

Abstract
β-lactamase inhibitory protein (BLIP) binds and inhibits a diverse collection of class A β-lactamases with a wide range of affinities. Alanine-scanning mutagenesis was previously performed to identify the amino acid sequence requirements of BLIP for binding the TEM-1, SME-1, SHV-1 and Bla1 β-lactamases. Twenty-three BLIP residues that contact TEM-1 β-lactamase in the structure of the complex were mutated to alanine and assayed for inhibition (Ki) of β-lactamase to identify two hotspots of binding energy. These studies have been extended by the development of a genetic screen for BLIP function in E. coli. The blaTEM-1 gene encoding TEM-1 β-lactamase was inserted into the E. coli pyrF chromosomal locus. Expression of wild-type BLIP from a plasmid in this strain resulted in a large decrease in ampicillin resistance while introduction of the same plasmid lacking BLIP had no effect on ampicillin resistance. In addition, it was found that when the BLIP alanine-scanning mutants were tested in the strain, the level of ampicillin resistance was proportional to the Ki of the BLIP mutant. These results indicate that BLIP function can be monitored by the level of ampicillin resistance of the genetic test strain. Each of the 23 BLIP positions examined by alanine-scanning was randomized to create libraries containing all possible substitutions at each position. The genetic screen for BLIP function was used to sort the libraries for active mutants and DNA sequence analysis of functional BLIP mutants identified the sequences required for binding TEM-1 β-lactamase. The results indicated the BLIP surface is tolerant of substitutions in that many contact positions can be substituted with other amino acid types and retain wild type levels of function.
Keywords: enzyme inhibition, molecular recognition, antibiotic resistance, random mutagenesis
Introduction
Protein-protein interactions are critical for most biological processes and a detailed understanding of the molecular basis of these interactions may lead to accurate methods to predict existing interactions and engineer new interactions. Computational prediction and design of protein-protein interaction sites is difficult because they are large (>900Å2) and relatively flat 1. In addition, the binding energy within the interaction site between proteins is not evenly distributed among contact residues. Several protein interfaces have been shown to be made up of “hotspot” residues that make a large contribution to binding energy along with other residues that contribute little to the binding energy of an interface 2; 3; 4. It is not currently possible, however, to accurately predict the energetic contribution of interface residues or to design new protein-protein interfaces 5. The accumulation of detailed information on structure-function relationships in protein-protein interfaces should contribute to the understanding necessary for high resolution prediction and design of protein interfaces.
β-lactamases catalyze the hydrolysis of β-lactam antibiotics to provide bacteria with resistance to these drugs. They are grouped into 4 classes (A, B, C and D) according to amino acid sequence homology 6; 7. Enzymes from classes A, C and D are serine hydrolases while class B enzymes are zinc metallo-enzymes unrelated in structure to the other classes 8. Class A β-lactamases exhibit broad substrate hydrolysis profiles which include penicillins, cephalosporins and, for a few enzymes, carbapenems 8; 9. TEM-1 β-lactamase is a class A enzyme and is the most common plasmid-encoded β-lactamase in Gram-negative bacteria and as a consequence is a widespread source of antibiotic resistance 10; 11. TEM-1 β-lactamase efficiently hydrolyzes pencillins and many cephalosporins but it is not an effective catalyst for extended-spectrum cephalosporins. In addition, mechanism-based inhibitors of TEM-1 β-lactamase such as clavulalic acid are used in conjuction with penicillins to avoid this resistance mechanism. Nevertheless, TEM-1 β-lactamase-mediated resistance has evolved over the past several years due to mutations in the blaTEM-1 gene which result in amino acid substitutions that allow the TEM enzyme to hydrolyze extended-spectrum cephalosporins or to avoid the action of mechanism-based inhibitors 12.
BLIP is an acromyn for β-lactamase inhibitory protein 13. BLIP is a 165 amino acid protein which binds and inhibits several class A β-lactamases including TEM-1 β-lactamase 4; 13; 14. BLIP is produced by the soil bacterium Streptomyces clavuligerus 13. This organism also produces the small molecule, mechanism-based inhibitor clavulanic acid 15. Gene knockout studies indicate BLIP is not essential for growth and the exact role of BLIP in the biology of S. clavuligerus remains unknown 16.
BLIP is known to bind to a diverse set of class A β-lactamases with a wide range of affinities 4; 14. For example, it binds to TEM-1 β-lactamase with a Ki of 0.5 nM but binds to the SHV-1 β-lactamase, which is 68% identical in amino acid sequence to TEM-1, with a Ki of 1.1 μM 17. In addition, high-resolution co-crystal structures of BLIP in complex with TEM-1 and SHV-1 β-lactamase are available 18; 19. The BLIP-β-lactamase interface has been extensively studied using structural, computational and biochemical approaches 18; 19; 20; 21; 22; 23; 24; 25. Extensive site-directed mutagenesis experiments by Schreiber and colleagues suggest the interface consists of modules of TEM-1 and BLIP residues where cooperative interactions occur between residues within modules and additive interactions occur among residues in different modules 26; 27.
In addition to the above studies, we previously performed alanine-scanning mutagenesis to identify the amino acid sequence requirements of BLIP for binding the TEM-1, SME-1, SHV-1 and Bla1 β-lactamases 4; 17; 28. Twenty-three BLIP residues that contact TEM-1 β-lactamase based on the X-ray structure were mutated to alanine and assayed for inhibition (Ki) of β-lactamase. This work identified three regions, including two loops that insert into the active site of TEM-1 β-lactamase and a Glu73-Lys74 buried charge motif as being important determinants of the specificity of binding to various β-lactamases 4; 28. In addition, two sets of hotspot residues were found to be common sources of binding energy for BLIP interactions with several class A β-lactamases 4.
Alanine-scanning mutagenesis provides information on the contribution of residue positions to protein-protein interactions but does not provide a detailed view of the sequence requirements with respect to the twenty naturally occurring amino acids found in proteins 29. However, constructing, purifying and testing all 19 substitutions at each of the 23 BLIP residues that contact β-lactamase would be a daunting task. An alternative to testing individual substitutions is to randomize the codon of a residue position to form a library of substitutions that can then be tested in batch for those substitutions that retain function. The codon randomization approach requires a rapid method to sort the libraries for functional clones such as a genetic selection or a powerful screen such as phage display 23; 30; 31. In this study, a genetic screen for BLIP function in E. coli was developed and used to identify BLIP mutants that retain the ability to bind TEM-1 β-lactamase. A total of 23 libraries were generated by randomizing the codon for each of the BLIP residues that contact TEM-1 β-lactamase and the libraries were screened for binders. Two data sets were established. One set included mutants that had partial function while the other set were more stringently screened for high levels of function. The results indicate that a correlation between previous alanine scanning results and the randomization results exists for the stringently selected set and is apparent but weak for the partial function mutants. The randomization approach coupled with the genetic screen is complementary to alanine scanning and taken together the results provide a detailed description of the sequence requirements for BLIP function.
Results
Development of a genetic screen for BLIP function
The goal of this study was to use saturation mutagenesis to obtain detailed information on the sequence requirements at BLIP residues that contact TEM-1 β-lactamase. In order to rapidly test large numbers of mutants it is very useful to have a genetic selection or screen with which to evaluate function in vivo. In order to develop a genetic screen for BLIP function, the gene and promoter for TEM-1 β-lactamase was placed on the chromosome of E. coli within the pyrF gene using recombineering methods to create strain E. coli TP112 32 (Materials and Methods) (Figure 1). The advantage of placing the blaTEM-1 gene on the chromosome with its constitutive, weak promoter is that it limits the copy number of the gene to one. It is important for the performance of the screen that the total amount of BLIP be greater than the amount of β-lactamase in the cell. If the number of β-lactamase molecules is higher than the number of BLIP molecules, even very tight binding BLIP variants will not result in a large reduction in β-lactam resistance levels because excess β-lactamase will be available to hydrolyze antibiotic. The insertion within the pyrF gene, which encodes orotidine 5′-monophosphate decarboxylase and is involved in pyrimidine biosynthesis, results in the E. coli strain being unable to grow on minimal medium. This phenotype can be used as a secondary screen to test for insertion of the blaTEM-1 gene into the chromosome.
Figure 1.

Schematic illustration of position of the blaTEM-1 gene and promoter inserted into the chromosome of E. coli TP112 at the pyrF locus.
In order to establish the screen, the pGR32 plasmid 17 encoding BLIP fused to the β-lactamase signal sequence and containing a His-tag sequence and under the transcriptional control of the trc promoter was introduced into E. coli TP112 by transformation. This plasmid was previously used for alanine-scanning mutagenesis of BLIP and the protein is known to be expressed in a functional form 4; 17; 28. As a negative control, the parent plasmid, pTP123, which lacks the BLIP gene, was also introduced into E. coli TP112 17. Since both BLIP and TEM-1 β-lactamase contain signal sequences and have previously been shown to be exported to the periplasm in E. coli 17, it was expected that they would be secreted to the periplasm in this construct. In this system, if BLIP is functional, it will bind β-lactamase and impair the ability of the enzyme to hydrolyze ampicillin, thus reducing the ampicillin resistance levels of E. coli. The minimal inhibitory concentration (MIC) for ampicillin was determined for E. coli TP112 containing pGR32 and, independently, pTP123. It was found that the ampicillin MIC was greatly reduced for the strain containing pGR32 encoding BLIP (1.5μg/ml) versus that containing pTP123 with no BLIP (256 μg/ml) (Table 1). These results suggest that the BLIP is functional in vivo and that a genetic screen for that function has been established.
Table 1.
Comparison of the minimum inhibitory concentration of ampicillin for growth of E. coli TP112 and TP132 strains containing plasmids encoding BLIP alanine-scanning mutants versus the Ki value for inhibition of TEM-1 β-lactamase by the purified BLIP derivatives. “No BLIP” refers to the E. coli strain containing the pTP123 plasmid that does not encode BLIP. “Wt BLIP” refers to the E. coli strain containing the pGR32 plasmid that encodes BLIP. The Ki data is from Zhang and Palzkill28 and the ΔΔG values are calculated from the Ki values (Materials and Methods).
| Ampicillin MIC (μg/ml) | ||||
|---|---|---|---|---|
| BLIP mutant | E. coli TP112 | E. coli TP132 | Ki (nM) | ΔΔG (kJ/mol) |
| No BLIP | 256 | >256 | - | - |
| Wt BLIP | 1.5 | 4 | 0.5 | - |
| E31A | 8 | 12 | 2.0 | 3.4 |
| S35A | 1.5 | 4 | 0.5 | 0 |
| F36A | 24 | 24 | 40 | 10.8 |
| S39A | 1.5 | 4 | 0.3 | −1.3 |
| H41A | 64 | 64 | 34 | 10.4 |
| G48A | 2 | 12 | 0.7 | 0.8 |
| D49A | 3 | 16 | 20 | 9.1 |
| Y50A | 1 | 4 | 0.01 | −9.7 |
| Y51A | 1.5 | 4 | 0.5 | 0 |
| Y53A | 4 | 24 | 21 | 9.3 |
| S71A | 3 | 6 | 0.2 | −2.3 |
| E73A | 1.5 | 6 | 0.4 | −0.5 |
| K74A | 3 | 24 | 46 | 11.2 |
| W112A | 1.5 | 16 | 13 | 8.0 |
| S113A | 1 | 4 | 0.1 | −4.0 |
| G141A | 3 | 8 | 1.8 | 3.18 |
| F142A | 2 | 16 | 16 | 8.58 |
| Y143A | 1 | 4 | 0.6 | 0.5 |
| R144A | 1 | 6 | 0.6 | 0.5 |
| H148A | 24 | 32 | 21 | 9.3 |
| W150A | 96 | 256 | 184 | 14.6 |
| R160A | 2 | 24 | 11 | 7.7 |
In previous studies, several alanine substituted BLIP mutants were constructed in the pGR32 plasmid and the expressed proteins were purified and tested by measuring the inhibition constant, Ki, for TEM-1 β-lactamase mediated antibiotic hydrolysis 4; 17; 28. These mutants were introduced into E. coli TP112 to test the relationship between the change in binding free energy of each alanine mutant (ΔΔG, derived from the Ki values) and the ampicillin resistance levels provided by the mutant (expressed as log MIC in Fig. 2). As seen in Table 1 and Figure 2A, there is a correlation (r2 = 0.49) between ampicillin resistance levels and the ΔΔG of the BLIP mutants, with tight binding mutants exhibiting low ampicillin MICs and weaker binding mutants exhibiting progressively higher ampicillin MICs. These results suggest that the ampicillin resistance level is inversely proportional to BLIP affinity for binding β-lactamase and therefore that the genetic screen can be used to sort mutant libraries for BLIP variants that retain tight binding to β-lactamase (ΔΔG ~ 0).
Figure 2.

BLIP alanine mutant ampicillin MIC versus binding energy. Panel A shows a comparison of the log of the minimum inhibitory concentration of ampicillin for growth of E. coli TP112 containing plasmids encoding the BLIP alanine mutants versus change in binding energy of the BLIP mutant compared to wild type BLIP (ΔΔG (kJ/mol)) for binding TEM-1 β-lactamase. The ΔΔG values were calculated from the inhibition constants (Ki) and are listed in Table 1. Panel B shows the analogous data using E. coli TP132 as the host strain for MIC determinations. Panels C and D use data for 14 BLIP alanine mutants whose ΔΔG values were determined by Reichmann et al. using surface plasmon resonance 26. These alanine mutants are a subset of those in Panels A and B and are compared with the corresponding ampicillin MIC data for each mutant in E. coli TP112 and TP132. Panel C represents log ampicillin MICs for E. coli TP112 and Panel D is log ampicillin MICs for E. coli TP132. r2 is the goodness of the linear regression fit of the indicated line.
Construction and screening of BLIP random libraries
As stated above, alanine scanning mutagenesis of 23 BLIP residues was previously used to identify those residues that contribute the most to binding energy in the complex 4; 28. In order to gain more detailed information about the chemical side chain requirements for binding, the codons for each of these residues were randomized individually by overlap extension PCR to create 23 libraries, each of which contains all possible substitutions for the position randomized 33(Materials and Methods). Each library was sorted using the genetic screen for those mutants that retain BLIP function. Because the goal is to find clones with high levels of BLIP function, it was necessary to screen a large number of mutants to identify those exhibiting low ampicillin resistances, which is reflective of strong inhibition of β-lactamase. This was accomplished by transforming each library into E. coli TP112 containing TEM-1 β-lactamase encoded on the chromosome. Approximately ten colonies were picked from each library without screening for ampicillin resistance to ensure the libraries were not obviously biased (Fig. 3A). In order to screen for ampicillin resistance, individual transformants were picked and inoculated into growth medium containing various concentration of ampicillin in 96-well microtiter plates. Mutant clones that exhibited low levels of ampicillin resistance were chosen and tested for their ampicillin minimal inhibitory concentration (MIC) (Materials and Methods). Those mutants that exhibited resistance levels similar to E. coli TP112 containing the wild type His-tagged BLIP (<4 μg/ml MIC) were analyzed by DNA sequencing to determine the identity of the amino acid substitution at that position. This process was repeated for each of the 23 libraries and the results are shown in Figure 3B.
Figure 3.

Results of DNA sequencing of clones from BLIP libraries before and after screening for ampicillin resistance. A. Nucleotide sequences and corresponding amino acid sequences of clones from each library picked after transformation of libraries into E. coli TP112. These clones were picked without screening for ampicillin resistance and are representative of diversity in the libraries. The fs label stands for frameshift mutation. The number before fs indicates the number of nucleotides deleted in the clone. B. Amino acid sequence replacements at BLIP contact positions that retain BLIP binding to TEM-1 β-lactamase as indicated by the genetic screen. Each of the 23 residues shown was individually randomized to NNN where N is any of the 4 nucleotides by site directed mutagenesis to create libraries containing all possible substitutions at each position. Each library was transformed into E. coli TP112 (lower panel) and screening was performed using 96-well plates as described in Materials and Methods to identify clones with low ampicillin resistance. Clones with resistance similar to the strain containing wild type BLIP were sequenced and the substitutions are shown below the listed amino acid residue positions. The superscript indicates the number of times a mutant with the listed amino acid was found. The number following the amino acids is the ampicillin MIC (μg/ml) of the clone. The columns above the listed residue positions indicate the substitutions that retain high level function when the ampicillin MIC determinations of the clones from the lower panel were performed in E. coli TP132. The number following the amino acids is the ampicillin MIC (μg/ml) of the clone.
It is apparent from DNA sequencing results that the majority of the 23 contact positions are quite tolerant of amino acid substitutions. For example, Asp49 is a key BLIP residue whose side chain is positioned in the TEM-1 β-lactamase active site where it makes several contacts with active site residues 19. When functional clones from the Asp49 library were identified and sequenced it was found that several types of hydrophilic substitutions were consistent with function (Fig. 3B). In addition, an alanine substitution was identified at this position among the functional clones. It is known from previous studies, however, that that Asp49Ala substitution has a Ki of 20 nM, which is 40 times higher that the wild type BLIP Ki of 0.5 nM (Table 1)28. This result suggested that the genetic screen identifies mutants with wild type function as well as mutants with partial function. Examination of the correlation between Ki and ampicillin MIC of the alanine mutants in Table 1 suggests that mutants with ampicillin MIC values in the same range as wild type BLIP (1–4 μg/ml) show a range of Ki values from 0.01 to 46 nM. These mutants could all be considered tight binding in that the binding constants are in the low nanomolar range, however, they do not all function as efficiently as wild type. This finding was further confirmed by purifying and determining the Ki value for inhibition of TEM-1 β-lactamase by several of the BLIP mutants listed in Figure 3 including the Glu31Gln, Asp49Ser, Glu73Tyr, Trp112Arg, and Phe142Gly proteins (Table 2). The Ki values ranged from 1.4 to 16 nM, consistent with the hypothesis that these mutants bind tightly to TEM-1 β-lactamase (<20 nM) but have partial function relative to wild type His-tagged BLIP (Ki = 0.5 nM).
Table 2.
Comparison of the minimum inhibitory concentration of ampicillin for growth of E. coli TP112 and TP132 strains containing plasmids encoding the various BLIP mutants versus the Ki value for inhibition of TEM-1 β-lactamase by the purified BLIP derivatives.
| Ampicillin MIC (μg/ml) | |||
|---|---|---|---|
| BLIP mutant | E. coli TP112 | E. coli TP132 | Ki (nM) |
| No BLIP | 256 | >256 | - |
| Wt BLIP | 1.5 | 4 | 0.5 |
| E31Q | 1.5 | 6 | 1.4 |
| D49S | 2 | 32 | 16 |
| E73Y | 1.5 | 12 | 3.7 |
| W112R | 1.5 | 12 | 3.9 |
| F142G | 1.5 | 16 | 12 |
It is expected that the ampicillin MIC of an E. coli strain containing BLIP and TEM-1 β-lactamase would be sensitive to the relative amounts of these proteins which, in turn, is determined by the expression levels of these proteins. The pGR32 plasmid used for BLIP expression in this and previous studies is under the transcriptional control of the trc promoter 17. This promoter is a fusion of the trp and lac promoters and is IPTG-inducible 34. The pGR32 expression plasmid also encodes the lacI repressor which down-regulates the promoter in the absence of IPTG 34. The genetic screen experiments described above were performed in the absence of IPTG and therefore the BLIP expressed is due to basal or “leaky” expression. The E. coli TP112 strain used for the screen contains the lacX74 deletion which removes the entire lac operon, including lacI. Therefore, the levels of basal expression of BLIP from the trc promoter could be relatively high in the E. coli TP112 strain. In an effort to gain tighter control of BLIP expression in the absence of IPTG, the blaTEM-1 gene was moved to the chromosome of E. coli MG1655 by P1 transduction and the new strain was named E. coli TP132. This strain contains no mutations in the lac operon or control region and it was reasoned that basal BLIP expression levels relative to β-lactamase may be reduced in this strain, which would result in a more stringent requirement for BLIP function.
The relationship between ampicillin MIC and Ki for inhibition of TEM-1 in E. coli strain TP132 was examined using the set of BLIP alanine-scanning mutants as described above. It is apparent from the results in Figure 2B and Table 1 that there is a stronger correlation (r2 = 0.76) between ampicillin MIC levels and the Ki inhibition constants and the corresponding ΔΔG values. The stronger correlation is also indicated by more stringent sequence requirements found for BLIP mutants in E. coli TP132. For example, among the Asp49 mutants identified in the screen using E. coli TP112, only the Asp49Glu mutant exhibited ampicillin MICs similar to wild type in the E. coli TP132 strain (Figure 3B). In addition, the ampicillin MIC of 12μg/ml of the W112R mutant with a Ki of 3.9 nM is higher than the 4 μg/ml ampicillin MIC for wild type BLIP that has a Ki of 0.5 nM (Table 2). Furthermore, the ampicillin MIC results for 13 BLIP alanine mutants in E. coli TP112 and TP132 were compared to ΔΔG binding energy determinations for these mutants published by Reichmann et al. 26 (Materials and Methods). This comparison is of interest because the mutants are a subset of those listed in Table 1 and because the binding energies were evaluated by surface plasmon resonance (SPR) rather than inhibition assays 26. As seen in Figure 2C and D, the correlations between the change in binding energy of the mutants determined by SPR versus the ampicillin MIC values are very similar to those obtained using ΔΔG values derived from Ki values for both E. coli TP112 (r2= 0.46) and TP132 (r2= 0.79). Taken together, the results indicate that determining the ampicillin MIC of mutants in the E. coli TP132 strain is a useful method to identify mutants with binding constants similar to wild type BLIP.
In order to gain more detailed information on the sequence requirements for BLIP function, each of the mutants identified in the initial genetic screen in E. coli TP112 (bottom panel of Fig. 3B) were introduced into the E. coli TP132 strain and the ampicillin MIC of the resultant strains was determined to more accurately identify those mutants with wild type levels of function (Fig. 3, 4). Those mutants that exhibited wild type levels of ampicillin resistance in E. coli TP112 but increased levels of resistance in E. coli TP132 are assumed to have sub-wild type BLIP inhibition function and are not listed in the top panel of Figure 3B. As seen in the top panel of Figure 3B, the sequence requirements for high level function are, in general, more stringent than those required for partial function. The tolerance of the BLIP residue positions to amino acid substitutions follow the same general trends in both experiments but, for several positions, fewer substitutions are consistent with the very tight binding of the wild type BLIP (0.5 nM). It is also of interest that some positions did not exhibit a large difference in substitution patterns in the screen for partial or wild type levels of function. For example, residues Tyr53 and Trp150 were found to be relatively intolerant of substitutions in both screens suggesting that nearly all substitutions at these positions result in a large decrease in function and that there is a precise requirement for the physical-chemical properties of the side chains at these positions. The sequence requirements at residues Glu31, Ser35, Tyr50, Tyr51, Ser71, Trp112, Ser113, Tyr143, Arg160 and Trp162 also did not change significantly between screens in that several different amino acids can substitute at these positions without impacting function. This finding suggests that a wide range of chemical properties of side chains are consistent with function at these positions. Finally, several positions did exhibit a significant difference in sequence requirements for partial versus wild type levels of function. These positions include Asp49, where only Asp and Glu exhibit wild type levels of function in the stringent screen while several different substitutions are consistent with partial function (Fig. 3B). Similarly, positions Phe36, His41, Ser39, Gly48 and Phe142 exhibit stringent sequence requirements for wild type (E. coli TP132) versus partial levels (E. coli TP112) of function. The pattern of substitutions between the screens for these residues suggests that there are stringent requirements on the chemical nature of the side chains for the tight binding characteristic of wild type BLIP but that more diverse chemical properties are consistent with partial function (< 20 nM Ki).
Figure 4.

Structure of BLIP with randomized residues highlighted. Panel A summarizes the results from alanine scanning of BLIP and measurement of binding to TEM-1 β-lactamase 28. Residues colored red represent positions where the alanine substitution resulted in a greater than 10-fold loss in binding affinity. Residues colored green represent positions where an alanine substitution had a less than 10-fold effect on binding and the residue colored cyan represents a position where the alanine substitution resulted in a greater than 10-fold increase in binding affinity 28. In Panel B the results of the saturation mutagenesis studies are summarized. Positions where 2 or less amino acids substitutions (including wild type) are consistent with function are colored red. Positions where 4 or less amino acids are consistent with function are colored yellow. Positions were greater than 4 amino acid substitutions are consistent with function are colored green.
Discussion
In this study, a genetic screen for BLIP function was developed and validated using a set of previously characterized alanine substitution mutants of BLIP 4. The availability of quantitative enzyme inhibition data for the alanine mutants allowed optimization of the system to modulate the levels of BLIP function required for mutants to pass the screen. The 23 BLIP residue positions that are in contact with TEM-1 β-lactamase based on the crystal structure and for which alanine mutants had previously been examined were randomized individually to create libraries containing all possible amino acid substitutions at each position. Each library of mutants was examined using the genetic screen to identify clones with ampicillin MIC values similar to a control strain encoding wild type BLIP. The clones with wild type levels indicate sequences that are consistent with tight binding to β-lactamase. The results revealed a wide range in sequence requirements among the 23 contact positions.
Eight residue positions in BLIP including Phe36, Ser39, His41, Gly48 Asp49, Tyr53, Phe142 and Trp150, exhibited very stringent sequence requirements with only one other amino acid besides wild type being consistent with high level function. For all of these positions, the one substitution was chemically conservative compared to the wild type residue (Figure 3B). Therefore, these residue positions have precise sequence requirements in order to provide tight binding BLIP function. It should be noted, however, that the sequence requirements could be due to protein stability requirements as well as binding requirements. The positions mutated are on the surface of the protein and so destabilizing substitutions are less likely but the in vivo screen does not distinguish binding versus stability defects. The randomization and selection results for positions Phe36, His 41, Asp49, Tyr53, Phe142 and Tyr150 are consistent with the previous alanine scanning mutagenesis results in that alanine substitutions at each of these residues resulted in a greater than 20-fold loss in binding affinity as indicated by the inhibition constant for TEM-1 β-lactamase 4; 28. Among these residues, Asp49 and Phe142 are present on loop structures in BLIP and both insert into the active site of TEM-1 β-lactamase. Asp49 makes numerous interactions with TEM-1 active site residues and appears to mimic the interactions of the carboxylate group found in β-lactam antibiotics 19. Phe142 fills a section of the active site in a region similar to that occupied by the benzyl group of benzylpenicillin 19. Therefore, there appear to be precise volume and chemical composition restrictions on these positions which greatly restrict the amino acids that can substitute for the wild type residues. Residues Phe36 and Trp150 are large aromatic residues that are near the periphery of the binding site and flank each side of a TEM-1 β-lactamase loop region containing residues 99-112 and appear to act as important “clamps” on the loop. Tyr53 is buried in the BLIP-TEM-1 β-lactamase interface approximately 6 Å from Phe36 where it interacts with Pro107 from TEM-1 presumably to also stabilize interactions with the 99-112 loop of TEM-1. Previous results indicated that an alanine substitution at His41 resulted in a greater than 50-fold loss in binding affinity as measured by the inhibition constant 4; 28 and the stringent sequence requirements are consistent with this observation. His41 is situated between the critical BLIP residues Phe36 and Tyr53 where it contributes to the surface of BLIP that interacts with the 99-112 loop of TEM-1 in the vicinity of TEM-1 Pro107.
BLIP positions Ser39, Gly48 and Gly141 also exhibited stringent sequence requirements. Interestingly, alanine substitutions at Ser39, Gly48 and Gly141 had little effect on binding affinity and yet these positions are not tolerant of substitutions 4; 28. This is due the ability of small volume amino acids to substitute these positions (Fig. 3B). Ser39 forms a cluster with Phe36, Tyr53 and His41 and interacts with side chain of Lys111 in the 99-112 loop of TEM-1. Gly48 and Gly141 appear to play similar roles in that they are adjacent to Asp49 and Phe142 that are on the BLIP loops that insert into the β-lactamase active site. The sequence constraint to small amino acids may be due to steric factors on the turn structures on which Asp49 and Phe142 reside. It is unclear why alanine mutants were not observed among the functional mutants for Ser39 and Gly141 since alanine did not greatly affect the Ki values. This could be due to a failure to sample the alanine mutant or these could be false negatives due to altered protein expression levels of BLIP mutants.
Several residues in BLIP were previously found to contribute strongly to binding via alanine substitution experiments and yet could be substituted by several different amino acids in the randomization experiments and retain function. These positions include Lys74, Trp112, Arg160 and Trp162. Lys74 is an interesting position in that it appears to play a key role in mediating the binding specificity of BLIP because substitution of this residue by alanine results in a greater than 50-fold loss in binding affinity for TEM-1 but a 10-fold increase in binding affinity for SME-1 β-lactamase 4; 28. In the BLIP-TEM-1 β-lactamase structure, Lys74 interacts with Glu104 from TEM-1 to form a key buried salt bridge which is consistent with the finding that substitution by alanine reduces affinity 19. Nevertheless, several different amino acid types are found at position 74 among the random mutants with high levels of BLIP function (Fig. 3B). The arginine and histidine substitutions may be able to approximate the positive charge of the lysine interaction to retain function. However, charge does not appear to be essential since leucine and tyrosine are also found among the functional mutants.
The Trp112 residue in BLIP is buried in the complex near the center of the binding interface between BLIP and the 99-112 loop region of TEM-1 β-lactamase 19. Substitution of Trp112 with alanine results in a loss of binding affinity with TEM-1 as well as with all other class A β-lactamases tested including SME-1, SHV-1 and Bla1 4. Despite this fact, several different amino acid types were found at position 112 among random mutants with BLIP function (Fig. 3B). Several of the substitutions such as leucine, isoleucine and valine are conservative and may retain similar interactions; however the serine substitution is more difficult to rationalize.
Substitution of BLIP Arg160 with alanine also results in a large loss of binding affinity for TEM-1 as well as SME-1, SHV-1 and Bla1 β-lactamases 4. However, several amino acid types at position 160 are consistent with BLIP binding to TEM-1 β-lactamase. Similarly, substitution of Trp162 in BLIP with alanine results in a greater than 20-fold loss in binding affinity to TEM-1 β-lactamase but the position is substituted by several residue types among functional mutants (Fig. 3B). Trp162 is near the periphery of the interface between BLIP and the TEM-1 99-112 loop region where it packs against TEM-1 residues Val103 and Pro167 19. Substitution of Trp162 with phenylalanine and histidine could reconstitute these interactions but the arginine and glutamine substitutions may work by a different mechanism (Fig. 3).
As is apparent from the above discussion, there is general agreement between the results obtained by alanine scanning mutagenesis versus the randomization and selection strategy but there are several exceptions. This is indicated in Fig. 5, which shows the ΔΔG of the alanine mutant for each position versus the number of amino acids that can be substituted at each position in the more stringent E. coli TP132 strain. Note that the exceptions, including positions Lys74, Trp112, Arg160 and Trp162, lie above the correlation line, i.e., ΔΔG for the alanine substitution is large and positive but the position can be substituted by several amino acids. From these results it is apparent that the saturation mutagenesis results provide a more fine-grained view of the sequence requirements for protein function.
Figure 5.
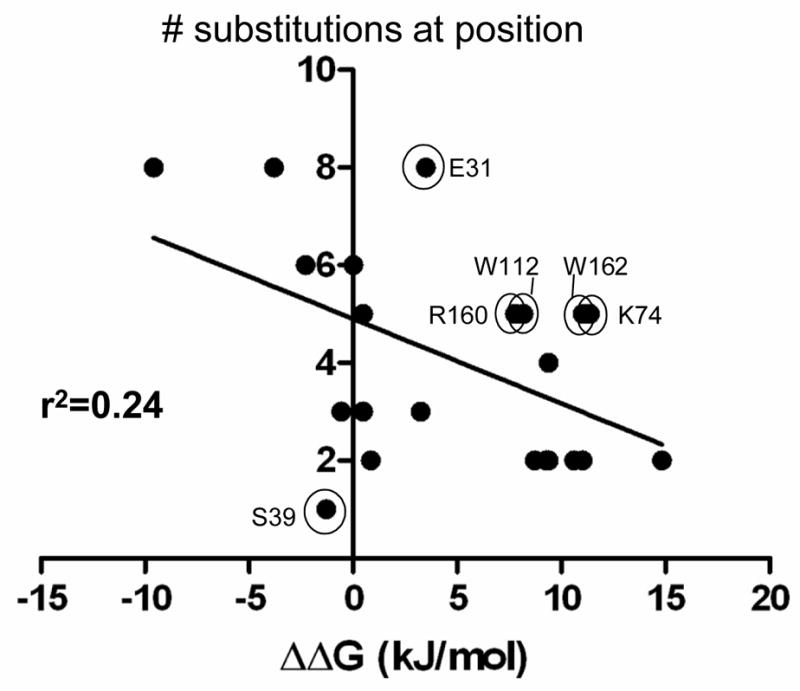
The graph shows the correlation between the randomization and selection results and the ΔΔG values of the alanine scanning mutants at the same positions using Ki data from Zhang and Palzkill 28. Each data point represents one of the 23 residues randomized and shown in Fig. 3. The X-axis indicates the ΔΔG (kJ/mol) values for the BLIP alanine mutants for binding TEM-1 β-lactamase 28. The Y-axis indicates the number of amino acids (including wild type) consistent with wild type levels of BLIP function in E. coli TP132 (top panel sequences in Fig. 3B). The circled and labeled positions are those where there is a lack of correlation between the alanine substitution result and the randomization result. For example, the W112 position exhibits a large loss of binding affinity when mutated to alanine suggesting the position is important for binding, however, five different amino acids can reside at residue 112 and provide high level function. r2 is the goodness of the linear regression fit of the indicated line.
Taken together, the randomization results in Fig. 3 indicate that the BLIP binding surface is quite tolerant of mutations. With the exception of one position, the residues analyzed are tolerant of at least one amino acid substitution and for most positions the presence of any of several different amino acids is consistent with function. A recent comprehensive saturation mutagenesis study to examine the sequence requirements for binding of human growth hormone (hGH) to human growth hormone receptor also found that residues in the hGH binding surface were tolerant of substitutions 31. In addition, the hGH study found that, for several positions, the substitutions observed were not conservative in chemical type 31. Similar observations were made in this work for a subset of residues including Lys74, Trp112, Arg160 and Trp162 as described above (Fig. 3). Therefore, the hGH-hGHR and BLIP-β-lactamase interfaces exhibit some similarities in terms of overall sequence requirements.
The accurate computational prediction of binding energy in protein-protein interactions is an important goal for protein engineering and design as well as the protein docking studies. Several structure-based computational methods have been developed to estimate free energy changes associated with mutations 35; 36; 37; 38; 39. As a first step towards comparing the BLIP experimental results with computational predictions, the results of the genetic screen in Fig. 3 were compared with free energy change predictions made using the FoldX force field 36; 40; 41. The free energy changes in the BLIP-TEM-1 β-lactamase complex were calculated using FoldX for all 19 mutants for each of the 23 BLIP positions that were randomized in this study (Materials and Methods). Although, there was variation in the extent of correlation from position to position, it was found that the average calculated ΔΔG for binding for the mutants from all 23 libraries with high levels of BLIP function (low ampicillin MIC) identified in the genetic screen in E. coli TP132 (Fig. 3B, top panel) was 2.93 kJ/mol. In contrast, the average calculated ΔΔG for all possible substitutions for all positions was 6.62 kJ/mol. Therefore, as a first approximation, FoldX appears to identify the functional tight binding BLIP mutants (low ampicillin MICs and low calculated ΔΔG) from the general pool of mutants. This point is illustrated for each of the 23 randomized positions in Figure 6. With the exception of positions E31, E73 and R160, the averaged FoldX calculated ΔΔG values for the functional mutants identified at each residue position in the genetic screen are lower than the averaged FoldX calculated ΔΔG values for all 19 mutants at each position (Supplemental Table 1). However, the results in Figure 6 also illustrate those positions where the FoldX calculated ΔΔG values for the functional mutants are relatively high and therefore predict weak binding including residues G48, E73, K74, R144 and R160. It will be of interest to study the binding characteristics of BLIP variants with substitutions at these positions in more detail.
Figure 6.

The graph shows the average ΔΔG values for binding for BLIP mutants containing amino acid substitutions at the indicated residue positions. The X-axis indicates the BLIP residue positions that were randomized in the study. The open bar for each residue position indicates the average change in free energy of the BLIP-TEM-1 β-lactamase complex calculated using FoldX for each mutant that was found in the genetic screen to exhibit high levels of BLIP function (top panel, Fig. 3B). The value for each mutant was calculated and then the values were averaged. The filled bars indicate the average change in binding free energy calculated using FoldX for each of the 19 substitutions. The value was calculated for each of the 19 substitutions and then the values were averaged.
BLIP is known to bind to a number of different class A β-lactamases with a wide range of affinities 4; 14. Previous studies of 23 contact residues by alanine substitution followed by quantitation of binding to the TEM-1, SME-1, SHV-1 and Bla1 β-lactamases distinguished three classes of positions, those where alanine substitution did not effect binding to any β-lactamase, which reflect positions whose side chains do not contribute to binding; those where alanine substitution decreased binding to all β-lactamases, which reflect positions that are important forβ-lactamase binding in general; and those where alanine substitutions had different effects depending on the β-lactamase being examined, which reflects positions that influence the substrate specificity of BLIP. The results in Figure 5 indicate that the alanine scanning results for BLIP binding TEM-1 do not always correlate with how freely a position can be substituted by saturation mutagenesis. It will be of interest to determine the sequence requirements for BLIP binding to other class A β-lactamases and how these requirements compare to those for binding TEM-1. Such studies should provide additional insights into mechanisms of molecular recognition between related proteins.
Materials and Methods
Bacterial strains and plasmids
The E. coli TP112 strain was constructed from the recombineering strain E. coli SW102 whose genotype is: F- mcrA Δ (mrr-hsdRMS-mcrBC) Φ80dlacZ M15 ΔlacX74 deoR recA1 endA1 araD139 Δ(ara, leu) 7649 galU galK rspL nupG [ λcI857 (cro-bioA) <> tet] 32. The blaTEM-1 gene was introduced on a PCR product containing 35 base pairs (bp) of flanking sequence from the pyrF gene on each end. The primers used for amplification were: pyrF-bla-top 5′-CACGCGATTGTCGTCTGAAGGTCGGCAAAGAGATGGTTTCTTAGACGTCAGGTGGCACTTTTCGGGGAAATG-3′ pyrF-bla-bot 5′-GCGGTGCATCTTTGCCAAACGGAACCAGTGCCTCTTACCAATGCTTAATCAGTGAGGCACCTATCTCAGCG-3′. The E. coli SW102 strain was grown and concentrated for electroporation at 30°C. The pyrF-bla PCR product was electroporated into E. coli SW102 and the cells were spread on agar plates containing 50 μg/ml ampicillin and 5 μg/ml tetracycline. The presence of the blaTEM-1 gene on the chromosome inserted into the pyrF gene was validated by PCR amplification of the region followed by DNA sequencing. The genotype of the E. coli TP112 strain is: F- mcrA Δ(mrr-hsdRMS-mcrBC) Φ80dlacZ M15 ΔlacX74 deoR recA1 endA1 araD139 Δ (ara, leu) 7649 galU galK rspL nupG pyrF::blaTEM-1 [ λcI857 (cro-bioA) <> tet]. The pyrF-blaTEM-1 gene region was moved from E. coli TP112 to the chromosome of E. coli MG1655 (F- lambda- ilvG- rfb-50 rph-1) by P1 transduction to create E. coli TP132. The genotype of E. coli TP132 is: F- lambda- ilvG- rfb-50 rph-1 pyrF::blaTEM-1. The BLIP random libraries were introduced into E. coli XL1-Blue (recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac [F′proAB lacIqZΔM15 Tn10 (Tetr)]) 42. E. coli RB791 43 was used for expression of BLIP mutants for protein purification.
Library construction and screening
The BLIP random libraries were constructed in the pGR32 plasmid which contains BLIP fused to the TEM-1 signal sequence under the transcriptional control of the trc promoter 17. The BLIP protein is tagged with a 6x His-tag at the N-terminus of the protein 17. Each of the 23 random libraries was constructed by overlap extension PCR whereby the codon to be randomized was converted to NNN where N is any of the 4 nucleotides 44. Briefly, primers complementary to internal and external sequences in the blip gene were used to PCR-amplify two DNA fragments with overlapping ends. Specific mutations in the nucleotide sequence of the internal complementary primers incorporated the NNN sequence at the codon of interest 33. The PCR products were examined on an agarose gel to ensure the reaction occurred to create the expected sized DNA products. The two PCR reactions containing the overlapping fragments were then combined into a single PCR reaction and amplified with the external primers TP123-top and blip-XbaI-4 to create the final PCR fusion product. The PCR product was digested with the SacI and XbaI restriction endonucleases and ligated into the pTP123 plasmid 17 that had been digested with the SacI and XbaI enzymes. The ligation products (library DNA) were used to transform E. coli XL1-Blue and cells were spread on agar plates containing 12.5 μg/ml chloramphenicol. A minimum of 1000 colonies were pooled for each library. Several clones were picked from each library for DNA sequence analysis to assess the mutagenesis procedure for the libraries. Each random library was used to transform E. coli TP112 and cells were spread on agar plates containing 12.5 μg/ml chloramphenicol. Approximately 10 colonies were picked for each library from the chloramphenicol containing agar plates and sequenced to test for any obvious sequence bias in each randomized library in the TP112 strain. Mutant clones with low ampicillin minimum inhibitory concentrations (MICs) were identified by picking individual colonies with toothpicks and inoculating growth media in 96-well microtiter plates. The 96-mini-cultures were diluted into additional 96-well plates containing various concentrations of ampicillin and the plates were grown overnight at 37°C with shaking. Those clones that exhibited low ampicillin resistance in this assay were tested to determine an exact ampicillin MIC using the E-test strip method according to manufacturer’s instructions (AB Biodisk). Those clones that exhibited ampicillin MIC values similar to the wild-type His-tagged BLIP control in pGR32 were examined by DNA sequencing to determine the identity of each mutant. The plasmid DNA from each of the sequenced clones with low ampicillin MICs from the E. coli TP112 screen was isolated and used to transform E. coli TP132. The ampicillin MIC from each of the clones in E. coli TP132 was determined by the E-test strip method.
Protein purification and inhibition assays
The wild type, His-tagged BLIP as well as the E31Q, D49S, E73Y, W112R, and F142G mutants were introduced into E. coli RB791 and expressed by induction with IPTG. The BLIP proteins were purified by metal-affinity chromatography as previously described 17. TEM-1 β-lactamase was purified using a zinc chelating column as described previously 45.
The β-lactamase inhibition assays were performed as described previously with minor modifications 17. Briefly, 0.3 nM of TEM-1 β-lactamase was incubated with increasing concentrations purified BLIP and allowed to equilibrate for one hour at 30°C. The colorimetric β-lactam substrate nitrocefin was added to a concentration of 10 μM and the initial velocity of the reaction was monitored at 482 nM. The initial velocity of nitrocefin hydrolysis was plotted versus BLIP concentration and the resulting curve was fit to the equation for a tight binding inhibitor to obtain the inhibition constant Ki as described previously with the modification that the Ki was calculated from the apparent Ki using the equation below as described18
Calculation of ΔΔG and correlation with ampicillin resistance of E. coli strains
The Ki values for the 23 BLIP alanine mutants listed in Table 1 were converted to ΔΔG values using the equation ΔΔG= −RT ln(Kiwt/Kimut). The kcal/mol values thus obtained were converted to kJoules/mol using 1 calorie equals 4.184 joules. The ΔΔG values for BLIP alanine mutants determined by surface plasmon resonance used for the correlation studies in Fig. 2C and 2D were from Table 1 of Reichmann et al. 26. The values used for Fig. 2C and 2D were for BLIP alanine mutants F36A, H41A, D49A, Y50A, Y53A, S71A, K74A, W112A, S113A, F142A, H148A, W150A and R160A 26.
Calculation of changes in binding free energy using FoldX
The FoldX 3.0 program package was obtained from the FoldX website (http://foldx.crg.es) 41. The 3-D structure of the BLIP-TEM-1 β-lactamase complex (1JTG) was first optimized using the RepairPDB command in FoldX. Each of the 23 BLIP residue positions was mutated to all 19 amino acid substitutions in the A-B complex within 1JTG using the BuildModel command to create a wild type and corresponding mutant PDB file for each substitution at each position. Each PDB file was then evaluated using the AnalyseComplex command to generate a binding interaction energy for each mutant and corresponding wild type structure. The calculated interaction energy of each wild type complex was subtracted from its corresponding mutant to generate a ΔΔG value.
Supplementary Material
Acknowledgments
This work was supported by NIH grant AI32956 to T.P. We thank the NCI-Frederick for E. coli strain SW102.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bahadur RP, Chakrabarti P, Rodier F, Janin J. A dissection of specific and non-specific protein-protein interfaces. J Mol Biol. 2004;336:943–955. doi: 10.1016/j.jmb.2003.12.073. [DOI] [PubMed] [Google Scholar]
- 2.Clackson T, Wells JA. A hot spot of binding energy in a hormone-receptor interface. Science. 1995;267:383–386. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 3.Keskin O, Ma B, Rogale K, Gunasekaran K, Nussinov R. Protein-protein interactions: organization, cooperativity and mapping in a bottom-up systems biology approach. Phys Biol. 2005;2:S24–S35. doi: 10.1088/1478-3975/2/2/S03. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Z, Palzkill T. Dissecting the protein-protein interface between beta-lactamase inhibitory protein and class A beta-lactamases. J Biol Chem. 2004;279:42860–42866. doi: 10.1074/jbc.M406157200. [DOI] [PubMed] [Google Scholar]
- 5.Reichmann D, Rahat O, Cohen M, Neuvirth H, Schreiber G. The molecular architecture of protein-protein binding sites. Curr Opin Struct Biol. 2007;17:67–76. doi: 10.1016/j.sbi.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 6.Ambler RP, Coulson FW, Frere JM, Ghuysen JM, Joris B, Forsman M, Levesque RC, Tiraby G, Waley SG. A standard numbering scheme for the class A β-lactamases. Biochem J. 1991;276:269–272. doi: 10.1042/bj2760269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bush K, Jacoby GA, Medeiros AA. A functional classification scheme for b-lactamases and its correlation with molecular structure. Antimicrob Agents Chemother. 1995;39:1211–1233. doi: 10.1128/aac.39.6.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Majiduddin FK, Materon IC, Palzkill TG. Molecular analysis of beta-lactamase structure and function. Int J Med Microbiol. 2002 doi: 10.1078/1438-4221-00198. [DOI] [PubMed] [Google Scholar]
- 9.Queenan AM, Bush K. Carbapenemases: the versatile beta-lactamases. Clin Microbiol Rev. 2007;20:440–458. doi: 10.1128/CMR.00001-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petrosino J, Cantu C, III, Palzkill T. b-lactamases: protein evolution in real time. Trends Microbiol. 1998;6:323–327. doi: 10.1016/s0966-842x(98)01317-1. [DOI] [PubMed] [Google Scholar]
- 11.Wiedemann B, Kliebe C, Kresken M. The epidemiology of β-lactamases. J Antimicrob Chemother. 1989;24:1–24. doi: 10.1093/jac/24.suppl_b.1. [DOI] [PubMed] [Google Scholar]
- 12.Perez F, Endimiani A, Hujer KM, Bonomo RA. The continuing challenge of ESBLs. Curr Opin Pharmacol. 2007;7:459–469. doi: 10.1016/j.coph.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doran JL, Leskiw BK, Aippersbach S, Jensen SE. Isolation and characterization of a b-lactamase-inhibitory protein from Streptomyces clavuligerus and cloning and analysis of the corresponding gene. J Bacteriol. 1990;172:4909–4918. doi: 10.1128/jb.172.9.4909-4918.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Strynadka NCJ, Jensen SE, Johns K, Blanchard H, Page M, Matagne A, Frere JM, James MNG. Structural and kinetic characterization of a b-lactamase-inhibitor protein. Nature. 1994;368:657–660. doi: 10.1038/368657a0. [DOI] [PubMed] [Google Scholar]
- 15.Liras P, Rodriguez-Garcia A. Clavulanic acid, a beta-lactamase inhibitor: biosynthesis and molecular genetics. Appl Microbiol Biotechnol. 2000;54:467–475. doi: 10.1007/s002530000420. [DOI] [PubMed] [Google Scholar]
- 16.Thai W, Paradkar AS, Jensen SE. Construction and analysis of beta-lactamase-inhibitory protein (BLIP) non-producer mutants of Streptomyces clavuligerus. Microbiology. 2001;147:325–335. doi: 10.1099/00221287-147-2-325. [DOI] [PubMed] [Google Scholar]
- 17.Petrosino J, Rudgers G, Gilbert H, Palzkill T. Contributions of aspartate 49 and phenylalanine 142 residues of a tight binding inhibitory protein of β-lactamases. J Biol Chem. 1999;274:2394–2400. doi: 10.1074/jbc.274.4.2394. [DOI] [PubMed] [Google Scholar]
- 18.Reynolds KA, Thomson JM, Corbett KD, Bethel CR, Berger JM, Kirsch JF, Bonomo RA, Handel TM. Structural and computational characterization of the SHV-1 beta-lactamase-beta-lactamase inhibitor protein interface. J Biol Chem. 2006;281:26745–26753. doi: 10.1074/jbc.M603878200. [DOI] [PubMed] [Google Scholar]
- 19.Strynadka NCJ, Jensen SE, Alzari PM, James MNG. A potent new mode of b-lactamase inhibition revealed by the 1.7Å X-ray crystallographic structure of the TEM-1-BLIP complex. Nature Struct Biol. 1996;3:290–297. doi: 10.1038/nsb0396-290. [DOI] [PubMed] [Google Scholar]
- 20.Albeck S, Schreiber G. Biophysical characterization of the interaction of the β–lactamase TEM-1 with its protein inhibitor BLIP. Biochemistry. 1999;38:11–21. doi: 10.1021/bi981772z. [DOI] [PubMed] [Google Scholar]
- 21.Joughin BA, Green DF, Tidor B. Action-at-a-distance interactions enhance protein binding affinity. Protein Sci. 2005;14:1363–1369. doi: 10.1110/ps.041283105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reynolds KA, Hanes MS, Thomson JM, Antczak AJ, Berger JM, Bonomo RA, Kirsch JF, Handel TM. Computational redesign of the SHV-1 β-lactamase/β-lactamase inhibitor protein interface. J Mol Biol. 2008;382:1265–1275. doi: 10.1016/j.jmb.2008.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rudgers GW, Palzkill T. Identification of residues in β-lactamase critical for binding β-lactamase inhibitory protein. J Biol Chem. 1999;274:6963–6971. doi: 10.1074/jbc.274.11.6963. [DOI] [PubMed] [Google Scholar]
- 24.Selzer T, Albeck S, Schreiber G. Rational design of faster associating and tighter binding protein complexes. Nat Struc Biol. 2000;7:537–541. doi: 10.1038/76744. [DOI] [PubMed] [Google Scholar]
- 25.Wang J, Zhang Z, Palzkill T, Chow DC. Thermodynamic investigation of the role of contact residues of b-lactamase-inhibitory protein for binding to TEM-1 β-lactamase. J Biol Chem. 2007;282:17676–17684. doi: 10.1074/jbc.M611548200. [DOI] [PubMed] [Google Scholar]
- 26.Reichmann D, Cohen M, Abramovich R, Dym O, Lim D, Strynadka NCJ, Schreiber G. Binding hot spots in the TEM1-BLIP interface in light of its modular architecture. J Mol Biol. 2007;365:663–679. doi: 10.1016/j.jmb.2006.09.076. [DOI] [PubMed] [Google Scholar]
- 27.Reichmann D, Rahat O, Albeck S, Meged R, Dym O, Schreiber G. The modular architecture of protein-protein binding interfaces. 2005;102:57–62. doi: 10.1073/pnas.0407280102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Z, Palzkill T. Determinants of binding affinity and specificity for the interaction of TEM-1 and SME-1 β-lactamase with β-lactamase inhibitory protein. J Biol Chem. 2003;278:45706–45712. doi: 10.1074/jbc.M308572200. [DOI] [PubMed] [Google Scholar]
- 29.Cunningham BC, Wells JA. High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science. 1989;244:1081–1085. doi: 10.1126/science.2471267. [DOI] [PubMed] [Google Scholar]
- 30.Huang W, Petrosino J, Hirsch M, Shenkin PS, Palzkill T. Amino acid sequence determinants of β-lactamase structure and activity. J Mol Biol. 1996;258:688–703. doi: 10.1006/jmbi.1996.0279. [DOI] [PubMed] [Google Scholar]
- 31.Pal G, Kouadio JLK, Artis DR, Kossiakoff AA, Sidhu SS. Comprehensive and quantitative mapping of energy landscapes for protein-protein interactions by rapid combinatorial scanning. J Biol Chem. 2006;281:22378–22385. doi: 10.1074/jbc.M603826200. [DOI] [PubMed] [Google Scholar]
- 32.Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 2005;33:e36. doi: 10.1093/nar/gni035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Materon IC, Palzkill T. Identification of residues critical for metallo-β-lactamase function by codon randomization and selection. Protein Sci. 2001;10:2556–2565. doi: 10.1110/ps.40884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brosius J, Erfle M, Storella J. Spacing of the -10 and -35 regions in the tac promoter. J Biol Chem. 1985;260:3539–3541. [PubMed] [Google Scholar]
- 35.Benedix A, Becker CM, de Groot BL, Caflisch A, Bockmann RA. Predicting free energy changes using structural enxembles. Nature Methods. 2009;6:3–4. doi: 10.1038/nmeth0109-3. [DOI] [PubMed] [Google Scholar]
- 36.Guerois R, Nielsen JE, Serrano L. Predicting changes in the stability of proteins and protein complexes: A study of more than 1000 mutations. J Mol Biol. 2002;320:369–387. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- 37.Kortemme T, Baker D. A simple physical model for binding energy hot spots in protein-protein complexes. Proc Natl Acad Sci U S A. 2002;99:14116–14121. doi: 10.1073/pnas.202485799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pokala N, Handel TM. Energy functions for protein design: adjustment with protein-protein complex affinities, models for the unfolded state, and negative design of solubility and specificity. J Mol Biol. 2005;347:203–227. doi: 10.1016/j.jmb.2004.12.019. [DOI] [PubMed] [Google Scholar]
- 39.Potapov V, Reichmann D, Abramovich R, Filchtinski D, Zohar N, Ben Halevy D, Edelman M, Sobolev V, Schreiber G. Computational redesign of a protein-protein interface for high affinity and binding sepcificity using modular architecture and naturally occurring template fragments. J Mol Biol. 2008;384:109–119. doi: 10.1016/j.jmb.2008.08.078. [DOI] [PubMed] [Google Scholar]
- 40.Kiel C, Wohlgemuth S, Rousseau F, Schymkowitz J, Ferkinghoff-Borg J, Wittenghofer F, Serrano L. Recognizing and defining true Ras binding domains II: In Silico prediction based on homology modelling and energy calculations. J Mol Biol. 2005;348:759–775. doi: 10.1016/j.jmb.2005.02.046. [DOI] [PubMed] [Google Scholar]
- 41.Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic Acids Res. 2005;33:W382–388. doi: 10.1093/nar/gki387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bullock WO, Fernandez JM, Short JM. XL1-Blue: a high efficiency plasmid transforming recA Escherichia coli strain with beta-galactosidase selection. BioTechniques. 1987;5:376–379. [Google Scholar]
- 43.Amann E, Brosius J, Ptashne M. Vectors bearing a hybrid trp-lac promoter useful for regulated expression of cloned genes in Escherichia coli. Gene. 1983;25:167–178. doi: 10.1016/0378-1119(83)90222-6. [DOI] [PubMed] [Google Scholar]
- 44.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 45.Cantu C, III, Huang W, Palzkill T. Selection and characterization of amino acid substitutions at residues 237–240 of TEM-1 β-lactamase with altered substrate specificity for aztreonam and ceftazidime. J Biol Chem. 1996;271:22538–22545. doi: 10.1074/jbc.271.37.22538. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.