other reactions and processes required for reducing transmission of the leptin signal to the effector molecules. There is significant but indirect evidence to suggest that one or more calcium-dependent PKC may play a major role in influencing leptin signaling. These two mechanisms may also operate in a synergistic fashion to simultaneously influence leptin expression and signaling. In addition, PKCβ−/−mice showed a slight increase in adiponectin levels, without significantly increasing adiponectin mRNA levels in either WAT or BAT. It is not known if the modest increase in adiponectin in PKCβ−/−mice functionally accounts for increased fatty acid oxidation and decreased hepatic steatosis.
It is interesting to note that in contrast to human subjects and several mouse models of lipodystrophy43–45, the loss of adiposity in the PKCβ−/−mice also led to increased insulin sensitivity. The reduced TG content in PKCβ−/−mice skeletal muscle or liver may enhance insulin sensitivity in these tissues66,67. Several additional features of the PKCβ−/− phenotype may contribute to increased insulin sensitivity in these mice. In particular, decreased adipocyte size, as we have observed in PKCβ−/−mice, is associated with increased insulin sensitivity. Also, hormonal effects of PKCβ deficiency may alter insulin sensitivity. Altered plasma levels of adipokines are frequently seen in patients with type 2 diabetes68. Reduced adipose tissue in PKCβ−/−mice is expected to alter plasma adipokines levels, thereby affecting glucose metabolism. In support of this possibility, dramatic reduction of leptin levels were observed in PKCβ−/−mice, and lower leptin levels have been correlated with insulin sensitivity69. In view of insulin regulating leptin gene expression70, it is likely that low insulin levels in PKCβ−/−mice accounts for markedly reduced leptin levels. The increase in plasma adiponectin levels may have additionally contributed to improvements in insulin sensitivity directly or through the enhancement of insulin action71. What is also interesting is the observation that, despite the significant disparity in fat mass, PKCβ−/−mice displayed slightly higher levels of adiponectin compared to WT mice on a HFD. We suggest that PKCβ deficiency might increase plasma adiponectin levels in the context of a lean phenotype, to levels slightly higher than those observed in WT mice, due to posttranslational control of protein secretion and degradation within the secretary pathway72,73. It appears that, in the setting of HFD-induced obesity, PKCβ may be a potential intermediary molecule linking obesity to liver damage and insulin resistance.
In conclusion, our study has revealed a novel role for PKCβ in diet-induced obesity. Our study suggests that a consequence of PKCβ deficiency is an activation of lipid oxidation in addition to reduced fatty acid synthesis and storage. It appears reasonable to propose that PKCβ is required in a signaling pathway that facilitate energy storage in response to a HFD, in part by triggering biological responses designed to paradoxically increase the efficiency of energy storage. This would represent an example of the “thrifty gene” phenotype and represent an advantage during times of food shortage74. However, in times of feast and sedentary lifestyle, such as today, PKCβ appears to provide a survival disadvantage, causing an epidemic of hypertrophic obesity. It is possible that upregulation of PKCβ expression and desensitization of insulin signaling in accumulated fat may serve as one of the mechanisms for maintaining obesity. Another intriguing finding is that few insulin signaling mediators have been reported to be diet-regulated at the transcriptional level in adipose tissue75. Our study provides the first evidence that an insulin signaling kinase, PKCβ, is physiologically regulated at the transcriptional level in adipose tissue by dietary fat. Our data provide support for the hypothesis that adipose PKCβ is controlled specifically by factors responding to the consumption of dietary fat and that the expression of PKCβ is linked to the development of the obesity. Future studies will determine whether induction of adipose PKCβ expression constitutes an early defect in the pathogenesis of obesity. It is anticipated that suppression of PKCβ expression or activity could be therapeutically effective for the management and prevention of obesity and related disorders.
Fig. 7.
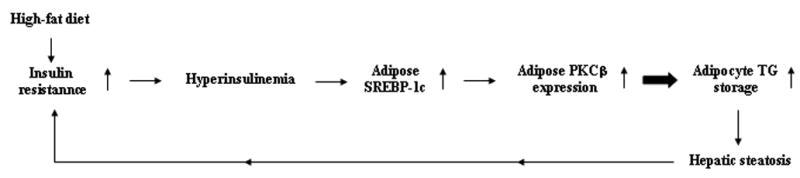
Schematic diagram of the hypothetical sequence of events whereby HFD-induced hyperinsulinemia stimulates expression of SREBP-1c, which in turn increases PKCβ expression and promotes TG storage and hepatic steatosis.
Acknowledgments
Financial support: This work was supported by National Institutes of Health Grant HL079091.
Abbreviations
- WAT
white adipose tissue
- PKCβ
protein kinase Cβ
- PKCβ−/−
PKCβ-deficient mice
- HFD
high-fat diet
- WT
wild-type mice
- BAT
brown adipose tissue
- TG
triglyceride
- SREBP
sterol response element-binding protein
- ITT
insulin tolerance test
- GTT
glucose tolerance test
- UCP
uncoupling protein
References
- 1.Kopelman PG. Obesity as a medical problem. Nature. 2000;404:635–643. doi: 10.1038/35007508. [DOI] [PubMed] [Google Scholar]
- 2.Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature. 2001;414:782–787. doi: 10.1038/414782a. [DOI] [PubMed] [Google Scholar]
- 3.Young LR, Nestle M. The contribution of expanding portion sizes to the US obesity epidemic. Am J Public Health. 2002;92:246–249. doi: 10.2105/ajph.92.2.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest. 2000;106:171–176. doi: 10.1172/JCI10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tang EY, Parker PJ, Beattie J, Houslay MD. Diabetes induces selective alterations in the expression of protein kinase C isoforms in hepatocytes. FEBS Lett. 1993;326:117–123. doi: 10.1016/0014-5793(93)81774-t. [DOI] [PubMed] [Google Scholar]
- 6.Xia P, Inoguchi T, Kern TS, Engerman RL, Oates PJ, King GL. Characterization of the mechanism for the chronic activation of diacylglycerol-protein kinase C pathway in diabetes and hypergalactosemia. Diabetes. 1994;43:1122–1129. doi: 10.2337/diab.43.9.1122. [DOI] [PubMed] [Google Scholar]
- 7.Considine RV, Nyce MR, Allen LE, Morales LM, Triester S, Serrano J, Colberg J, Lanza-Jacoby S, Caro JF. Protein kinase C is increased in the liver of humans and rat with non-insulin-dependent diabetes mellitus: an alteration not due to hyperglycemia. J Clin Invest. 1995;95:2938–2944. doi: 10.1172/JCI118001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shmueli E, Alberti KGEM, Record CO. Diacylglycerol/protein kinase C signaling: a mechanism for insulin resistance? J Int Med. 1993;234:397–400. doi: 10.1111/j.1365-2796.1993.tb00761.x. [DOI] [PubMed] [Google Scholar]
- 9.Inoguchi T, Battan R, Handler E, Sportsman JR, Heath W, King GL. Preferential elevation of protein kinase C isoform beta II and diacylglycerol levels in the aorta and heart of diabetic rats: differential reversibility to glycemic control by islet cell transplantation. Proc Natl Acad Sci USA. 1992;89:11059–11063. doi: 10.1073/pnas.89.22.11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Itani SI, Zhou Q, Pories WJ, MacDonald KG, Dohm GL. Involvement of protein kinase C in human skeletal muscle insulin resistance and obesity. Diabetes. 2000;49:1353–1358. doi: 10.2337/diabetes.49.8.1353. [DOI] [PubMed] [Google Scholar]
- 11.Schmitz-Peiffer C, Biden TJ. Protein kinase C function in muscle, liver, and β-cells and its therapeutic implications for type 2 diabetes. Diabetes. 2008;57:1774–1783. doi: 10.2337/db07-1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rask-Madsen C, King GL. Proatherosclerotic mechanisms involving protein kinase C in diabetes and insulin resistance. Atheroscler Thromb Vasc Biol. 2005;25:487–496. doi: 10.1161/01.ATV.0000155325.41507.e0. [DOI] [PubMed] [Google Scholar]
- 13.Mellor H, Parker P. The extended protein kinase C superfamily. Biochem J. 1998;332:281–292. doi: 10.1042/bj3320281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nishizuka Y. The molecular heterogeneity of protein kinase C and its implications for cellular regulation. Nature. 1998;344:661–665. doi: 10.1038/334661a0. [DOI] [PubMed] [Google Scholar]
- 15.Blobe GC, Khan WA, Halpern AE, Obeid LM, Hannun YA. Selective regulation of expression of protein kinase C beta isoenzymes occurs via alternative splicing. J Biol Chem. 1993;268:10627–10635. [PubMed] [Google Scholar]
- 16.Dempsey EC, Newton AC, Mochly-Rosen D, Fields AP, Reyland ME, Insel PA, Messing RO. Protein kinase C isozymes and the regulation of diverse cell responses. Am J Physiol Lung Cell Mol Physiol. 2000;279:L429–L438. doi: 10.1152/ajplung.2000.279.3.L429. [DOI] [PubMed] [Google Scholar]
- 17.Patel NA, Apostolatos HS, Mebert K, Chalfant CE, Watson JE, Pillay TS, Sparks J, Cooper DR. Insulin regulates PKCβII alternative splicing in multiple target tissues: Development of a hormonally responsive heterologous minigene. Mol Endocrinol. 2004;18:899–911. doi: 10.1210/me.2003-0391. [DOI] [PubMed] [Google Scholar]
- 18.Schmitz-Peiffer C. Protein kinase C and lipid-induced insulin resistance in skeletal muscle. Ann N Y Acad Sci. 2002;967:146–157. doi: 10.1111/j.1749-6632.2002.tb04272.x. [DOI] [PubMed] [Google Scholar]
- 19.Su TT, Guo B, Kawakami Y, Sommer K, Chae K, Humphries LA, Kato RM, Kang S, Patrone L, Wall R, Teitell M, Leitges M, Kawakami T, Rawlings DJ. PKC-beta controls I kappa B kinase lipid raft recruitment and activation in response to BCR signaling. Nature Immunol. 2002;3:780–786. doi: 10.1038/ni823. [DOI] [PubMed] [Google Scholar]
- 20.Aguirre V, Werner ED, Giraud J, Lee YH, Shoelson SE, White MF. Phosphorylation of Ser(307) in insulin receptor substrate-1 blocks interaction with the insulin receptor and inhibits insulin action. J Biol Chem. 2002;277:1531–1537. doi: 10.1074/jbc.M101521200. [DOI] [PubMed] [Google Scholar]
- 21.Liu GK, Zhou H, Dai Z, Zhang J, Sun R, Chen J, Sun Q, Lu W, Kang X, Chen P. Involvement of protein kinase C beta-extracellular signal-regulating kinase 1/2/p38 mitogen-activated protein kinase-heat shock protein 27 activation in hepatocellular carcinoma cell motility and invasion. Cancer Sci. 2008;99:486–496. doi: 10.1111/j.1349-7006.2007.00702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Fea K, Roth RA. Protein kinase C modulation of insulin receptor substrate-1 tyrosine phosphorylation requires serine 612. Biochem. 1997;36:12939–12947. doi: 10.1021/bi971157f. [DOI] [PubMed] [Google Scholar]
- 23.De Fea K, Roth RA. Modulation of insulin receptor substrate-1 tyrosine phosphorylation and function by mitogen-activated protein kinase. J Biol Chem. 1997;272:31400–31406. doi: 10.1074/jbc.272.50.31400. [DOI] [PubMed] [Google Scholar]
- 24.Osterhoff MA, Heuer S, Pfeiffer M, Tasic J, Kaiser S, Isken F, Spranger J, Weickert MO, Möhlig M, Pfeiffer AF. Identification of a functional protein kinase Cβ promoter polymorphism in humans related to insulin resistance. Mol Genet Metab. 2008;93:210–215. doi: 10.1016/j.ymgme.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 25.Ikeda Y, Suchiro T, Osaki F, Tsuzura S, Kumon Y, Hashimoto K. Polymorphisms in the 5′-upstream region of the PKCbeta gene in Japanese patients with Type 2 diabetes. Diabet Med. 2004;21:1113–1120. doi: 10.1111/j.1464-5491.2004.01304.x. [DOI] [PubMed] [Google Scholar]
- 26.Ishii H, Jirousek MR, Koya D, Takagi C, Xia P, Clermont A, Bursell SE, Kern TS, Ballas LM, Heath WF, Stramm LE, Feener EP, King GL. Amelioration of vascular dysfunctions in diabetic rats by an oral PKCβ inhibitor. Science. 1996;272:728–731. doi: 10.1126/science.272.5262.728. [DOI] [PubMed] [Google Scholar]
- 27.Avignon A, Yamada K, Zhou X, Spencer B, Cardona O, Saba-Siddique S, Galloway L, Standaert ML, Farese RV. Chronic activation of protein kinase C in soleus muscle and other tissues of insulin-resistant type II diabetic Goto-Kakizaki (GK), obese/aged, and obese/Zucker rats: a mechanism for inhibiting glycogen synthesis. Diabetes. 1996;45:1396–1404. doi: 10.2337/diab.45.10.1396. [DOI] [PubMed] [Google Scholar]
- 28.Choe M, Kris ES, Luthra R, Copenhaver J, Pelling JC, Donnelly TE, Birt DF. Protein kinase C is activated and diacylglycerol is elevated in epidermal cells from Sencar mice fed high fat diets. J Nutr. 1992;122:2322–2329. doi: 10.1093/jn/122.12.2322. [DOI] [PubMed] [Google Scholar]
- 29.Pajari AM, Rasilo ML, Mutanen M. Protein kinase C activation in rat colonic mucosa after diets differing in their fatty acid composition. Cancer Lett. 1997;114:101–103. doi: 10.1016/s0304-3835(97)04635-1. [DOI] [PubMed] [Google Scholar]
- 30.Pajari AM, Mutanen M. Phospholipid fatty acid composition and protein kinase C activity in the large intestine of rats fed on butter and coconut-oil diets. Br J Nutr. 1999;82:411–418. doi: 10.1017/s0007114599001658. [DOI] [PubMed] [Google Scholar]
- 31.Jalili T, Manning J, Kim S. Increased translocation of cardiac protein kinase Cβ2 accompanis mild cardiac hypertrophy in rats fed saturated fat. J Nutr. 2003;133:358–361. doi: 10.1093/jn/133.2.358. [DOI] [PubMed] [Google Scholar]
- 32.Bansode RR, Huang W, Roy SK, Mehta M, Mehta KD. Protein kinase Cβ deficiency increases fatty acid oxidation and reduces fat storage. J Biol Chem. 2008;283:231–236. doi: 10.1074/jbc.M707268200. [DOI] [PubMed] [Google Scholar]
- 33.Kumar A, Chambers TC, Cloud-Heflin BA, Mehta KD. Phorbol ester-induced low density lipoprotein receptor gene expression in HepG2 cells involves protein kinase C-mediated p42/44 MAP kinase activation. J Lipid Res. 1997;38:2240–2248. [PubMed] [Google Scholar]
- 34.Huang W, Batra S, Atkins BA, Mishra V, Mehta KD. Selective repression of LDL receptor expression by SP600125: Coupling of histone H3-Ser10 phosphorylation and Sp1 occupancy. Mol Cell Biol. 2006;26:1307–1317. doi: 10.1128/MCB.26.4.1307-1317.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reitman ML. Metabolic lessons from genetically lean mice. Annu Rev Nutr. 2002;22:459–482. doi: 10.1146/annurev.nutr.22.010402.102849. [DOI] [PubMed] [Google Scholar]
- 36.Hallas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, Lallone RL, Burley SK, Friedman JM. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269:543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- 37.Ishizuka T, Kajita K, Yamada K, Miura A, Kanoh Y, Ishizawa M, Wada H, Itaya S, Yamamoto M, Yasuda K, Nagata K, Okano Y. Insulin regulates PKC isoform mRNA in rat adipocytes. Diabetes Res Clin Practice. 1996;33:159–167. doi: 10.1016/0168-8227(96)01324-1. [DOI] [PubMed] [Google Scholar]
- 38.Kotzka J, Muller-Wieland D, Koponen A, Njamen D, Kremer L, Roth G, Munck M, Knebel B, Krone W. ADD1/SREBP-1c mediates insulin-induced gene expression linked to the MAP kinase pathway. Biochem Biophys Res Commun. 1998;249:375–379. doi: 10.1006/bbrc.1998.9161. [DOI] [PubMed] [Google Scholar]
- 39.Foretz M, Pacot C, Dugail I, Lemarchand P, Guichard C, Liepvre XL, Bertelier-Lubrano C, Spiegelman B, Kim JB, Ferre P, et al. ADD1/SREBP-1c required in the activation of hepatic lipogenic gene expression by glucose. Mol Cell Biol. 1999;19:3760–3768. doi: 10.1128/mcb.19.5.3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Niino YS, Ohno S, Suzuki K. Positive and negative regulation of the transcription of the human protein kinase Cβ gene. J Biol Chem. 1992;267:6158–6163. [PubMed] [Google Scholar]
- 41.Obeid LM, Blobe GC, Karolak LA, Hannun YA. Cloning and characterization of the major promoter of the human protein kinase C β gene. J Biol Chem. 1992;267:20804–20810. [PubMed] [Google Scholar]
- 42.Shimano H, Horton JD, Shimomura I, Hammer RE, Brown MS, Goldstein JL. Isoform 1c of sterol regulatory element binding protein is less active than isoform 1a in livers of transgenic mice and in cultured cells. J Clin Invest. 1997;99:846–854. doi: 10.1172/JCI119248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shimomura I, Hammer RE, Richardson JA, Ikemoto S, Bashmakov Y, Goldstein JL, Brown MS. Insulin resistance and diabetes mellitus in transgenic mice expressing nuclear SREBP-1c in adipose tissue: model for congenital generalized lipodystrophy. Genes & Dev. 1998;12:3182–3194. doi: 10.1101/gad.12.20.3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McKnight SL. WAT-free mice: diabetes without obesity. Genes & Dev. 1998;12:3145–3148. [PubMed] [Google Scholar]
- 45.Kim JK, Gavrilova O, Chen Y, Reitman ML, Shulman GI. Mechanism of insulin resistance in A-ZIP/F-1 fatless mice. J Biol Chem. 2000;275:8456–8460. doi: 10.1074/jbc.275.12.8456. [DOI] [PubMed] [Google Scholar]
- 46.Qiu J, Ogus S, Lu R, Chehab FF. Transgenic mice overexpressing leptin accumulate adipose mass at an older, but not younger, age. Endocrinol. 2001;142:348–358. doi: 10.1210/endo.142.1.7909. [DOI] [PubMed] [Google Scholar]
- 47.Shimada M, Tritos NA, Lowell BB, Flier JS, Maratos-Flier E. Mice lacking melanin-concentrating hormone are hypophagic and lean. Nature. 1998;396:670–674. doi: 10.1038/25341. [DOI] [PubMed] [Google Scholar]
- 48.Matsui M, Motomura D, Karasawa H, Fujikawa T, Jiang J, Komiya Y, Takahashi S, Taketo MM. Multiple functional defects in peripheral autonomic organs in mice lacking muscarinic acetylcholine receptor gene for the M3 subtype. Proc Natl Acad Sci USA. 2000;97:9579–9584. doi: 10.1073/pnas.97.17.9579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamada M, Miyakawa T, Duttaroy A, Yamanaka A, Moriguchi T, Makita R, Ogawa M, Chou CJ, Xia B, Crawley JN, Felder CC, Deng CX, Wess J. Mice lacking the M3 muscarinic acetycholine receptor are hypophagic and lean. Nature. 2001;410:207–212. doi: 10.1038/35065604. [DOI] [PubMed] [Google Scholar]
- 50.Abu-Elheiga L, Matzuk MM, Abo-Hashema KA, Wakil SJ. Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science. 2001;291:2613–2616. doi: 10.1126/science.1056843. [DOI] [PubMed] [Google Scholar]
- 51.Smith SJ, Cases S, Jensen DR, Chen HC, Sande E, Tow B, Sanan DA, Raber J, Eckel RH, Farese RV., Jr Obesity resistance and multiple mechanisms of triglyceride synthesis in mice lacking Dgat. Nat Genet. 2000;25:87–90. doi: 10.1038/75651. [DOI] [PubMed] [Google Scholar]
- 52.Xia Z, Stanhope, et al. Acylation stimulating protein (ASP)/complement C3adesArg deficiency results in increased energy expenditure in mice. J Biol Chem. 2004;279:4051–4057. doi: 10.1074/jbc.M311319200. [DOI] [PubMed] [Google Scholar]
- 53.Ferrannini E. Physiological and metabolic consequences of obesity. Metabolism. 1995;44:15–17. doi: 10.1016/0026-0495(95)90313-5. [DOI] [PubMed] [Google Scholar]
- 54.Saggerson D. Malonyl-CoA, a key signaling molecule in mammalian cells. Ann Rev Nutr. 2008;28:253–272. doi: 10.1146/annurev.nutr.28.061807.155434. [DOI] [PubMed] [Google Scholar]
- 55.Ruderman NB, Saha AK, Vavvas D, Witters LA. Malonyl-CoA, fuel sensing, and insulin resistance. Am J Physiol Endocrinol Metab. 1999;276:E1–E18. doi: 10.1152/ajpendo.1999.276.1.E1. [DOI] [PubMed] [Google Scholar]
- 56.Witters LA, Kemp BE. Insulin activation of acetyl-CoA carboxylase accompanied by inhibition of the 5′-AMP-activated protein kinase. J Biol Chem. 1992;267:2864–2867. [PubMed] [Google Scholar]
- 57.Vidal H, Langin D, Andreelli F, Millet L, Larrouy D, Laville M. Lack of uncoupling protein 2 and 3 mRNA induction during fasting in type 2 diabetic subjects. Am J Physiol. 1999;277:E830–E837. doi: 10.1152/ajpendo.1999.277.5.E830. [DOI] [PubMed] [Google Scholar]
- 58.Bao S, Kennedy A, Wojciechowski B, Wallace P, Ganaway E, Garvey WT. Expression of mRNAs encoding uncoupling proteins in human skeletal muscle: effects of obesity and diabetes. Diabetes. 1998;47:1935–1940. doi: 10.2337/diabetes.47.12.1935. [DOI] [PubMed] [Google Scholar]
- 59.Nakamura J. Protein kinase CβI interacts with the β1-adrenergic signaling pathway to attenuate lipolysis in rat adipocytes. Biochim Biophys Acta. 2008;1781:277–281. doi: 10.1016/j.bbalip.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 60.Czaja MJ. Liver injury in the setting of steatosis: cross-talk between adipokine and cytokine. Hepatology. 2004;40:19–22. doi: 10.1002/hep.20328. [DOI] [PubMed] [Google Scholar]
- 61.Rajala MW, Scherer PE. The adipocyte-at the crossroads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology. 2003;144:3765–3773. doi: 10.1210/en.2003-0580. [DOI] [PubMed] [Google Scholar]
- 62.Pittas AG, Joseph NA, Greenberg AS. Adipocytokines and insulin resistance. J Clin Endocrinol & Metab. 2004;89:447–452. doi: 10.1210/jc.2003-031005. [DOI] [PubMed] [Google Scholar]
- 63.Yu YH, Ginsberg HN. Adipocyte signaling and lipid homeostasis: Sequelae of insulin-resistant adipose tissue. Circulation Res. 2005;96:1042–1052. doi: 10.1161/01.RES.0000165803.47776.38. [DOI] [PubMed] [Google Scholar]
- 64.Dyck DJ, Heigenhauser GJF, Bruce CR. The role of adipokines as regulators of skeletal muscle fatty acid metabolism and insulin sensitivity. Acta Physiol. 2006;186:5–16. doi: 10.1111/j.1748-1716.2005.01502.x. [DOI] [PubMed] [Google Scholar]
- 65.Bartella V, Cascio S, Fiorio E, Auriemma A, Russo A, Surmacz E. Insulin-dependent leptin expression in breast cancer cells. Cancer Res. 2008;68:4919–4927. doi: 10.1158/0008-5472.CAN-08-0642. [DOI] [PubMed] [Google Scholar]
- 66.Phillips DI, Caddy S, Ilic V, Fielding BA, Frayn KN, Borthwick AC, Taylor R. Intramuscular triglyceride and muscle insulin sensitivity: evidence for a relationship in nondiabetic subjects. Metabolism. 1996;45:947–950. doi: 10.1016/s0026-0495(96)90260-7. [DOI] [PubMed] [Google Scholar]
- 67.Pan DA, Lillioja S, Kriketos AD, Milner MR, Baur LA, Bogardus C, Jenkins AB, Storlien LH. Skeletal muscle triglyceride levels are inversely related to insulin action. Diabetes. 1997;46:983–988. doi: 10.2337/diab.46.6.983. [DOI] [PubMed] [Google Scholar]
- 68.Antuna-Puente B, Feve B, Fellahi S, Bastard JP. Adipokines: the missing link between insulin resistance and obesity. Diabetes Metab. 2008;34:2–11. doi: 10.1016/j.diabet.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 69.Zimmet PZ, Collins VR, de Courten MP, Hodge AM, Collier GR, Dowse GK, Alberti KG, Tuomilehto J, Hemraj F, Gareeboo H, Chitson P, Fareed D. Is there a relationship between leptin and insulin sensitivity independent of obesity? A population-based study in the Indian ocean nation of Mauritius. Int J Obes Relat Metab Disord. 1998;22:171–177. doi: 10.1038/sj.ijo.0800559. [DOI] [PubMed] [Google Scholar]
- 70.Kolaczynski J, Nyce MR, Considine RV, Boden G, Nolan JJ, Henry R, Mudaliar SR, Olefsky J, Caro JF. Acute and chronic effects of insulin on leptin production in humans. Studies in vivo and in vitro. Diabetes. 1996;45:699–701. doi: 10.2337/diab.45.5.699. [DOI] [PubMed] [Google Scholar]
- 71.Hotta K, Funahashi T, Bodkin NL, Ortmeyer HK, Arita Y, Hansen BC, Matsuzawa Y. Circulating concentrations of the adipocyte protein adiponectin are decreased in parallel with reduced insulin sensitivity during the progression to type 2 diabetes in rhesus monkeys. Diabetes. 2001;50:1126–1133. doi: 10.2337/diabetes.50.5.1126. [DOI] [PubMed] [Google Scholar]
- 72.Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka N, Ezaki O, Akanuma Y, Gavrilova O, Vinson C, Reitman ML, Kagechika H, Shudo K, Yoda M, Nakano Y, Tobe K, Nagai R, Kimura S, Tomita M, Froguel P, Kadowaki T. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. 2001;7:941–946. doi: 10.1038/90984. [DOI] [PubMed] [Google Scholar]
- 73.Combs TP, Wagner JA, Berger J, Doebber T, Wang WJ, Zhang BB, Tanen M, Berg AH, O’Rahilly S, Savage DB, Chatterjee K, Weiss S, Larson PJ, Gottesdiener KM, Gertz BJ, Charron MJ, Scherer PE, Moller DE. Induction of adipocyte complement-related protein of 30 kilodaltons by PPAR-gamma agonists: a potential mechanism of insulin sensitization. Endocrinol. 2002;143:998–1007. doi: 10.1210/endo.143.3.8662. [DOI] [PubMed] [Google Scholar]
- 74.Neel JV. Diabetes mellitus: a “thrifty” genotype rendered detrimental by “progress”? 1962. Bull World Heath Organ. 1999;77:694–703. [PMC free article] [PubMed] [Google Scholar]
- 75.Shimomura I, Matsuda M, Hammer RE, Bashmakov Y, Brown MS, Goldstein JL. Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol Cell. 2000;6:77–86. [PubMed] [Google Scholar]