

Abstract
The co-chaperone Hop [heat shock protein (HSP) organising protein] is known to bind both Hsp70 and Hsp90. Hop comprises three repeats of a tetratricopeptide repeat (TPR) domain, each consisting of three TPR motifs. The first and last TPR domains are followed by a domain containing several dipeptide (DP) repeats called the DP domain. These analyses suggest that the hop genes result from successive recombination events of an ancestral TPR–DP module. From a hydrophobic cluster analysis of homologous Hop protein sequences derived from gene families, we can postulate that shifts in the open reading frames are at the origin of the present sequences. Moreover, these shifts can be related to the presence or absence of biological function. We propose to extend the family of Hop co-chaperons into the kingdom of bacteria, as several structurally related genes have been identified by hydrophobic cluster analysis. We also provide evidence of common structural characteristics between hop and hip genes, suggesting a shared precursor of ancestral TPR–DP domains.
Electronic supplementary material
The online version of this article (doi:10.1007/s12192-008-0083-8) contains supplementary material, which is available to authorized users.
Keywords: Hop, HSP organizing protein, Hsp70 interacting protein, Hydrophobic cluster analysis, Tetratricopeptide repeat, TPR protein
Introduction
The first sequenced hop gene was an open reading frame (ORF) coding for a protein related to stress response in Saccharomyces cerevisiae (STI1); however, the function was not clearly established, although some experiments suggested it was involved in activating heat shock proteins (HSPs; Nicolet and Craig 1989). Homologous proteins were successively characterized in Homo sapiens (IEF SSP 3521; Honoré et al. 1992) and soybean (Glycine max sti, GMSTI; Torres et al. 1995). Lately, two soybean ESTs, GmHop1 and GmHop2, were reported; neither were homologous to GMSTI (Zhang et al. 2003). The human IEF SSP 3521 was described as interacting with Hsp70 and Hsp90 and was renamed Hsp70/90 organizing protein (Hop).
Numerous alternative names of Hop exist and are: p60 (Chen et al. 1996); RF-HSP70 (Gross and Hessefort 1992); Stip1 (Mizrak et al. 2006); and mSTI1 (Blatch et al. 1997). Two genes have been described in the plant Arabidopsis thaliana and Interpro (Mulder and Apweiler 2008); in addition to those in soybean, the presence of several hop genes is mentioned for various plant species.
Structural analysis of GMSTI protein revealed the presence of a tetratricopeptide repeat (TPR) domain, the first known in a plant. It is composed of a 34 amino acid (aa) motif, called TPR motif (for tetratricopeptide repeat), which is repeated several times with some degeneracy. Throughout this paper, we will call TPR motif this 34 aa repeat and TPR domain several successive TPR motifs forming a domain. Scheufler et al. (2000) were able to define three TPR domains in human Hop, namely, TPR1 (4–118), TPR2A (223–352), and TPR2B (353–477), with a fairly homologous sequence identity of 29% between TPR1 and TPR2A and of 34% between TPR1 and TPR2B. The C-terminal 70 aa of Hop is termed DP due to a number of dipeptide repeats (Chen and Smith 1998). The 100 aa long region separating TPR1 from TPR2A is composed of a duplication of this DP domain, followed by a hinge region. Recently, the two domains following TPR1 and TPR2B have been called DP1 and DP2, respectively (Carrigan et al. 2005), and they share 27% sequence identity.
TPR domains are likely to be ancient since they are found in Eukarya, Bacteria, and Archaea in many different subfamilies, such as kinesin light chains, SNAP secretory proteins, and clathrin heavy chains (Andrade et al. 2001). The TPR and DP domains are, however, only found together in Hop and Hip (Hsp70 interacting protein) homologs (Nelson et al. 2003). Each TPR domain contains three TPR motifs (34 aa), and each motif gives rise to a structure composed of two successive alpha helices. Therefore, a TPR domain comprises six helices, named A1, B1, A2, B2, A3, and B3. A seventh helix, called C, can sometimes be found at the C-terminal end, but its sequence is highly divergent from the six other helices. A global propensity to build a TPR motif has been derived from a multiple alignment of 1,827 TPR motifs (not domains; Main et al. 2003), and one third of the consensus is composed of topologically conserved hydrophobic residues, even if non-hydrophobic residues in TPR domains are involved in the binding to the HSP chaperons (Liu et al. 1999).
TPR domains are involved in many protein–protein interactions and act as co-chaperons (Blatch and Lässle 1999). The structure of human Hop has been solved for the TPR1 and TPR2A domains complexed with C-terminal peptides containing the EEVD motif from Hsp70 and Hsp90 (PDB codes 1ELW and 1ELR, respectively). Currently, there is no structure available for any DP domain, although complete models of human Hop are proposed in ModBase (code P31948) ranging from 6% to 22% sequence identity with the templates. A complex is formed between Hop, Hsp70, and Hsp90 in order to produce steroid receptor maturation (Kimmins and MacRae 2000). Hop interacts simultaneously with the EEVD C-terminal motif of Hsp70 through its TPR1 domain and with the same highly conserved motif of Hsp90 through its TPR2A motif (Demand et al. 1998; Carrello et al. 1999), thus, acting as an adaptor protein (Chen and Smith 1998; Odunuga et al. 2004).
Several Hop-containing multiprotein complexes are present in eukaryotes with different functions (reviewed in Odunuga et al. 2004) such as: oligomerization of HSF1; protein folding for Gcn2, Hsp104, CDC37, and Harc complexes; translocation of prion protein complexes; activation of hepatitis B virus reverse transcriptase complex; activation of Drosophila ecdysone EcR/USP receptor; and cell division with the 20S cytosome complex. In humans, the Hop protein described (IEF SSP 3521) mentioned the presence of one single functional gene with five variations, “corresponding more to charge variants than to a family of related proteins” (Honoré et al. 1992), allowing diversity in subcellular location. Isoforms can be due to post translational modifications, which depend on subcellular location or to different sequences. The latter point can be determined by HCA analysis.
In this paper, we explain the absence of DP1 domain in some species, such as Drosophila, or the absence of typical signature in GMSTI-DP1 and TcSTI1-DP2 (Trypanosoma cruzi STI). For the latter situation, we propose that shifts in the open reading frames of the genes result in the absence of DP1 or DP2, respectively. The behavior of DP domains as a potential globular domain, instead of a simple hinge region, will be discussed.
We will focus on the fact that hop genes are composed of several modules, defined as a set of exons surrounded by introns of the same phase. Hop is composed of three main modules surrounded by introns of phase 0. To date, Hip is the only known protein to contain TPR domains, located in the C-terminal region in proximity to a DP domain. Like Hop, Hip has been reported to bind to Hsp70 proteins. An alignment of TPR1-DP from Hop with the TPR–DP domains of Hip has been performed by means of HCA plots, and a genomic analysis is performed.
It has long been believed that Hop is ubiquitous in the eukaryotic world but absent in prokaryotes. The presence of a short Hop-like protein in a bacterium has been revealed from homology between DP domains in the Interpro database. In this article, we describe the GerD sequence from Bacillus subtilis (CAB11931) as a novel Hop-like protein. We propose that the GerD sequence is similar to one TPR–DP unit, revealing, therefore, the existence of a TPR domain in GerD. This discovery then suggests the presence of a common ancestor of Hop and GerD proteins.
Materials and methods
Hydrophobic cluster analysis
HCA is a method of drawing a sequence on a scaffold corresponding to an alpha helix. The sequence is written along the surface of an infinite cylinder, projected into 2D space; the image is duplicated in order to display the local environment for each amino acid resulting in clustering of neighboring hydrophobic residues. HCA realizes tightening of the sequence around hydrophobic amino acids. It has been statistically demonstrated that centers of the clusters correspond to the centers of regular secondary structures (Woodcock et al. 1992). The shapes of the clusters are keen indication of the nature of the secondary structure (Callebaut et al. 1997), due to the fact that the sequence is threaded along a fixed scaffold, a helix. As distribution of hydrophobic residues is different in strands (all residues are hydrophobic if the strand is buried and half of them if the strand faces the solvent) and in helices (most of helices are not buried, and there are 3.6 residues per turn), the clustering of the hydrophobic amino acids produces different patterns. Clusters are roughly vertical when they code for a strand, while they are fairly horizontal when they code for a helix. When analyzing a pair of HCA plots, one does not pay much attention to the exact conservation of the residues inside the clusters but rather to the conserved shapes of the clusters, keeping in mind the underlying idea that shape is a testimony of the secondary structure. Thus, it allows alignments between very distantly related proteins, with as low as 10% sequence identity. Guidelines for the method and access to the HCA program are available from http://www.lmcp.jussieu.fr/%7Emornon/hca.html.
Most interacting residue simulations
Lattice simulations have been performed in order to predict, from the sequence, the residues involved in the folding nucleus. Briefly, an initial conformation is chosen so that the C-alpha carbons are distributed at random on the nodes of a lattice, provided continuity of the backbone and self avoiding walk are accomplished. Displacements of the residues on empty nodes of the lattice are allowed following the Monte Carlo criterion until the chain reaches a sufficiently compact state. The mean number of neighbors is recorded, and the most interacting residues (MIRs) correspond to structurally important residues as they match the folding nucleus (Papandreou et al. 1998, 2004). Therefore, the density of MIR is a powerful tool for estimating the probability of a folded domain.
Intron phase definition
As stated by Patthy (1987), introns of protein genes are grouped following their position relative to the reading frame of the genes: phase 0 introns are those lying between two codons, e.g., 5′GAC CAG:GT—intron—AG:GTC ATG3′; phase 1 introns are those lying between the first and second nucleotides of a codon, e.g., 5′GA CCA G:GT—intron—AG:GT CAT G3′, and phase 2 introns are those lying between the second and third nucleotides of a codon, e.g., 5′G ACC AG:GT—intron—AG:G TCA TG3′.
Results
Hop orthologs lack DP1 or DP2 domains
Results from deletion mutants have been inconclusive, regarding the importance of DP1 domain for Hop activity (Chen and Smith 1998; Carrigan et al. 2004; Flom et al. 2007). DP2 has recently been demonstrated to be essential for the in vivo function (Flom et al. 2006). To further understand the role that DP domains may play in Hop, we examined two atypical orthologs from soybean (G. max) and T. cruzi with numerous alterations in aa composition of DP1 or DP2 sequences, presumably producing inactivity.
Several soybean genes have been reported to code for Hop protein: gmsti, which is convergently transcribed with nuclear fus genes (Torres et al. 1995) and two closely related genes (GmHop-1 and GmHop-2), which share nearly 90% nucleotide identity (Zhang et al. 2003). The unaltered GMSTI sequence (CAA56165) is called GMSTI-rf1 in Fig. 1a. Comparison between the HCA profiles of GmHop-1 and GMSTI-rf1 reveal conserved shaped hydrophobic clusters at both ends of DP1 domain between aa 120–137 and aa 173–190 (Fig. 1a). The central region of DP1 lacks secondary structure and long hydrophobic regions (Fig. 1a); suggesting that the assumption of one third of a protein density is composed of hydrophobic residues is incorrect for GMSTI-rf1. The second and third reading frames of gmsti gene give rise to the sequences named GMSTI-rf2 and GMSTI-rf3, respectively. The central region of rf2 and the N-terminal sequence of rf3 from the DP1 domain are equivalent to those of GmHop-1 and AtHop. Thus, rf3 contains the residues (120–145) and rf2 residues (146–172) of the missing fragment caused by the mutation described for GMSTI-rf1. This suggests that multiple mutations in the gmsti gene have occurred in the DP1 domain, thus, shifting the ORF to the reading frames 2 and 3 then returning to the original ORF near aa 173. The limits of the shifted regions are delineated by broken lines with up and down arrows in Fig. 1. In order to guarantee reliability of the gmsti cDNA sequence, an exhaustive reading of sequencing gels was performed with great care (Torres et al. 1995), confirming the hypothesis of a double shift, thus, leading to the globular nature of the GmHop1-DP1 domain.
Fig. 1.

HCA plots of the DP domains. Conserved hydrophobic clusters are grey shaded. Relevant nonhydrophobic identities are indicated on a black background and DP repeats by squares. The three reading frames are shown from the same gene sequence. Arrows delineate the location of the residues lost because of the frame shifts and that are recovered in the noncoding reading frames. Vertical lines connect the occurrences of the DP repeats. a DP1 domain HCA plots. AtHop putative A. thaliana Hop (NP_176461); GmHop-1 G. max Hop-1 (Zhang et al. 2003); GMSTI-rf1, rf2, and rf3 reading frames 1 (coding), 2, and 3 (noncoding) from G. max GMSTI (Torres et al. 1995). b DP2 domain HCA plots. TbHop putative T. brucei Hop (XP_825513); TcHop1 putative T. cruzi Hop1 (EAN97552); TcHop2-rf1 and TcHop2-rf3 correspond to the reading frames 1 (coding) and 3 (noncoding) from T. cruzi Hop2 (AAC97378), respectively. The onset helps interpreting the HCA plots. Because of the duplication (see “Materials and methods”), sequence is read vertically, one line over two, and the secondary structure is read horizontally, a cluster corresponding statistically to a regular secondary structure
The second case of a possible frameshift concerns the DP2 domain of the putative T. cruzi gene TcHop2 (AAC97378) in relation to its paralog (EAN97552). The actual sequence of TcHop2 gives rise to clusters compatible with Hop in the DP1 and hinge regions (data not shown); however, after the first 20 aa of DP2 (position 485 of Tc Hop2-rf1 in Fig. 1b), the ORF (TcHop2-rf1) lacks the clusters seen in the putative T. brucei TbHop (XP_825513) and T. cruzi TcHop1 (EAN97552). The end of DP2 is not compatible with the end of Hop, as the hydrophobic clusters are completely different; the correct cluster shapes (as seen in TcHop1 and TbHop) can be recovered by shifting the frame of TcHop2 gene to the position seen in rf3. As shown in Fig. 1b, the reading frame 3 (TcHop2-rf3) encodes the C-terminal 485–553 aa of TcHop2 with hydrophobic clusters of very similar shapes to the ones from TbHop and TcHop1. It is then feasible to hypothesize, once more, that a frameshift mutation (actually a G nucleotide deletion) has distorted the C-terminal end of TcHop2. TcHop1 and TcHop2 have a 98% identity, excluding the DP2 domain. A multiple alignment of DP1 with DP2 domains for human, soybean, and T. cruzi is available in Electronic supplementary material.
Genomic coevolution of TPR and DP domains, as a whole recombination unit
To understand why there are internal repeats in TPR proteins, it has been argued that convergent evolution is not feasible because of the high conservation of these domains (Andrade et al. 2001). Thus, one can imagine the occurrence of chromosomal rearrangements and recombination events such as gene duplication, exon duplication, and exon shuffling (Long 2001).
Evidence of rearrangements can be seen in Hop proteins coded for multigene families, the result of gene duplication. In the case of the A. thaliana genome (Rhee et al. 2003), at least two loci are involved, in chromosomes 1 (F23N19.10) and 4 (T1P17.2). Several hop genes have also been identified by southern hybridization in the soybean genome (unpublished data). We have found, by screening genomic databases (HGP bank) with BlastN (Altschul et al. 1990), two loci in human, on chromosomes 11 and X (Hs11_34058 and HsX_11808, respectively), with 94% sequence identity, revealing that duplication of hop genes is a widespread phenomenon.
The hop gene contains multiple introns; this is a typical feature found in recombination (Long et al. 1995; Fedorova and Fedorov 2003). In human intron VIII, the presence of an Alu element of type B2 (Batzer et al. 1996) is present, and this is often involved in recombination events which produce genomic diversity (Batzer and Deininger 2002). Human hop gene includes multiple introns of phase 0, which is the most favorable for exon duplication or shuffling without modifying the reading frame (Fig. 2a). According to Patthy (1985), genes can exchange modules. In this paper, we define a module as a gene fragment surrounded by introns of phase 0 (Fig. 2). Three major modules are present in the human hop gene: module 1 corresponds to coding regions for TPR1, DP1, and a hinge of charged residues; module 2 corresponds to the largest part of TPR2A (the C-terminal 7 aa are missing in this module and predicted by homology modeling to rest outside the last helix); module 3 corresponds (including the 7 aa of the previous domain) to the TPR2B domain (although missing the 15 C-terminal aa, corresponding to the whole C helix). TPR2A does not include a DP domain and is surrounded by phase 0 introns. As there is no DP domain at the C-terminal end of the TPR2A domain, one can argue that TPR2A might originate from a partial duplication of an ancestral module present in prokaryotes and containing both the TPR and the DP domains, such as TPR1. In some species such as Drosophila melanogaster, the duplication may have lost the largest part of the DP1 domain (Carrigan et al. 2005).
Fig. 2.

Schematic representation of the human hop (a) and hip (b) genes, at the same scale (in base pairs). Rectangles symbolize the various exons coding for TPR1, TPR2A, TPR2B, and DP domains; grey code is given in the onset. Introns located in between the exons are represented by broken lines. Arabic numbers indicate the phase of each intron and Latin numbers indicate introns. The extent of the modules is indicated by horizontal lines. In (c), the various domain distribution is represented for GmHop, TcHop2, and GerD
Hip (Hsp70 interacting protein), another DP domain-containing protein
Hip, another co-chaperon that contains an ATPase binding domain, binds the ATPase domain of Hsc70 but does not bind Hsc90. Hip induces the Hsc70-ADP state that presents a high affinity for the peptide substrate (Irmer and Höhfeld 1997). When the Hop TPR1 and DP1 domains are used as query sequences in a BLAST search, the Hip C-terminal sequence is retrieved with a level of sequence identity around 22%. It is known that hip gene contains one TPR and one DP domain, connected by a highly charged 50 aa hinge region. In Fig. 3, we show an HCA alignment of Hop and Hip TPR–DP domains. The Human TPR1, TPR2B, and Hip all end with a DP domain containing well-conserved hydrophobic clusters. A hinge region comprised of highly charged aa (E, K, D, R) with a high content of prolines is found at the end of TPR1 as well as between Hip TPR and DP domains. Nothing is currently known about the importance of these domains for Hop or Hip activity.
Fig. 3.

HCA alignment of human TPR1-DP1, TPR2A, and TPR2B-DP2 Hop domains, B. subtilis spore germination D protein, GerD (CAB11931), and human Hip-DP domains. Hip is the Hsp70 interacting protein (NP_003923). The secondary structures from both TPR1 (PDB code 1ELW) and TPR2A (PDB code 1ELR) are represented below the corresponding plots. Helices are called A1, B1….Conserved hydrophobic clusters are grey shaded
We performed an analysis of the exon–intron structure of hip, as shown in Fig. 2b. Hip gene is long and complex in human, composed of 12 small exons and 11 large introns altogether extending over 32 kb. The TPR-containing module is surrounded by phase 0 introns, potentially arising from exon shuffling in evolution (Patthy 1987, 1999, 2003). The highly charged hinge region and possibly one DP domain are also surrounded by phase 0 introns, suggesting that they can also recombine separately from the TPR domain. Nevertheless, it is plausible to consider that the entire TPR–DP module is able to recombine as a single unit, which is bordered by phase 0 introns (introns IV to XI or a downstream point of recombination). Sequence identity between human Hop (NP_006810) and mouse Hop (NM_016737) is 97%, and 93% between human Hip (NP_003923) and mouse Hip (NM_133726). Gene organization, in terms of phase, is strictly identical between human and mouse for both Hop and Hip.
Do GerD and the eukaryotic Hop and Hip proteins share a common ancestor?
Since the discovery of TPR proteins, it was established that tetratricopeptide motifs are also present in bacteria (D’Andrea and Regan 2004); nevertheless, a prokaryotic counterpart of Hop or Hip proteins has not been reported. However, GerD protein (germination response), whose activity is essential for the germination of B. subtilis spores in media-containing asparagine, glucose, and fructose, is associated to Hop in Interpro database because of the presence of homologous DP domains. We show in Fig. 3 a HCA the alignment of human Hop and Hip TPR–DP domains with GerD from B. subtilis. We propose that GerD contains putative TPR–DP–hinge domains seen from the hydrophobic cluster shape and number similarities between DP domains linked to TPR1 and TPR2B (the conserved regions of the clusters are grey-shaded on Fig. 3). Even with a low sequence identity, the conserved shapes of the clusters, in conjunction with the strict sequence identities in close proximity to the clusters, are a clear indicator of the correct alignment. From this alignment, we found significant sequence identities between the N-terminal part of GerD and the TPR domains of Hop (14% with TPR1, 19% with TPR2A, 14% with TPR2B) and 16% with that of Hip.
The highly charged or polar hinges located at the end of DP1 of Hop, between TPR and DP domains of Hip and at the end of DP domain of GerD, have a preference for residues E, K, D, Q, S, and T. Although the B. subtilis hinge is short (20 aa), it can, in other Bacillus species, be up to 10 aa longer. We can postulate that GerD represents an evolutionary reminiscence of a single TPR–DP–hinge module and that Hip and particularly Hop proteins, originated from successive duplications or recombinations of that recombinable multidomain array. The domain distribution for G. max, T. cruzi, and GerD is provided as a scheme in Fig. 2c.
Discussion
Are DP1 and DP2 essential for Hop activity?
The first intriguing evidence that a DP domain is not necessary for Hop activity came from D. melanogaster, which lacks DP1 (Carrigan et al. 2005). Recently, an exhaustive genetic analysis of the minimal fragments of STI1 required for in vivo activity resulted in three main conclusions: (1) “results indicate that TPR1 and DP1 are dispensable for some but not all in vivo functions of Sti1”; (2) “TPR1, DP1, and DP2 are dispensable for Hsp90 interaction,” and (3) “In contrast with previous studies with Hop, deletion of DP2 or a point mutation within DP2 did not inhibit the Hsp70 interaction. The reason for this difference is unknown and may reflect species-specific differences” (Flom et al. 2007). It is clear that multiple isoforms of Hop exist in the eukaryotic cell (Honoré et al. 1992) with diverse functions that remain poorly characterized (Odunuga et al. 2004). The hop gene from Cenorhabditis elegans does not contain TPR1 + DP1 domains (Flom et al. 2006); thus, it is possible to conclude that Hop isoforms are responsible for independent functions and that each of these functions does not require the presence of the entire TPR and DP domains. This assumption could explain the existence of mutant alleles of Hop such as in G. Max and T. cruzy which lack DP1 or DP2 but are expressed and functional.
Another argument that can derive from these observations is that DP segments have a structural role rather than a functional one, such as linking successive TPR domains. It may be argued that the DP domain is actually a globular domain, containing standard density of hydrophobic residues with a presumable nucleus, as our MIR simulations show. Although the respective DP regions of Hop and Hip are similar, they are not functionally interchangeable. For instance, a double point mutation that converts the second DP domain of Hop into the sequence found in Hip disrupts Hop function, while the corresponding mutation in Hip does not alter its function. DP domains are, therefore, structural elements within a C-terminal domain since their truncation arrests maturation of receptor complexes (Nelson et al. 2003). In the same work, chimeras were constructed by swapping the DP domains of Hop and Hip; these tail swap chimeras are impaired in their ability to assemble into complexes with their receptors. Interestingly, point mutations in the Hop DP repeats render C-terminal regions hypersensitive to proteolysis. We can conclude that the DP domains play an important structural role in maintaining the compact folding of a C-terminal domain. The function of DP is now unclear, although the globular nature is presumably correct as a large number of residues are predicted to be included in the folding nucleus, as predicted MIR simulations (data not shown).
TPR–DP–hinge module as a precursor of Hop, Hip, and GerD
Domain combination in a single protein and development of new genes have been explained by genetic events such as exon shuffling, in particular, by modules surrounded by phase 0 introns (Long 2001; Patthy 2003). Taking into account the exceptional similarities in domain composition among hop, hip, and GerD, it is reasonable to assume that a common ancestor is at the origin of the three proteins. From the presented results, we are now in a position to propose an evolutionary model. We propose that there were two domains, called TPR and DP–hinge that were able to constitute a single recombinant module. The new open reading frame by itself was able to acquire new functions in bacteria, giving rise to the protein we know in the present day as GerD. At a given moment in the evolution, the ancient module gained multiple introns, mainly of phase 0. A series of gene duplication, incomplete recombination, and exon shuffling events between homologous modules occurred resulting in a protein of three repeated units in the primitive eukaryote. As a consequence of partial recombination of the DNA segments surrounded by introns of the same phase, the primitive Hop suffered a loss of the DP–hinge domain in module 2. Introns sharing the same phase, i.e., phase 0, were crucial to maintain in frame the new coding sequence. In the case of Hip, it gained the entire module and included a tandemly repeated stretch of FPGG between the two domains.
Now, it is clear that Hop proteins exhibit an unusual variability between species. GMSTI and TcHop2 have lost the DP1 and DP2 domains, respectively, because of frame shifts. D. melanogaster Hop protein does not contain DP1 domain, and in C. elegans Hop, TPR1 and DP1 are absent. It is possible that the Hop protein today has not lost all unnecessary sequences, and then it may continue to evolve and lose some domains such as DP1 or DP2. If this is the case, we will be able to discover in nature new intriguing Hop functional isoforms and alternatively, we could produce them in the laboratory and verify whether or not they maintain their activity intact.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
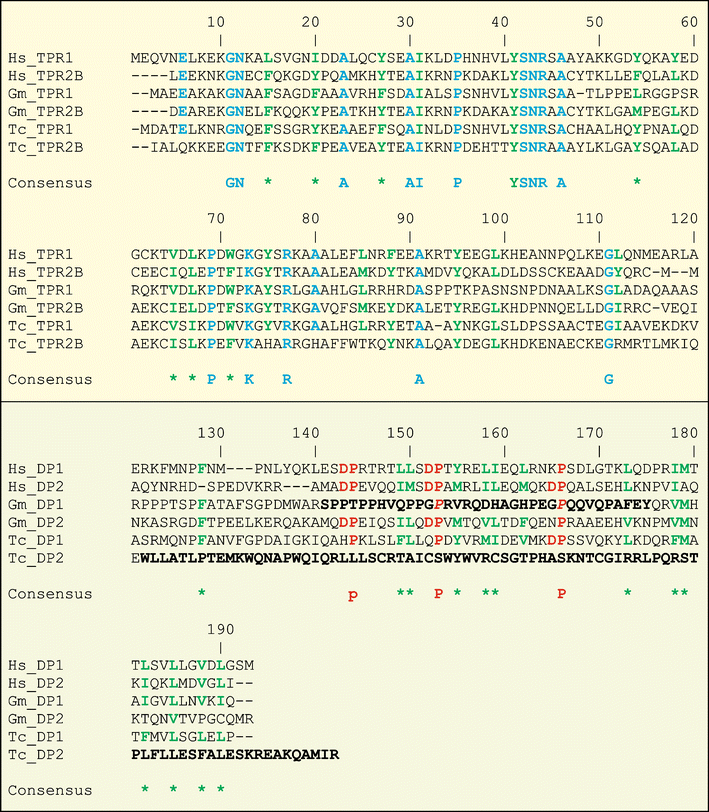
Multiple alignment of TPR–DP domains (top and bottom boxes, respectively) for H. sapiens (Hs), G. max (Gm) and T. cruzi (Tc). To help to recover, the names are as follows: Hs_TPR1 and Hs_DP1 (Hs_TPR2B and Hs_DP2) are TPR1 (TPR2B) from Fig. 3. Gm_DP1 is GMSTI-rf1 and Tc_DP2 is TcHop2-rf1 from Fig. 1. Identical nonhydrophobic residues are colored in blue, and hydrophobic residues identical in at least five sequences (four in the case of DP domains) are colored in green. Stars under the alignment indicate positions only occupied by hydrophobic amino acids. Prolines involved in DP dipeptides are red, and they are also in italic when they are mutated in G. max. In black, the residues mutated by frameshifts (GIF 222 kb)
Acknowledgments
Many thanks are due to Kirsty MacLellan for careful reading of this manuscript. This work has benefited of a grant from ECOS-Nord French and Colombian program (A0502). J.H.T. has been invited as research fellow by CNRS. RPBS bioinformatics resources have been used for this project (http://bioserv.rpbs.univ-paris-diderot.fr/). We also aknowledge the Vicerrectoría de Investigaciones y Extensión of the Universidad Industrial de Santander, by its financial aid. Part of this work is an application of the Protein Folding Fragments project, supported by European Union, under the grant number QLG2-CT-2002-01298.
Abbreviations
- aa
amino acid
- HCA
hydrophobic cluster analysis
- Hip
Hsp70 interacting protein
- Hop
HSP organizing protein
- HSPs
heat shock proteins
- TPR
tetratricopeptide repeat
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Andrade MA, Perez-Iratxeta C, Ponting CP. Protein repeats: structures, functions, and evolution. J Struct Biol. 2001;134:117–131. doi: 10.1006/jsbi.2001.4392. [DOI] [PubMed] [Google Scholar]
- Batzer MA, Deininger PL. Alu repeats and human genomic diversity. Nat Rev Genet. 2002;5:370–379. doi: 10.1038/nrg798. [DOI] [PubMed] [Google Scholar]
- Batzer MA, Deininger PL, Hellmann-Blumberg U, Jurka J, Labuda D, Rubin CM, Schmid CW, Zietkiewicz E, Zuckerkandl E. Standardized nomenclature for Alu repeats. J Mol Evol. 1996;42:3–6. doi: 10.1007/BF00163204. [DOI] [PubMed] [Google Scholar]
- Blatch GL, Lässle M. The tetratricopeptide repeat: a structural motif mediating protein–protein interactions. BioEssays. 1999;21:932–939. doi: 10.1002/(SICI)1521-1878(199911)21:11<932::AID-BIES5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Blatch GL, Lässle M, Zetter BR, Kundra V. Isolation of a mouse cDNA encoding mSTI1, a stress-inducible protein containing the TPR motif. Gene. 1997;194:277–282. doi: 10.1016/S0378-1119(97)00206-0. [DOI] [PubMed] [Google Scholar]
- Callebaut I, Labesse G, Durand P, Poupon A, Canard L, Chomilier J, Henrissat B. Deciphering protein sequence information through Hydrophobic Cluster Analysis (HCA): current status and perspectives. Cell Mol Life Sci. 1997;53:621–645. doi: 10.1007/s000180050082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrello A, Ingley E, Minchin RF, Tsay S, Ratajczak T. The common tetratricopeptide repeat acceptor site for steroid receptor-associated immunophilins and Hop is located in the dimerization domain of Hsp90. J Biol Chem. 1999;274:2682–2689. doi: 10.1074/jbc.274.5.2682. [DOI] [PubMed] [Google Scholar]
- Carrigan PE, Nelson GM, Roberts PJ, Stoffer J, Riggs DL, Smith DF. Multiple domains of the co-chaperone Hop are important for Hsp70 binding. J Biol Chem. 2004;279:16185–16193. doi: 10.1074/jbc.M314130200. [DOI] [PubMed] [Google Scholar]
- Carrigan PE, Riggs DL, Chinkers M, Smith DF. Functional comparison of human and Drosophila Hop reveals novel role in steroid receptor maturation. J Biol Chem. 2005;280:8906–8911. doi: 10.1074/jbc.M414245200. [DOI] [PubMed] [Google Scholar]
- Chen S, Smith DF. Hop as an adaptor in the heat shock protein 70 (Hsp70) and Hsp90 chaperone machinery. J Biol Chem. 1998;273:35194–35200. doi: 10.1074/jbc.273.52.35194. [DOI] [PubMed] [Google Scholar]
- Chen S, Prapapanich V, Rimerman RA, Honoré B, Smith DF. Interactions of p60, a mediator of progesterone receptor assembly, with heat shock proteins Hsp90 and Hsp70. Mol Endocrinol. 1996;10:682–693. doi: 10.1210/me.10.6.682. [DOI] [PubMed] [Google Scholar]
- D’Andrea LD, Regan L. TPR proteins: the versatile helix. Trends Biochem Sci. 2004;28:655–662. doi: 10.1016/j.tibs.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Demand J, Lüders J, Höhfeld J. The carboxy-terminal domain of Hsc70 provides binding sites for a distinct set of chaperone cofactors. Mol Cell Biol. 1998;18:2023–2028. doi: 10.1128/mcb.18.4.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorova L, Fedorov A. Introns in gene evolution. Genetica. 2003;118:123–131. doi: 10.1023/A:1024145407467. [DOI] [PubMed] [Google Scholar]
- Flom G, Weekes J, Williams JJ, Johnson JL. Effect of mutation of the tetratricopeptide repeat and aspartate-proline 2 domains of Sti1 on Hsp90 signaling and interaction in Saccharomyces cerevisiae. Genetics. 2006;172:41–51. doi: 10.1534/genetics.105.045815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flom G, Behal RH, Rosen L, Cole DG, Johnson JL. Definition of the minimal fragments of Sti1 required for dimerization, interaction with Hsp70 and Hsp90 and in vivo functions. Biochem J. 2007;404:159–167. doi: 10.1042/BJ20070084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross M, Hessefort S. Purification and characterization of a 66-kDa protein from rabbit reticulocyte lysate which promotes the recycling of Hsp70. J Biol Chem. 1992;271:16833–16841. doi: 10.1074/jbc.271.28.16833. [DOI] [PubMed] [Google Scholar]
- Honoré B, Leffers H, Madsen P, Rasmussen HH, Vandekerckhove J, Celis JE. Molecular cloning and expression of a transformation-sensitive human protein containing the TPR motif and sharing identity to the stress-inducible yeast protein STI1. J Biol Chem. 1992;267:8485–8491. [PubMed] [Google Scholar]
- Irmer H, Höhfeld J. Characterization of functional domains of the eukaryotic co-chaperone Hip. J Biol Chem. 1997;272:2230–2235. doi: 10.1074/jbc.272.4.2230. [DOI] [PubMed] [Google Scholar]
- Kimmins S, MacRae TH. Maturation of steroid receptors: an example of functional cooperation among molecular chaperones and their associated proteins. Cell Stress Chaperon. 2000;5:76–86. doi: 10.1379/1466-1268(2000)005<0076:MOSRAE>2.0.CO;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu FH, Wu SJ, Hu SM, Hsiao CD, Wang C. Specific interaction of the 70-kDa heat shock cognate protein with the tetratricopeptide repeats. J Biol Chem. 1999;274:34425–34432. doi: 10.1074/jbc.274.48.34425. [DOI] [PubMed] [Google Scholar]
- Long M. Evolution of novel genes. Curr Opin Genet Dev. 2001;11:673–680. doi: 10.1016/S0959-437X(00)00252-5. [DOI] [PubMed] [Google Scholar]
- Long M, Souza SJ, Gilbert W. Evolution of the intron-exon structure of eukaryotic genes. Curr Opin Genet Dev. 1995;5:774–778. doi: 10.1016/0959-437X(95)80010-3. [DOI] [PubMed] [Google Scholar]
- Main ERG, Xiong Y, Cocco MJ, D’Andrea L, Regan L. Design of stable α-helical arrays from an idealized TPR motif. Structure. 2003;11:497–508. doi: 10.1016/S0969-2126(03)00076-5. [DOI] [PubMed] [Google Scholar]
- Mizrak SC, Bogerd J, Lopez-casas PP, Párraga M, Mazo J, Rooij DJ. Expression of stress inducible protein 1 (Stip1) in the mouse testis. Mol Reprod Dev. 2006;73:1361–1366. doi: 10.1002/mrd.20548. [DOI] [PubMed] [Google Scholar]
- Mulder NJ, Apweiler R. The Interpro database and tools for protein domain analysis. Curr Protoc Bioinformatics. 2008;21:2.7.1–2.7.18. doi: 10.1002/0471250953.bi0207s21. [DOI] [PubMed] [Google Scholar]
- Nelson GM, Huffman H, Smith DF. Comparison of the carboxy-terminal DP-repeat region in the co-chaperones Hop and Hip. Cell Stress Chaperon. 2003;8:125–133. doi: 10.1379/1466-1268(2003)008<0125:COTCDR>2.0.CO;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolet CM, Craig EA. Isolation and characterization of STI1, a stress-inducible gene from Saccharomyces cerevisiae. Mol Cell Biol. 1989;9:3638–3646. doi: 10.1128/mcb.9.9.3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odunuga OO, Longshaw VM, Blatch GL. Hop: more than an Hsp70/Hsp90 adaptor protein. BioEssays. 2004;26:1058–1068. doi: 10.1002/bies.20107. [DOI] [PubMed] [Google Scholar]
- Papandreou N, Kanehisaa M, Chomilier J. Folding of the human protein FKBP. Lattice Monte-Carlo simulations. CR Biol. 1998;321:835–843. doi: 10.1016/s0764-4469(99)80023-7. [DOI] [PubMed] [Google Scholar]
- Papandreou N, Berezovsky IN, Lopes A, Eliopoulos E, Chomilier J. Universal positions in globular proteins. Eur J Biochem. 2004;271:4762–4768. doi: 10.1111/j.1432-1033.2004.04440.x. [DOI] [PubMed] [Google Scholar]
- Patthy L. Evolution of the proteases of blood coagulation and fibrinolysis by assemble from modules. Cell. 1985;41:657–663. doi: 10.1016/S0092-8674(85)80046-5. [DOI] [PubMed] [Google Scholar]
- Patthy L. Intron-dependent evolution: preferred types of exons and introns. FEBS lett. 1987;214:1–7. doi: 10.1016/0014-5793(87)80002-9. [DOI] [PubMed] [Google Scholar]
- Patthy L. Genome evolution and the evolution of exon-shuffling––a review. Gene. 1999;238:103–114. doi: 10.1016/S0378-1119(99)00228-0. [DOI] [PubMed] [Google Scholar]
- Patthy L. Modular assembly of genes and the evolution of new functions. Genetica. 2003;118:217–231. doi: 10.1023/A:1024182432483. [DOI] [PubMed] [Google Scholar]
- Rhee Y, Beavis W, Berardini TZ, et al. The Arabidopsis Information Resource (TAIR): a model organism database providing a centralized, curated gateway to Arabidopsis biology, research materials and community. Nucleic Acids Res. 2003;31:224–228. doi: 10.1093/nar/gkg076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheufler C, Brinker A, Bourenkov G, Pegoraro S, Moroder L, Bartunik H, Hartl FU, Moarefi I. Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell. 2000;101:199–210. doi: 10.1016/S0092-8674(00)80830-2. [DOI] [PubMed] [Google Scholar]
- Torres JH, Chatellard P, Stutz E. Isolation and characterization of gmsti, a stress-inducible gene from soybean (Glycine max) coding for a protein belonging to the TPR (tetratricopeptide repeats) family. Plant Mol Biol. 1995;27:1221–1226. doi: 10.1007/BF00020896. [DOI] [PubMed] [Google Scholar]
- Woodcock S, Mornon J-P, Henrissat B. Detection of secondary structure elements in proteins by Hydrophobic Cluster Analysis. Prot Eng. 1992;5:629–635. doi: 10.1093/protein/5.7.629. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Quick MK, Kanelakis KC, Gijzen M, Krishna P. Characterization of a plant homolog of Hop, a co-chaperone of Hsp90. Plant Physiol. 2003;131:525–535. doi: 10.1104/pp.011940. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Below is the link to the electronic supplementary material.
Multiple alignment of TPR–DP domains (top and bottom boxes, respectively) for H. sapiens (Hs), G. max (Gm) and T. cruzi (Tc). To help to recover, the names are as follows: Hs_TPR1 and Hs_DP1 (Hs_TPR2B and Hs_DP2) are TPR1 (TPR2B) from Fig. 3. Gm_DP1 is GMSTI-rf1 and Tc_DP2 is TcHop2-rf1 from Fig. 1. Identical nonhydrophobic residues are colored in blue, and hydrophobic residues identical in at least five sequences (four in the case of DP domains) are colored in green. Stars under the alignment indicate positions only occupied by hydrophobic amino acids. Prolines involved in DP dipeptides are red, and they are also in italic when they are mutated in G. max. In black, the residues mutated by frameshifts (GIF 222 kb)