

Abstract
Huntington’s disease (HD) is one of the few neurodegenerative diseases with a known genetic cause, knowledge that has enabled the creation of animal models using genetic manipulations that aim to recapitulate HD pathology. The study of behavioral and neuropathological phenotypes of these HD models, however, has been plagued by inconsistent results across laboratories stemming from the lack of standardized husbandry and testing conditions, in addition to the intrinsic differences between the models. We have compared different HD models using standardized conditions to identify the most robust phenotypic differences, best suited for preclinical therapeutic efficacy studies. With a battery of tests of sensory-motor function, such as the open field and prepulse inhibition tests, we replicate previous results showing a strong and progressive behavioral deficit in the R6/2 line with an average of 129 CAG repeats in a mixed CBA/J and C57BL/6J background. We present the first behavioral characterization of a new model, an R6/2 line with an average of 248 CAG repeats in a pure C57BL/6J background, which also showed a progressive and robust phenotype. The BACHD in a FVB/N background showed robust and progressive behavioral phenotype, while the YAC128 full-length model on either an FVB/N or a C57BL/6J background generally showed milder deficits. Finally, the HdhQ111 knock-in mouse on a CD1 background showed very mild deficits. This first extensive standardized cross-characterization of several HD animal models under standardized conditions highlights several behavioral outcomes, such as hypoactivity, amenable to standardized preclinical therapeutic drug screening.
Introduction
The genetic factors underlying onset, penetrance and progression of most neurodegenerative and psychiatric diseases are poorly understood. In 1983 a genetic marker for Huntington’s disease, a dominantly inherited disorder featuring early loss of neurons in the striatum, with onset of movements and behavioral and cognitive decline, was discovered (Gusella et al., 1983). In 1993, the expanded CAG repeat mutation, extending a polyglutamine tract in the huntingtin protein from the normal range (8–37) to the HD range (>38), responsible for this fatal disease was isolated (The Huntington’s Disease Collaborative Research Group, 1993), enabling the creation of a number of different HD animal models (Menalled and Chesselet, 2002). These models are broadly distinguished by distinct initiating insults. Transgenic mice express transgenes with the first exon of the HD gene (R6/2) or the entire human HD gene (BACHD, YAC128). By contrast, HdhQ111 knock-in mice, generated by inserting expanded CAG repeats into the murine HD gene homologue, express endogenous murine huntingtin with an extended polyglutamine tract.
In humans, overt HD phenotypes reflect an accrued plethora of changes, including loss of brain architecture, which develops over many decades, consequent to the expression of the HD gene mutation, so proper expression of mutant huntingtin may provide a window into the earliest consequences of the HD-initiating mechanism that manifest in the mouse. By contrast, ectopic expression of a mutant HD gene, or a portion of it, produce rapid onset of overt abnormalities, which may expedite evaluation of candidate therapeutics, though the underlying transgene insults do not exactly recapitulate the HD mutation. While transgenic and knock-in HD mouse models have been evaluated extensively, differences in genetic background, breeding, housing, dosing, and testing protocols increase variability within any given model. Taken together, these genetic, environmental, and technical differences have frustrated efforts to characterize behavioral phenotypes that may provide efficient and standardized testing of putative therapies.
Attempts to tease out the factors that influence phenotype in HD and other models of neurodegenerative disease have been only partly successful for HD and for other neurodegenerative diseases (Argmann and Auwerx, 2006; Hockly et al., 2003). It is now clear that the genetic background on which the model is maintained is one of many crucial factors as dominant and recessive genetic modifiers can have a profound influence on the penetrance and progression of pathology (Gerlai, 1996; Lloret et al., 2006; Nadeau, 2003; Van Raamsdonk et al., 2007). External factors that influence the model’s phenotype include husbandry (in particular the presence or absence of environmental enrichment) and the specific protocols used for the model’s characterization (Glass et al., 2004; Hockly et al., 2002; Hockly et al., 2003; Lazic et al., 2006; Schilling et al., 2004; Spires et al., 2004; van Dellen et al., 2000; van Dellen et al., 2008). Moreover, in drug-efficacy studies, variability is amplified by the use of different doses, formulations, routes of administration, timing and duration of dosing. Because of the many factors affecting phenotype, the ensuing variability in the results from different laboratories is one confound to efforts to advance potential therapies from laboratory to clinic.
The complexity of preclinical research in HD is not unique. For many diseases, such as Muscular Dystrophy, Spinal Muscular Atrophy and Amyotrophic Lateral Sclerosis, various animal models are available that differ in their pathogenesis, pathophysiology and progression, and in other modifiers such as the genetic background of the model. Institutional efforts (ALS Association; SMA Foundation; TREAT-NMD) are under way to standardize testing protocols to allow proper comparison across parallel preclinical research.
To establish a preclinical testing program for HD, we have developed standardized protocols for husbandry and behavioral testing of different types of HD mouse models. Considering that different models may provide assessment of different aspects of disease, and that different behavioral tests may differentially tackle processes that are affected in varying degrees across models, we employed a battery of tests under standardized husbandry and testing conditions to assess the potential behavioral phenotypes of four genetic HD models – R6/2, HdhQ111, BACHD, and YAC128 – with two of the transgenic models – R6/2 and YAC128 – maintained on two different genetic backgrounds. The two R6/2 lines differed also in the number of CAG repeats, thus differences could be attributable to either factor or their interaction. Based on our findings, we describe here a comprehensive protocol for breeding, housing, and testing these animals, with attention to standardization of experimental conditions and the statistical models used for analysis.
Materials and methods
Subjects
Table 1 shows the characteristics of the different HD models. We used a sample size of 12 for all lines per gender and genotype. Littermates were used as controls.
Table 1.
Summary of characteristics of the 6 models studied.
| Line | Construct | Specific name |
CAG repeatsa (mean/range) |
Full name (vendor catalog number) |
Background (vendor) |
Refs. |
|---|---|---|---|---|---|---|
| R6/2 | Transgenic: human exon 1 under control of human HD promoter sequences |
R6/2 | 136 (128–142) CAG | B6CBATg (HD exon 1) 62Gpb/3J (Jax Stock No. 006494) |
CBAB6F1 (Jax) | (Mangiarini et al., 1997) |
| R6/2B | 260 (254–2276) CAG | Not commercially available | C57BL/6J (Jax) | NA | ||
| HdhQ111 | Knock-in: chimeric human/mouse exon 1 construct under control of mouse HD promoter |
HdhQ111 | 106 (97–114) CAG |
Htttm5Mem/J (Jax Stock No. 003456) |
CD1 (CRL)b | (Wheeler et al., 2000) |
| BACHD | Transgenic: full-length genomic construct under control of human HD promoter gene |
BACHD | 97–98 (CAG-CAA) | Not commercially available | FVB/NJ (Jax) | (Gray et al., 2008) |
| YAC128 | Transgenic: full-length genomic construct under control of human HD promoter |
YAC128 | 100 and 126 CAGs | FVB-Tg(YAC128)53Hay/J (Jax Stock No. 004938) |
FVB/NCrl (CRL)b | (Slow et al., 2003b) |
| YAC128B | 100 and 126 CAGs | Not commercially available | C57BL/6J (Jax) | (Van Raamsdonk et al., 2007) |
Note. Jax: Jackson Laboratories. CRL: Charles River Laboratories. NA=Not available, new line.
CAG repeat length correspond to the cohorts used in these studies.
Note that original background strain and current supplier differ, thus controls should be obtained from original vendor or backcrossing should be ensured.
R6/2
Heterozygous and wild type (WT) mice were generated crossing ovarian-transplanted females (from R6/2 CBAB6 female donors) with CBAB6F1 WT males. R6/2 congenic mice in C57BL/6J background (R6/2B) were generated by crossing R6/2B male mice with C57BL/6J females.
HdhQ111
Homozygous, heterozygous and WT mice were generated crossing heterozygous HdhQ111 on a CD1 background.
YAC128
Mutant and WT mice were generated crossing mutant male mice YAC128 to WT females in two different backgrounds (FVB/NCrl and C57BL/6J).
BACHD
Mutant and WT mice were generated crossing mutant BACHD males with WT FVB/NJ females.
Husbandry
Mice were group-housed (4–5/cage) in shoe-box cages with wood shavings and a filter top. The environment was enriched with a play tunnel, shredded paper, and a plastic bone. Breeder animals also received igloos, instead of play tunnels, and cotton nestlets. Food and water were available ad libitum. R6/2 and corresponding WT mice also received wet powdered food placed inside a cup on the floor of the cage; this additional food was replaced fresh daily and was provided starting at the time of weaning. Temperature (68–76 °C), humidity (30–70%) and the light–dark cycle (6:00–18:00 EST) were controlled and monitored daily.
Genotyping
Genotype was determined at 15 days of age by PCR of tail snips (Morton et al., 2000). CAG repeat lengths were measured by Laragen (USA). All CAG repeat numbers reported here are those determined directly by Genemapper software.
R6/2, R6/2B, BAC103, YAC128 and YAC128B genotyping protocol
Sequencing was performed with an ABI Prism 7700 Sequence Detector (Applied Biosystems) Taqman Master Mix (Taqman Universal PCR Master Mix – Part Number 4326708, Applied Biosystems). Mouse tail DNA was brought to room temperature, while the primer and probe sets were thawed and vortexed. For each reaction a master mix was prepared with R62for (GGGACTGCCGTGCCG, 1 μl), R62rev (GGCCTTCATCAGCTTTTCCAG, 1 μl), R62MGBprobe (6FM-CGGGAGACCGCCATG, 0.25 μl), BetaActinMGBFor CCGGGACCTGACAGACTACCT, 1 μl), BetaActinMGBRev (TGGTGGTGAAGCTGTAGCCA, 1 μl), BetaActinMGBProbe (VIC-ATGAAGATCCTGACCGAGC, 0.25 μl) and double distilled water (6.0 μl). The Taqman Master Mix (23 μl) and 2 μl of extracted mouse tail DNA (ca 80 ng) were added to each well of 96-Well Optical Reaction Plate. The plate was vortexed and centrifuged to spin down the reaction mix. Both FAM and VIC layers in the sample setup of sequence detector were selected. The following PCR parameters were used: 50 °C for 2 min, 95 °C for 10 min, and 40 cycles of 95 °C 15 s and 60 °C for 1 min. Both FAM and VIC thresholds were set at 0.05. The sample was considered positive for the transgenic gene if the Fam Ct0 was less than 30.
HdhQ111 genotyping protocol
The Taqman Master Mix for each reaction contained Q111MTMGBForPrimer (CTCACTTGGGTCTTCCCTTGTC, 1 μl), Q111MTMGBRevPrimer CAGAGACCCCGCAAGACG, 1 μ l), Q111MTMGBprobe (NED-CTGTCCTGAATTCGATG-MGBNFQ, 0.25 μl), Q111WTMGBForPrimer (CATGGCCAACACTGTTCCCT, 1 μl), Q111WTMGBRevPrimer (GGGAAAGCCTGGCCTCAG, 1 μl), Q111WTMGBProbe (FAM-TACCGCGACCCTCTGG-MGBNFQ, 0.25 μl), BetaActinMGBFor (2 μl), BetaActinMGBRev (2 μl), BetaActinMGBProbe (0.25 μl), DNA (2 μl), and double distilled water (1.75 μl). The NED and Vic thresholds were set at 0.05 and 0.025, respectively. All other experimental details followed the R6/2 protocol. The sample was considered homozygous mutant if the Fam Ct0 was larger than 34 and NED Ct0 was smaller than 28. The sample was considered WT if the Fam Ct0 was less than 28 and NED Ct0 was larger than 34. The sample was considered heterozygous mutant if the Fam Ct0 was less than 28 and NED Ct0 was larger than 28.
Body weight
Mice were weighed daily during testing weeks and once a week during non-testing weeks.
Survival
Mice were examined daily to determine if they were still alive. If mice were found moribund, unable to stand up, they were sacrificed by CO2 exposure.
Behavioral evaluation
Experimenters were blind to the genotype during all testing, at least until the appearance of a robust behavioral phenotype in the mutants (for the R6/2 and R6/2B line in particular). A longitudinal design was used to evaluate the phenotype of the different lines at different ages (Table 2). The same mice were followed for all tests, unless noted.
Table 2.
Testing age for all lines.
| Age (weeks) Line |
4 | 6 | 8 | 12 | 14 | 16 | 24–28 | 36 | 48 | 52–56 | 100 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R6/2 | ||||||||||||
| R6/2B | ||||||||||||
| HdhQ111 | Cohort 1 | |||||||||||
| Cohort 2 | ||||||||||||
| BACHD | ||||||||||||
| YAC128 | Cohort 1 | |||||||||||
| Cohort 2 | ||||||||||||
| YAC128B |
Mice were tested longitudinally design using a behavioral testing battery consisting of: rotarod, grip strength, open field during both light phases of the diurnal cycle, rearing–climbing test, paw-print analysis, dark-light choice and prepulse inhibition of startle. In HdhQ111 and YAC128 mice prepulse inhibition of startle and body weight was evaluated in an independent group also using a longitudinal design (Table 2, cohorts 2). Mice were transported from the colony in their home cages to the behavioral testing room and allowed to acclimate for at least 1 h prior to the experiment. Mice were tested no earlier than 1 h after lights on and up to 1 h before lights off for the light phase testing and after 15 to 30 min after lights off, for dark phase testing.
Rotarod (RR; AccuScan, OH)
Mice were tested over 3 consecutive days. Daily sessions included a 5-min training trial at 4 RPM. At least 1 h later, mice were tested in three 5 min-trials with an accelerating speed (from 0 to 40 RPM in 5 min) separated by a 30-min inter-trial interval. The latency to fall from the rod was recorded. Mice remaining on the rod for more than 300 s were removed and their time scored as 300 s. Data from the training trial is not included.
Open field (OF)
This test was performed both during the dark and light phases of the light cycle. Mice were placed in the center of activity chambers equipped with infrared beams (Med Associates, St. Albans, VT; 27×27×20.3 cm) and then covered by an acrylic transparent lid. Activity was recorded for 30 min under normal conditions of lighting (~300–500 lux), in the light phase of the diurnal cycle, or under red light conditions in the dark phase of the light cycle. Quantitative analysis was done on distance traveled (center and total) and rearing rate in the center.
Grip strength (GS)
Mice were held by the tail and placed on the apparatus (Ugo Basile, Italy) so that they grabbed the handle with both front paws and then gentle pulled back until they released it. Each session consisted of 5 trials. Data for the 12 weeks R6/2B test was lost due to computer malfunction, and it is not reported.
Rearing–climbing (RC) (Hickey et al., 2005)
A metal wire mesh pencil holder with circular mesh at the bottom was placed upside down over the mice for 5 min and videotaped for later analysis. Quantitative analysis was done on latency to climb.
Gait analysis
Hindpaws of the animals were painted with different colors of nontoxic paint, and the animals are allowed to walk across an 84-cm uncovered corridor (L×W×H: 84×5×5 cm) lined with paper. Measurements of stride length (the distance between one step to the next, for the same hindpaw), base length (the orthogonal distance between the two hindpaws), and splay (the diagonal distance between contralateral hindpaws as the animal walks) were determined. Measurements were made for three continuous strides, right and left, and averaged for each animal.
Light–dark choice (LD)
Black plastic inserts were used to separate a dark and a well-lit area (~900 lux) in infrared-beam-equipped chambers, covered with a transparent lid (Med Associates, St. Albans, VT). Mice were placed in the center of the arena, facing the dark area entrance. Mice that failed to move for 3 min or more in the light area (freezing) were not included in the analysis. Freezing behavior was observed only in the YAC128 line and increased with age. About 20% of the mutants and almost 70% of the WT mice showed freezing at 110 weeks of age. Sessions lasted 15 min but only the first 6 min were used to calculate the initial preference for the dark zone (% time in the dark). For the 12 weeks time point for the R6/2 line, zone preference was based on the first 5 min of the session as detailed min by min data was not available due to computer malfunction.
Prepulse Inhibition of Startle (PPI)
Mice were placed in plastic holders and in the startle chamber for a 10-min habituation period. Background noise was 70 dB. Each session consisted of 56 trials. Each trial consisted of a 50-ms null period, followed by a 20-ms prepulse period during which a prepulse white noise sound of 72, 74, or 78 dB (white noise) could be presented. After a 100-ms delay, the startle stimulus was presented (40-ms, 120-dB white noise). The total duration of the trial was 500 ms. Eight types of trials were given: prepulse-plus startle (10 trials per prepulse intensity), prepulse alone (4 trials per prepulse intensity), startle alone (10 trials), and no stimulation (4 trials). In the startle-alone trials, the basic auditory startle was measured, and, in the prepulse-plus-startle trials, the amount of inhibition of normal startle was measured and was expressed as a percentage of the basic startle. To ensure that startle responses were within physiologically possible ranges a set of strict criteria was used. Trials were excluded in which the time to the startle peak was less than 10 ms or longer than 38 ms, the latency to startle was less than 5 ms, and the minimum peak amplitude was less than 50. Mice with a mean startle under 100 units were also excluded from the analysis.
Data analysis
An alpha level of 0.05 was selected for all inferential statistics. The repeated measures analysis was carried out with SAS (SAS Institute Inc.) using Mixed Effect Models. This approach is based on likelihood estimation which is more robust to missing values than moment estimation. The models were fitted using the procedure PROC MIXED (Singer, 1998). For this study, genotype, gender, age and their interactions were considered in all the models. Prepulse level was used as a factor for the prepulse inhibition of startle data. Slight variations of those models were tested and the best model based on the AIC/BIC criteria was selected.
Significant genotype×age or genotype×gender×age interactions were followed up with simple main effects to find at which age the genotype differences reached significance. For the HdhQ111 line, significant simple main effects were followed by Fisher LSD for the comparisons of WT versus heterozygous and WT versus homozygous mice.
For the rearing climbing test, the latency to climb was categorized in 4 different bins: less than 75 s, between 75 and 150 s, between 150 and 300 s, and not climbing. Differences between genotypes were analyzed with a Chi Square at each age separately. Gender differences were not apparent so the data was pooled across genders. Survival data were analyzed with Kaplan–Meier analysis (with the p values derived from the Mantel–Cox Log-rank statistic).
All experiments were carried out in accordance with the US Public Health Service Guide for Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the New York Medical College (Valhalla, NY).
Results and discussion
All models show deficits in the rotarod test of motor skill learning and coordination but the onset of deficits and the severity differ amongst the lines
To assess motor coordination we tested mice in the rotarod test. Starting at 4 weeks of age R6/2 mice showed a reduced latency to fall compared to the WT controls (Fig. 1A). The differences became more pronounced with age (genotype: F(1,44)=328.1, p<0.0001; age: F(4,161)=42.5, p<0.0001; genotype×age: F(4,161)=19.3, p<0.0001; simple main effects: ps<0.0008).
Fig. 1.

Average latency to fall (±SEM) from the rotarod as a function of genotype, gender and age. Data points show latency averaged over 3 daily trials and 3 daily sessions. A: R6/2; B: R6/2B; C: HdhQ111; D: BACHD; E: YAC128 and F: YAC128B.
The R6/2B mice showed a progressive impairment, falling significantly faster than WT mice starting at 8 weeks of age (genotype: F(1,44)=24.7, p<0.0001; age: F(4,176)=100.0, p<0.0001; genotype×age interaction: F(4,176)=70.0, p<0.0001; simple main effects: ps<0.04; Fig. 1B). Gender did not show any significant main effect or interactions.
Homozygous male mutant HdhQ111 mice performed better than their WT controls at early ages, and homozygous females slightly worse at the oldest age (genotype: F(2,62)=9.3, p<0.0003; age: F(6,331)=28.6, p<0.0001; genotype×age: F(12,331)=3.0, p<0.0007; genotype×age×gender: F(18,331)=2.1, p<0.006; simple main effects: ps<0.05; Fisher LSD: ps<0.05; Fig. 1C).
BACHD mice (Fig. 1D) performance was worse than that of the WT controls at 4 weeks of age and progressively worsened as they aged, independently of gender (genotype: F(1,42)=176.9, p<0.0001; age: F(5,202)=19.5, p<0.0001; genotype×age: F(5,202)=7.9, p<0.0001; simple main effects: ps<0.0004).
YAC128 mice performance was worse than that of the WT controls although this did not progress with age (genotype: F(1,44)=42.4, p<0.0001; age: F(7,265)=26.5, p<0.0001; Fig. 1E). There was no significant effect or interaction with gender.
YAC128B mice performance was also generally worse than that of the WT controls, with the genotype effect being slightly larger and more consistent in females. As with the YAC128 mice, the deficits in the YAC128B mice did not progress with age. Females were significantly worse starting at 34 weeks, whereas males were worse than WT males only at 16 weeks of age (genotype: F(1,43)=11.8, p<0.002; age: F(6,258)=84.7, p<0.0001; genotype×age: F(6,258)=3.8, p<0.0002; age×genotype×gender: F(12,258)=2.8, p<0.002; simple main effects: p<0.03; Fig. 1F).
Summary and discussion of rotarod results
The R6/2 line in a mixed background showed progressive, robust deficits, consistent with published results (Carter et al., 1999). We have extended these findings to the R6/2B on a pure C57BL/6J background with 240 CAGs. The deficits in this new line were also robust, although delayed by 2 weeks compared to the R6/2 mice on the mixed background. The full-length models showed significant deficits in the rotarod, although the pattern was different than that seen in the two R6/2 lines. Whereas the knock-in model HdhQ111 showed impairment only at very late ages and only in females, the transgenic YAC128 lines in both backgrounds showed an early deficit that did not seem to deteriorate with age. This is not dissimilar to published results that show fixed, non-progressive deficits after 6 months of age in the YAC128 (Slow et al., 2003). The observed effects in the BACHD mice found are consistent with published findings showing a strong progressive phenotype in the BACHD mice starting at 2 months of age (Gray et al., 2008). Our results showed an earlier phenotype.
Both R6/2 lines and the BACHD line are hypoactive in the open field test in both light phases, whereas YAC128 lines show mild hypoactivity during the dark phase and HdhQ111 knock-in mice do not show this deficit
To assess motor function, reaction to a novel environment, general activity and exploration we tested mice in the open field test during the light and dark phases of the light cycle. R6/2 transgenic mice were in general hypoactive, as measured by the total distance covered, in both phases of the light cycle. In the light phase, although there was no significant main effect of gender, female R6/2 were only significantly different from WT mice at 8 and 14 weeks of age whereas males were significantly less active than WT males starting at 6 weeks of age (genotype: F(1,44)=19.3, p<0.0001; age: F(4,158)=21.2, p<0.0001; genotype×age: F(4,158)=6.7, p<0.0001; genotype×age×gender: F(8,158)=5.0; p<0.0001; simple main effects: ps<0.03; not shown). In the dark phase R6/2 mice, both female and males, were significantly hypoactive from 12 weeks of age (genotype: F(1,44)=32.7, p<0.0001; age: F(4,161)=9.6, p<0.0001; genotype×age: F(4,161)=9.7, p<. 0001; simple main effects: ps<0.0001; Fig. 2A).
Fig. 2.

Average total distance traveled (±SEM) as a function of age, gender, and genotype during the dark phase of the light cycle in the open field. A: R6/2; B: R6/2B; C: HdhQ111;D: BACHD; E: YAC128 and F: YAC128B.
R6/2 mice showed progressive hypoactivity in the center of the open field starting at 4 weeks in both light phases (light phase, genotype: F(1,44)=49.2, p<0.0001; age: F(4,158)=5.0, p<0.0001; genotype×age: F(4,158)=3.0, p<0.02; dark phase, genotype: F(1,44)=61.1, p<0.0001; age: F(4,161)=3.6, p<0.008; genotype×age: F(4,161)=5.2, p<0.0006; simple main effects: ps<0.002; not shown).
R6/2 mice also reared less in the center than WT mice at all ages in the light phase, and starting at 6 weeks of age in the dark phase (light phase, genotype: F(1,44)=34.7, p<0.0001; dark phase, genotype: F(1,44)=35.7, p<0.0001; age: F(4,161)=3.2, p<0.01; genotype×age: F(4,161)=5.0, p<0.0008; simple main effects: ps<0.02; Fig. 3A for dark phase results).
Fig. 3.

Mean rearing rate in the center of the open field as a function of age, gender, and genotype during the dark phase of the light cycle. A: R6/2; B: R6/2B; C: HdhQ111; D: BACHD; E: YAC128 and F: YAC128B.
R6/2B mice covered less distance in total than WT mice in the light phase with deficits starting at 6 weeks of age in females and 8 weeks of age in males (genotype: F(1,44)=38.7, p<0.0001; age: F(4,175)=134.0, p<0.0001; genotype×age: F(4,175)=37.3, p<0.0001; genotype×age×gender: F(8,175)=2.7, p<0.008; simple main effects: ps<0.0001; Fig. 2B). In the dark, the R6/2B hypoactivity was significant at 6 weeks of age in both genders (genotype: F(1,44)=46.7, p<0.0001; age: F(4,176)=98.0, p<0.0001; genotype×age: F(4,176)=10.2, p<0.0001; simple main effects: ps<0.001).
In the light and dark phases, R6/2B mice covered significantly less distance in the center starting at 12 and 6 weeks of age, respectively (light phase, genotype: F(1,44)=12.6, p<0.0009; age: F(4,175)=63.6, p<0.0001; genotype×age: F(4,175)=6.0, p<0.0001; simple main effects: ps<0.0001; dark phase, genotype: F(1,44)=46.7, p<0.0001; age: F(4,176)=69.5, p<0.0001; genotype×age: F(4,176)=8.1, p<0.0001; simple main effects: ps<0.006).
R6/2B mice reared less in the light but not in the dark phase, starting at 12 weeks of age (genotype: F(1,44)=1.9, p<0.0001; age: F(4,158)=21.2, p<0.0001; genotype×age: F(4,158)=6.7; genotype×gender×age: F(8,158)=5.0; p<0.0001; simple main effects: ps<0.02).
For the HdhQ111 line there were no consistent genotype effects. All groups showed less activity as they aged in both total and center distances, as well in rearing (age: Fs(6,330)>2.4, ps<0.03; Figs. 2C and 3C). There were a few minor interactions between gender and genotype at different ages that seem to result from non consistent differences found over the 2 years of testing and do not seem to reflect a general effect of genotype. For total distance, for example, there was a genotype effect in the females at 24 and 100 weeks of age, but it was not due to the homozygous knock-in mice being more or less active than the WT or heterozygous mice (genotype×gender×age: F(18,330)=6.5, p<0.0001; simple main effect, p<0.05; Fig. 2C).
BACHD mice were hypoactive as measured by the total distance in the light and dark phases, and distance in the center in the light, starting at 28 weeks of age for both genders (genotype in the light phase: Fs>7.7, ps<0.009; age: Fs>3.4, ps<0.006; genotype×age for total distance: Fs>8.7, ps<.0001; genotype×age×gender: Fs>2.0, ps<0.03; simple main effects: ps<0.03; Figs. 2D and 3D for dark phase, light phase not shown). The genotype effects, however, did not reach significance for distance in the center, in the dark phase.
BACHD mice reared in the center less frequently than WT mice both during the light and dark part of the light cycle at all ages, an effect that did not increase with age or depended on gender (Fig. 3D, genotype: Fs>12.1, ps<0.007).
During the light phase, YAC128 mutant mice behave like their WT controls (Figs. 2E and 3E). There were no genotype effects in total distance traveled, apart from some inconsistent fluctuations in activity as age progressed. Female mutant, for example, were significantly hyperactive compared to female WT at 52 weeks of age only (age: F(7,252):20.8, p<0.0001; genotype×age: F(7,252)=5.9, p<0.0001; genotype×age×gender: F(14,252)=3.2, p<0.0001, simple main effects: p<0.002). During the dark phase, YAC128 mice were hypoactive at older ages, with females showing a significant decrease in locomotion at ages 36 and 52 weeks, and males at 16 and older ages, although not at 100 weeks of age (genotype: F(1,43)=29.7, p<0.0001; age: F(7,259):14.1, p<0.0001; genotype×age: F(7,259)=6.9, p<0.0001; genotype×gender: F(1,43)=5.2, p<0.03; genotype×age×gender: F(14,259)=1.8, p<0.05, simple main effects: p<0.03).
For the YAC128 line, during the light phase, there was no significant genotype effect in distance covered in the center, apart from a general decrease in activity with age (age: F(7,251)=6.3, p<0.0001; not shown). In the dark phase, however, mutants covered more distance in the center (genotype: F(1,43)=6.3, p<0.02; gender: F(1,43)=6.3, p<0.02; not shown).
In the light phase mutant YAC128 mice reared more at 36 and 52 weeks (age: F(7,250):3.5, p<0.002; gender: F(1.43)=10.8, p<0.0002; genotype×age: F(7,250)=42.1, p<0.05; simple main effects: p<0.03). In the dark phase male YAC128 mutant mice also reared more but only at 52 weeks of age (age: F(7,260):6.0, p<0.0001; gender: F(1.43)=9.0, p<0.005; genotype×age: F(7,260)=2.1, p<0.05; genotype×age×gender: F(14,260)=2.0, p<0.03, simple main effects: p<0.002; Fig. 3E).
In the YAC128B line the transgenic mice were hypoactive in the open field, as measured by the total distance covered, with genotype differences reaching significance at 12 weeks in the light phase, and, at 8, 16 and 34 weeks in the dark phase (light phase, age: F(6,253):88.9, p<0.0001; gender: F(1.43)=5.5, p<0.03; genotype×age: F(6,253)=3.4, p<0.003; simple main effects: p<0.05; Dark phase, genotype: F(1,43)=5.8, p<0.02; age: F(6,252):98.4, p<0.0001; genotype×age: F(6,252)=5.8, p<0.0001; simple main effects: p<0.02; Fig. 2F for dark phase results). Whereas locomotion in the center showed no effects due to genotype in the light phase, in the dark phase it revealed hypoactivity of the mutants at 16 and 34 weeks of age (light phase, age: F(6,253):47.0, p<0.0001; Dark phase, genotype: F(1,43)=6.4, p<0.02; age: F(6,252)=30.9, p<0.0001; genotype×age: F(6,252)=3.2, p<0.005; simple main effects: ps<0.003). Rearing decreased with age but was similar for both genotypes, in both light phases (Fs>4.9, ps<0.0001; Fig. 3F for dark phase results).
Summary and discussion of the open field test
In the R6/2 lines, the open field test revealed robust hypoactivity expressed as reduced locomotion and rearing. The mixed background with 122–135 CAGs showed early deficits, as published before (Carter et al., 1999). The R6/2 line with and 243–264 CAG repeats in the C57BL/6J background also showed robust yet slightly later deficits, starting at 6 weeks. In the full-length models we could not detect any consistent difference between the homozygous and heterozygous HdhQ111 and the WT control in any of the measures taken. In a different cohort of HdhQ111 mice (data not shown) we detected hyperactivity at 4 weeks of age, using a cross-sectional design, consistently with other results in the knock-in mice with 140 CAG repeats suggesting an early phase of hyperkinesis (Menalled et al., 2002). Rearing and locomotion seemed to be consistently decreased in the BACHD mice in this and in a previous study (Gray et al., 2008). In contrast, the YAC128 showed hyperactivity during the light, and hypoactivity during the dark phase, paralleling aspects of published data (Slow et al., 2003). The differences during the light phase, however, could be attributed to the WT freezing behavior that developed at older ages. The lack of freezing behavior in the mutant YAC128 makes the hyperlocomotion during the light phase difficult to interpret. During the dark phase, however, WT mice did not freeze and thus the hypoactivity of the YAC128 was evident. This points to a difference between the two FVB/N strains, with the FVB/NCrl WT from Charles River (corresponding to the YAC128 line), but not the FVB/NJ WT from the Jackson Laboratories (corresponding to the BACHD mice), showing significant freezing behavior in a well-lit open field during the light phase. These differences in overt freezing behavior of the WT FVB/n mice need be taken into account when interpreting the open field behaviors of the transgenic mice in the light. YAC128B (on a C57BL/6J background) in contrast, showed mild hypolocomotion in both light phases, suggesting that this genetic background might be more suitable for assessment of activity and exploration.
N-terminal fragment R6/2 transgenic but not full-length models show deficits in the grip strength test of muscle tone
To assess muscle strength we tested mice in the grip strength test. The R6/2 mice showed a deterioration of their grip strength as they aged, an effect more robust in males, compared to their WT littermates with differences being significant at 12 and 14 weeks (genotype×gender: F(1,44)=4.4, p<0.05; genotype×age: F(3,118)=106.5, p<0.0001; simple main effects: p<0.0001; Fig. 8A). A separate cohort of mice showed a significant difference at 10 weeks. The R6/2B line showed a similar pattern, with a significant difference at 14 weeks, which was more pronounced in the males (genotype×age: F(3,132)=9.9, p<0.0001; genotype×age×gender interaction: F(6,132)=2.2, p<0.05; simple main effects for the genotype×age interaction: ps<0.0001).
Fig. 8.

Average startle magnitude (±SEM.) as a function of age, genotype, and gender. Data points are means±SEM. A: R6/2; B: R6/2B; C: HdhQ111; D: BACHD; E: YAC128 and F: YAC128B.
None of the other four lines (HdhQ111, YAC128, YAC128B and BACHD) showed any genotype or gender effects, although there is an overall effect of age (ps<0.0001), with weaker grip strength at both the youngest and oldest ages for all mouse lines. (Fig. 4).
Fig. 4.

Average grip strength (±SEM) as a function of age and genotype. A: R6/2; B: R6/2B; C: HdhQ111; D: BACHD; E: YAC128 and F: YAC128B.
Summary and discussion of the grip strength test
The grip strength data show a robust deficit in both R6/2 models, although the line with 240 CAG repeats in the C57BL/6J background shows the deficit at a later age. Interestingly, males showed a more robust deficit than females. This test differentiated between the R6/2 lines and the full-length HD transgenic or knock-in models, for which we did not find any significant difference.
The rearing–climbing test showed less vertical activity in R6/2 and BACHD mice, with minor or no deficits in YAC128, YAC128B, R6/2B and HdhQ111 lines
R6/2 mice showed a pronounced deficiency in this test, failing to climb or climbing later than the WT controls starting at age 4 weeks (χ2 test, p<0.031). As Fig. 5A shows, the deficit worsened with age as more and more mice failed to climb during the test. Gender effects were not apparent.
Fig. 5.

Average latency to climb (±SEM) in the rearing–climbing test as a function of age, genotype and gender. Each panel shows the percent of mice that climbed in less than 75 s (black), between 75 and 150 s (dark grey), between 150 and 300 s (light grey), or did not climb (white). A: R6/2; B: R6/2B; C: HdhQ111; D: BACHD; E: YAC128 and F: YAC128B.
Surprisingly, in the R6/2B line there were no significant differences seen in climbing. This was probably because the C57B/6 WT mice did not show the typical rearing activity seen in this test (Fig. 5B; χ2 test, ps>0.2).
HdhQ111 heterozygous and homozygous mice did not show any significant difference at any age (χ2 test, ps>0.05) nor were gender effects apparent.
BACHD transgenic mice showed a consistent difference at all ages tested, with a much higher proportion of transgenic mice not climbing at all during the test period (white area, Fig. 5D; χ2 test, ps<0.05). Again, there were no apparent differences between genders.
YAC128 mice climbed less and showed longer latencies only at age 16 weeks (χ2 test, p<0.001). Gender effects were not consistent across ages (Fig. 5E).
In YAC128B (Fig. 5F), instead, there were no significant differences at any age (χ2 test, ps>0.05). As observed with the R6/2B mice, the WT C57BL/6J control mice did not show typical rearing behavior therefore, climbing deficits could not be measured in the YAC128B mice.
Summary and discussion of the rearing–climbing test
Background strain affected the performance in the rearing climbing test: the R6/2 transgenic mice, with both low CAG (R6/2) and higher (R6/2B) CAG repeat lengths show little or no climbing, whereas the WT of the mixed strain showed much more frequent climbing behavior as compared to the WT of the pure C57BL/6J strain, making this particular outcome measure a more robust measure only in the mixed background. This result replicates and extends that published by others using the R6/2 model (Hickey et al., 2005). This difference in the genetic background changed the statistical power of this particular test. From the four full-length models the BACHD model was the only one to show a consistent deficit in this test.
The paw-print test showed robust gait anomalies in R6/2 mice, with minor or no deficits in the other five lines
To assess motor function and gait we tested mice in the paw-print test. Gait can be assessed measuring the distance covered by a step (stride length, if ipsilateral or splay, if contralateral) and the separation between the legs while walking (base), among other measures. The analysis of the paw prints revealed shorter stride length (Fig. 6A) in R6/2 mice as compared to their WT controls starting at 8 weeks of age, differences in gait being larger for females (genotype: F(1,43)=43.02, p<0.0001; age: F(4,154)=15.8, p<0.0001; genotype×age: F(4,154)=14.0, p<0.0001; genotype×gender: F(1,43)=5.1, p<0.03; simple main effects for the genotype×age interaction: ps<0.006).
Fig. 6.

Average hindlimb stride length (±SEM). A: R6/2; B: R6/2B; C: HdhQ111; D: BACHD; E: YAC128 and F: YAC128B.
R6/2 mice splay was shorter at 6, 12 and 14 weeks of age, and base was significantly wider at 8 and 12 weeks of age in females and at 14 weeks in males (splay, genotype: (1,43)=16.0, p<0.0002; age: F(4,155)=14.6, p<0.0001; genotype×age: F(4,155)=18.2, p<0.0001; simple main effects: ps<0.02; base, genotype: (1,43)=8.7, p<0.005; genotype×age: F(4,155)=2.8, p<0.03; genotype×age×gender: F(8,155)=2.9, p<0.005; simple main effects: ps<0.02).
For the R6/2B line there were no differences in stride length, although splay was significantly shorter at 14 weeks of age and base was significantly narrower for the mutants at all ages (Fig. 6B; splay, genotype: F(1,44)=4.4, p<0.05; age: F(4,172)=60.6, p<0.0001; gender: F(1,44)=8.4, p<0.006; genotype×age: F(4,172)=2.9, p<0.03; simple main effects: ps<0.000; base, genotype: F(1,44)=7.2, p<0.01; age: F(4,172)=16.7, p<0.0001).
For the HdhQ111 mutant mice there were no effects or interactions of genotype with any other factor (Fig. 6C).
BACHD mice presented a shorter stride length and splay at 12 weeks, shorter splay at 12 and longer at 36 weeks, and wider base at 36 weeks (Fig. 6D; stride length, age: F(5,189)=17.3, p<0.0001; genotype×age: F(5,189)=3.1, p<0.02. Splay, age: F(5,189)=31.5, p<0.0001; genotype×age: F(5,189)=5.0, p<0.0003; base, age: F(5,189)=13.1, p<0.0001; gender: F(1,42)=18.9, p<0.0001; genotype×age: F(5,189)=3.1, p<0.02; simple main effects for the three measures: ps<0.0006).
For the YAC128 line there were no effects of genotype in any of the three measures.
Finally, the YAC128B mutants had shorter stride length and splay, and narrower base but only at 16 weeks of age (age: Fs(6,255)>28.5, ps<0.0001; genotype×age: Fs(6,255)>2.2, ps<0.05; simple main effects: ps<0.03).
Summary and discussion of the paw-print test
The gait analysis showed a robust deficit in the R6/2 line in the three measures of gait. The R6/2B line instead did not develop a deficit at the ages tested. From the full-length models only the BACHD showed statistically significant deficits, although at 36 weeks the differences – longer splay and wider base – differ from the deficit expected – shorter steps – based on the HD literature (Koller and Trimble, 1985).
The light–dark choice test showed anxiety-like behavior in the R6/2 lines and BACHD, with little or no differences in the YAC128, YAC128B or HdhQ111 line
To assess emotional reactivity to a novel anxiogenic environment, general activity and exploration we tested mice in the dark-light choice test. R6/2 mutant mice showed a preference for the dark area at 12 and 14 weeks of age that was significantly higher than the WT control mice irrespective of gender (genotype: F(1,43)=13.7, p<0.0006; age: F(4,153)=20.7, p<0.0001; genotype×age interaction: F(4,153)=6.9, p<0.0001; simple main effects: ps<0.02; Fig. 7A).
Fig. 7.

Percent time in the dark (±SEM) as a function of genotype, age and gender. The dotted line shows indifference. A: R6/2; B: R6/2B; C: HdhQ111; D: BACHD; E: YAC128 and F: YAC128B.
R6/2B mice showed a higher preference for the dark at 14 weeks (genotype×age interaction: F(4,174)=3.7, p<0.006; simple main effect: p<0.0004; Fig. 7B).
For the HdhQ111 (Fig. 7C) there was a brisk decrease in preference for the dark for all groups at the last age tested. Although there was a significant triple interaction with genotype, age and gender, a follow up simple mains effects analysis did not show any significant difference (age: F(6,329)=12.2, p<0.001; genotype×age×gender: F(18,329)=2.0, p<0.01).
BACHD mice showed a significantly greater preference for the dark starting at 12 weeks of age, with males preferring the dark at 4 weeks (genotype×age×gender: F(10,203)=2.6, p<0.006; simple main effects: ps<0.03; Fig. 7D; gender effects not shown).
In YAC128 there was a triple interaction between genotype, gender and age due to a significant increased preference for the dark at 52 weeks in male YAC128 mice (genotype×age×gender: F(14,229)=1.9, p<0.03; simple main effect: p<0.01; Figs. 7E).
For the YACB128 mutant lines (Fig. 7F) there were no significant genotype effects or interactions.
Summary and discussion of the light/dark choice test
Both R6/2 and R6/2B lines showed a progressive anxiety-like phenotype in this test. BACHD showed the strongest differences from all the lines with YAC128 lines and HdhQ111 showing little or no differences between genotypes.
The HdhQ111 heterozygous, homozygous and WT controls showed almost no preference at the oldest age tested, probably reflecting blindness in the CD1 albino background. All FVB mice (the background strain for both BACHD and YAC128) are homozygous for the Pde6brd1 (rd1) allele. The CBA/J parental line used for breeding for the R6/2 line by Jackson Laboratories in the US is also homozygous for the rd1 allele resulting in about 25% homozygocity levels for both WT and R6/2 mice on the mixed background. Mice homozygous for rd1 have little visual acuity after weaning, although enough sensitivity to discriminate between dark and lit areas (Kopp et al., 1999) but no vision altogether beyond 1 year of age (Bowes et al., 1990). The observed preference for the dark in all these lines suggests that mice were sensitive to the lighting levels and the differences between genotypes are probably due to anxiety-like behavior. Note that R6/2 mice bred in the UK did not carry the rd1 allele (Carter et al., 1999; Lione et al., 1999; Morton et al., 2005).
The FVB/NJ WT mice from Jackson Lab. (from the BACHD line) but not the FVB/NCrl WT mice from Charles Rivers Lab. (from the YAC128 line) showed indifference between the dark and light zones (Figs. 7D and E, respectively). The more robust preference of the BACHD mice compared to the YAC128 mice for the dark could be, therefore, a result of baseline differences in the corresponding WT mice.
The prepulse inhibition of startle test shows sensory-gating deficits in the R6/2 models, BACHD and YAC128B but not YAC128 and HdhQ111 models
To assess reactivity to a startling noise and sensory-motor gating, we tested mice in the prepulse inhibition of startle test. R6/2 mice startle less than WT mice at 4 weeks and older ages (genotype: F(1,44)=37.0, p<0.0001; age: F(4,160)=29.8, p<0.0001; genotype×age: F(4,160)=12.3, p<0.0001; simple main effects ps<0.03; Fig. 8A). Despite the decrease in startle, prepulse inhibition of startle (PPI) was normal up to 8 weeks of age, and deficient thereafter (age: F(4,165)=12.7, p<0.0001; prepulse: (F(2,88)=6.8, p<0.002; genotype×age: F(4,165)=12.3, p<0.0001; simple main effects ps<0.0001; Fig. 9A).
Fig. 9.

Average percent inhibition of startle (±SEM) for three prepulse intensities as a function of age, genotype, and gender. A: R6/2; B: R6/2B; C: HdhQ111; D: BACHD; E: YAC128 and F: YAC128B.
The R6/2B transgenic mice showed a tendency to startle less, males in particular, but this difference only reached significance at 6 and 14 weeks of age, for both genders (genotype: F(1,44)=11.0, p<0.0001; age: F(4,171)=30.1, p<0.0001; gender: F(1,44)=28.8, p<0.02; genotype×age: F(4,171)=12.4, p<0.0001; genotype×gender: F(1,44)=4.2, p<0.05; simple main effects of the genotype×age interaction: ps<0.03; Fig. 8B). R6/2B mice showed a significant PPI deficit at 14 weeks, which was more pronounced in the females (genotype: F(1,44)=8.5, p<0.006; age: F(4,171)=10.9, p<0.0001; prepulse: F(2,88)=77.1, p<0.0001; genotype×age: F(4,171)=27.0, p<0.0001; genotype×gender×age: F(8,171)=2.0, p≤0.05; simple main effects: ps<0.0001; Fig. 9B).
For the HdhQ111 there were no effects of genotype or gender for either the startle or PPI measure (age: Fs>18.4, ps<0.0001; prepulse for PPI: F(2,132)=45.3, p<0.0001; Figs. 8C and 9C).
BACHD mutant mice showed a decreased startle at 24 and 36 weeks in females, and at 36 in male mutants (age: F(5,199)=27.2, p<0.0001; genotype×gender: F(1,42)=4.7, p<0.04; genotype×age×gender: F(10,199)=2.1, p<0.02; simple main effects of the genotype×age interaction: ps<0.04; Fig. 8D). BACHD mutant mice showed an increase in PPI with age, as did the controls, but showed deficits at 36 and 52 weeks in females and 28 and 52 in males (genotype: F(1,42)=6.14, p<0.02; age: F(5,199)=25.5, p<0.0001; prepulse: F(2,84)=146.3, p<0.0001; genotype×age: F(5,199)=5.6, p<0.0001; genotype×gender×age: F(10,199)=1.9, p<0.05; simple main effects: ps<0.04; Fig. 9D).
For the YAC128 there were no effects of genotype or gender for either startle or PPI (age: Fs>31.7, ps<0.0001; prepulse for PPI: F(2,88)=31.4, p<0.0001; Figs. 8E and 9E).
In the YAC128B line there were also no consistent effects of genotype on startle although there was significant variability across age and gender levels (age: F(5,207)=24.7, p<0.0001; gender: F(1,43)=14.7, p<0.004; Fig. 8E). There was, however, a significant deficit in PPI at the 56 weeks of age (age: F(5,207)=6.2, p<0.0001; prepulse: F(2,86)=17.2, p<0.0001; genotype×age: F(5,207)=3.1; p<0.009; simple main effects: p>0.0002; Fig. 9E).
Prepulse level increased PPI in all six lines without changing other patterns due to the above-discussed factors (prepulse level: Fs>6.8, ps<0.002).
Summary and discussion of the prepulse inhibition of startle test
The deficit in PPI seen here extends the results found in the R6/2 mice (Carter et al., 1999) to the C57B/6 background. The deficit in PPI was independent of a reduced startle seen at earlier ages in the two R6/2 lines. The separation between the baseline startle and the PPI deficits in the N-terminal fragment models is interesting as it suggests the deficits correlate with different neuropathologies, consistently with published studies (Paylor and Crawley, 1997). These deficits in PPI parallel findings with HD patients, who exhibit less PPI (Swerdlow et al., 1995). BACHD and YAC128B models also showed a decrease in PPI at older ages, accompanied by normal baseline startle. The other full-length models, YAC128 and HdhQ111, showed no consistent differences, although some reduction in the startle is seen at the oldest age in HdhQ111 mice.
Body weight decreased in the R6/2 models and HdhQ111 heterozygous knock-in mice but is increased in the other full-length models
To assess general health we measure body weight throughout the lifespan of all mice. R6/2 mice weighed considerably less than their WT controls at 7 weeks, 9 weeks and older for females, and at 7 weeks and older for males (genotype: F(1,44)=41.7, p<0.0001; age: F(9,396)=131.0, p<0.0001; gender: F(1,44)=224.9, p<0.0001; genotype×age: F(9,396)=15.09, p<0.0001; genotype×age×gender: F(18,396)=12.6, p<0.0001; simple main effects: ps<0.05; Fig. 10A).
Fig. 10.

Average body weight (±SEM) as a function of age and gender. A: R6/2; B: R6/2B; C: HdhQ111; D: BACHD; E: YAC128 and F: YAC128B.
The R6/2B mutant mice weighed less than their WT controls at 10 weeks and older for females, and at 9 weeks and older for males (genotype: F(1,43)=64.9, p<0.0001; age: F(10,406)=92.1, p<0.0001; gender: F(1,43)=68.5, p<0.0001; genotype×age: F(10,406)=24.0, p<0.0001; genotype×age×gender: F(20,406)=2.1, p<0.003; simple main effects: ps<0.008; Fig. 10B).
In the HdhQ111 mice male homozygous mutants showed significantly lower body weight than WT and heterozygous mice at 28 weeks and older ages (genotype: F(2,61)=4.2, p<0.0; age: F(10,406)=92.1, p<0.02; age: F(21,1129)=154.4, p<0.0001; gender: F(1,61)=43.3, p<0.0001; genotype×age×gender: F(63,1129)=1.9, p<0.0001; simple main effects: ps<0.05; Fisher LSD: p<0.03 except at 60 and 64 weeks of age, when there were no genotype differences; Fig. 10C). An independent cohort of mice confirmed and extended this finding to the females, as differences became significant at 76 weeks of age for both female and male homozygous mutants as compared to the WT mice (genotype: F(2,66)=4.2, p<0.02; age: F(22,1294)=139.4, p<0.0001; gender: F(1,66)=92.0, p<0.0001; genotype×gender×age: F(66,1294)=2.2, p<0.0001; simple main effects for the triple interaction: ps<0.04, Fisher LSD: ps<0.02; not shown).
The BACHD transgenic mice became heavier than the controls with age, starting at 12 weeks in the females and 16 weeks in the males (genotype: F(1,42)=51.6, p<0.0001; age: F(12,484)=123.7, p<0.0001; gender: F(1,42)=31.8, p<0.0001; genotype×age: F(12,484)=9.0, p<0.0001; genotype×gender: F(1,42)=7.1, p<0.01; genotype×age×gender: F(24,484)=4.3, p<0.0001; simple main effects: ps<0.006; Fig. 10D).
YAC128 mice were heavier than the WT controls, especially the males, which were heavier starting at 32 weeks of age. Females were not consistently heavier, showing a significant difference at weeks 48–76 but not before or after (genotype: F(1,44)=21.5, p<0.0001; age: F(24,839)=54.6, p<0.0001; gender: F(1,44)=20.8, p<0.0001; genotype×age: F(24,839)=2.2, p<0.001; genotype×age×gender: F(48,839)=2.6, p<0.0001; simple main effects: ps<0.05; Fig. 10E). An independent cohort of mice confirmed this pattern (genotype: F(1,44)=28.2, p<0.0001; age: F(13,542)=2.7, p<0.002; gender: F(1,44)=52.4, p<0.0001; genotype×age: F(13,542)=2.7, p<0.002; simple main effects: ps<0.04; not shown).
YAC128B mice were heavier than the WT controls, starting at 16 weeks of age in females and 8 weeks in males, although the difference failed to reach significance in males at 56 weeks of age (genotype: F(1,43)=35.0, p<0.0001; age: F(13,559)=303.9, p<0.0001; gender: F(1,43)=141.8, p<0.0001; genotype×age: F(13,559)=3.4, p<0.0001; genotype×age×gender: F(26,559)=5.1, p<0.0001; simple main effects: ps<0.03; Fig. 10F).
To explore the effect of increased body weight on rotarod performance we followed up with an analysis of a subset of mice matched for body weight. For the BACHD, a subset of male mice that showed no body-weight differences still shows a significant deficit in rotarod performance (genotype: F(1,23)=83.1, p<0.0001; Figs. 11A and D). We could not form a subset of female BACHD mice matched for body weight so we did not perform this analysis on this gender. For YAC128 mice a subset of mice was found that showed no significant increase in body weight at any age in females and only at 4 and from 36 to 44 weeks in males (Fig. 11B). In this subset, rotarod deficits were not significant in females at any age, and in males only at 16 weeks of age (body weight and rotarod: genotype×age×gender: Fs>2.2, ps<0.002; simple main effects: ps<0.05; Fig. 11E). For the YAC128B line a subset of mice was found with no significant increase in body weight at any age in females and only at 16 and 20 weeks in males (Fig. 11C). In this subset, rotarod deficits were significant in females and males at most ages tested (4, 12, 16, 34–56 weeks and 4, 16, 34 and 56 weeks, respectively; genotype: F(1,24)=16.0, p<0.0005; genotype×age: F(6,114)=4.5, p<0.0004; genotype×age×gender: F(12,144)=3.5, p<0.0001; simple main effects: ps<0.05; Fig. 11F).
Fig. 11.

Subgroups of mice matched for body weight, A: BACHD; B: YAC128 and C: YAC128B and their rotarod performance, D: BACHD; E: YAC128 and F: YAC128B.
Summary and discussion of body weight results
R6/2 mice failed to gain, and then lost weight starting at about 8 weeks of age, consistent with other published studies (Hickey et al., 2005; Mangiarini et al., 1996). The R6/2B mice also showed a consistent, although milder body-weight loss phenotype with changes starting around 9 weeks of age. The HdhQ111 mice also showed reduced body weight, whereas all transgenic full-length models (YAC128 lines and BACHD line) showed increased body weight from an early age, replicating previous findings in full-length mutant transgenic mice, an effect of the genetic manipulation that has been interpreted as the result of an increased level of the full-length huntingtin protein (Gray et al., 2008; Van Raamsdonk et al., 2006b; Van Raamsdonk et al., 2005). A follow up analysis suggested independence between body weight and rotarod performance in BACHD males, replicating the original findings in this model (Gray et al., 2008) and extending it to YAC128B mice, although deficits in the weight-matched groups were not progressive (Fig. 11F). In BACHD female mice this follow up analysis was inconclusive because we could not match the body weight of subsets of WT and transgenic mice. In YAC128 and corresponding WT mice matched for body weight we did not detect significant rotarod deficits, either because there is no independence and rotarod deficits are caused by increased body weight, or because the sample size of the subset was too small and thus statistical power was lacking.
Only the two R6/2 models show decreased survival
R6/2 mice died significantly earlier than their WT counterparts with a median survival of 113 days (Mantel–Cox: p<0.0001; Fig. 12A). R6/2B mice also died prematurely, especially the females, with a median survival of 141 and 179 days for males and females, respectively (Mantel–Cox: p<0.05; Fig. 12B). All full-length models, showed similar survival to the corresponding WT, at least up to the ages tested (Table 3).
Fig. 12.
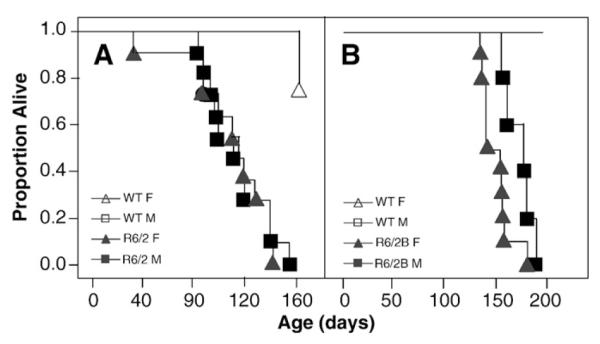
Kaplan–Meier survival curves as a function of genotype, age, and gender. A: R6/2; B: R6/2B.
Table 3.
Survival (median days) for all lines tested.
| Line | Genotype | Female | Male |
|---|---|---|---|
| R6/2 | Mutant | 109 | 113 |
| R6/2B | Mutant | 150 | 173 |
| HdhQ111 | Homo | 669 | 661 |
| Het | 661 | 605 | |
| WT | 674 | 509 | |
| BACHD | WT | a | |
| Mutant | |||
| YAC128 | WT | 399 | 556 |
| Mutant | 419 | 469 | |
| YAC128B | WT | a | |
| Mutant |
No death during period of observation.
Summary and discussion
R6/2 mice started to die after 11 weeks of age, and R6/2B mice started at 18 weeks of age. It is curious that all R6/2 mice died within 12 weeks from the first death, whereas R6/2B mice took only 9 weeks. If this finding is confirmed in different cohorts, it would suggest that the onset of lethality, or terminal pathogenic processes, may occur later in the C57BL/6J background with 240 CAGs, but the disease progression towards lethality may be faster. Unfortunately, all other measures apart from survival, including body weight were terminated around 14 weeks of age and thus we cannot test this hypothesis with other measures of progression in the present study. If future research confirms this observation, then a separation between disease onset and progression would have been found, at least for those processes resulting in lethality, and the underlying neuropathology could be tracked down, to either genetic modifiers (background strain) or CAG repeat length. Survival was normal in the models expressing full-length huntingtin, whether ectopic transgene-driven or endogenous huntingtin from the knock-in allele, although we do not discard increased mortality at much later ages than those monitored in this study.
General discussion
We have compared the transgenic line with randomly inserted short amino-terminal fragments of huntingtin with expanded CAG repeats, in two different backgrounds (the well-known R6/2, and the novel R6/2B), with four HD models that express either ectopic full-length mutant huntingtin from a YAC transgene on two different genetic backgrounds (YAC128 and YAV128B), or from a BAC transgene with CAA-CAG expansions (BACHD), or endogenous full-length huntingtin from an Hdh CAG knock-in allele, that extends the polyglutamine tract of murine huntingtin. These results, summarized in Table 4, pave the way for the use of standardized behavioral batteries of motor and sensory-motor function to obtain comprehensive, reproducible and comparable results across HD models used in preclinical research to the select clinical candidates. We show the importance of different background strains (Gerlai, 1996) and extend the findings to the source (vendor) of the genetic background strains.
Table 4.
Summary of phenotypic characteristics for all lines.
| Test | Measure | R6/2 |
R6/2B |
HdhQ111 (homo v WT) |
BACHD |
YAC128 |
YAC128B |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Effect | Start age |
Effect | Start age |
Effect | Start age |
Effect | Start age |
Effect | Start age |
Effect | Start age |
||
| Rotarod | Fall latency | ↓↓↓ | 4 | ↓↓↓ | 8 | ↑↓ | 4–16♂ | ↓↓↓ | 4 | ↓ | Through | ↓ | Through |
| 100♀ | |||||||||||||
| Open field | Total distance | ↓↓ | 6♂8, | ↓↓ | 8♂6♀ | - | - | ↓ | 28 | ↑ | 52♀ | ↓ | 12 |
| (light cycle) | 14♀ | ||||||||||||
| Distance center | ↓↓↓ | 4 | ↓↓↓ | 12 | - | - | ↓ | 28 | ↑ | 52 | - | - | |
| Rate of rearings | ↓↓↓ | 4 | ↓ | 12 | - | - | ↓↓ | 4 | ↑ | 32, 52 | - | - | |
| Open field | Total distance | ↓↓ | 12 | ↓↓ | 6 | - | - | ↓ | 28 | ↓↓ | 16–52♂32- | ↓ | 8,16,34 |
| (dark cycle) | 52♀ | ||||||||||||
| Distance center | ↓↓↓ | 4 | ↓↓↓ | 6 | - | - | - | - | ↑ | 4 | ↓ | 16–34 | |
| Rate of rearings | ↓↓↓ | 6 | - | - | - | - | ↓↓ | 4 | ↑ | 52 | - | - | |
| Grip strength | Force | ↓↓ | 10 | ↓ | 14 | - | - | - | - | - | - | - | - |
| Rearing–climbing | Latency to climb | ↓↓↓ | 4 | - | - | - | - | ↓↓ | 4 | ↓ | 16 | - | - |
| Paw print | Gait/splay | ↓↓↓ | 6 | ↓ | 14 | - | - | ↓,↑ | 12, 36 | - | - | ↓ | 16 |
| Base | ↑ | 14♂8, | - | - | - | - | ↑ | 36 | - | - | ↓ | 16 | |
| 12♀ | |||||||||||||
| Light–dark choice | Time in the dark | ↑↑ | 12 | ↑ | 14 | - | - | ↑↑↑ | 12 | ↑ | 52 | ↑ | 12,24 |
| Sensory gating | Startle | ↓↓ | 4 | ↓ | 6,14 | ↓ | 52 | ↓ | 36♂24, 36♀ | - | - | - | - |
| Prepulse | ↓↓ | 8 | ↓↓↓ | 14 | - | - | ↓↓ | 28, 52♂36, | - | - | ↓ | 56 | |
| inhibition | 52♂ | ||||||||||||
| Body weight | ↓↓↓ | 7 | ↓↓ | 9♂ | ↑ | 28♂76♀ | ↓↓ | 12♂16♀ | ↑↑ | 32♂48–76♀ | ↑ | 16♂8♀ | |
| 10♀ | |||||||||||||
| Survival | ↓↓↓ | 11 | ↓↓ | 18 | - | - | - | - | - | - | - | - | |
R6/2 models
The two R6/2 lines showed a robust and similar phenotype although there was a difference in the onset of their behavioral deficits as the R6/2B line showed deficits delayed by about 2 to 4 weeks (Table 4) and did not present seizures. Another interesting difference between the two R6/2 lines was the delayed onset of lethality in the R6/2B line with a seemingly more aggressive decline after onset of terminal events. The differences can be ascribed to either the CAG repeat count (122–135 in the R6/2 and 243–264 in the R6/2B) and/or to the genetic background (mixed CBAB6 versus pure C57Bl/6). In humans, longer CAG repeat lengths are associated with an earlier onset of the disease (The Huntington’s Disease Collaborative Research Group, 1993). However, at least in HD exon 1 R6/2 transgenic mice, spontaneous expansion to CAG numbers higher than 350 attenuates phenotypes observed at lower repeat numbers, perhaps because of altered sub-cellular localization of the polyglutamine transgene product (Goldowitz et al., 2009; Morton et al., 2009). Thus, it is also possible that the delayed behavioral onset in the R6/2B line compared to the R6/2 line in the mixed background, is due to the higher repeats, over and above genetic background differences. In humans, such levels of repeats are rare, with a child reported to have close to 250 repeats (Nance et al., 1999) and other cases with up to 121 (Telenius et al., 1993) and 180 CAG repeats (Sathasivam et al., 1997), though with phenotypes of severe juvenile-onset HD. A thorough comparison of different CAG repeat lengths on the same background is needed to elucidate these findings.
The R6/2 transgene models do not mimic the genetic disease-initiating mechanism of HD because the insult is an ectopically-inserted transgene expressing a polypeptide from the amino terminus of huntingtin that is composed mostly of polyglutamines. As a result, the R6/2 line may be more a model of polyglutamine disease rather than HD in particular. However, despite this, there has been found to be no difference between the R6/2 model and full-length models in the extent to which they recapitulate the transcriptional dysregulation that occurs in HD (Kuhn et al., 2007). Many researchers have shown a consistent and reproducible phenotype (Carter et al., 1999; Lione et al., 1999) and our results replicate and extend those findings to a novel genetic background. The model clearly has a robust phenotype, which develops rapidly but within a timeframe that allows for efficient drug testing. The differences in activity (locomotion and rearing), grip strength, body weight, and survival are robust and allow for a fast and economical drug-testing program.
Full-length huntingtin-expressing models
The full-length models showed milder deficits than the R6/2 models, consistently with published studies (Menalled and Chesselet, 2002). BACHD mice showed a stronger phenotype, followed by the YAC128 lines, with milder phenotypes, and the HdhQ111 line on the CD1 background, which showed a very mild phenotype. Although the full-length models studied here have shorter CAG repeat lengths (<120 CAGs, see Table 1) than the N-terminal fragment lines (>122 CAGs), other full-length models with higher repeats (~150) also show slower progression than the R6/2 and R6/1 lines (Lin et al., 2001; Woodman et al., 2007). The YAC46 and YAC72 mice show almost no behavioral deficit, therefore, within a same full-length model the relationship between behavioral deficits and CAG length is similar to that in humans, for at least a certain range (Slow et al., 2005).
Knock-in models have high construct validity for the pathophysiology of HD given the similarity of the genetic insult used in the mouse model and present in HD, and exhibit subtle behavioral and molecular abnormalities, that may pre-date more overt end-stage phenotypes (Menalled and Chesselet, 2002). In this first behavioral characterization of the HdhQ111 line, we found a mild behavioral phenotype, namely, body-weight loss and a slight decrease in rotarod performance and startle at the older ages. Congenic strains of mice carrying this HdhQ111 allele may, however, exhibit more robust behavioral differences on particular genetic backgrounds, such as a C57Bl/6J background, which has been shown to modify both nuclear mutant huntingtin and CAG repeat instability phenotypes in the striatum caused by the HdhQ111 allele (Lloret et al., 2006). Indeed, homozygote Q150 knock-in mice, for example, on a CBA/Ca×C57BL/6/J F1 background showed a strong phenotype, reaching within their lifetime an end-stage disease characterized by behavioral and molecular phenotypes similar to those found in the R6 exon 1 models (Woodman et al., 2007).
The BACHD line has a robust phenotype in a number of tests which replicates and extends published results (Gray et al., 2008). The model is amenable to drug testing, although, as behavioral deficits occur at an older age than in the R6/2 line, testing is more time consuming and expensive.
YAC128 and YAC128B mice show some clear behavioral differences (Table 4). It has been shown that the background strain affects the severity of the phenotype, with the YAC128 in an FVB/N background mice being most affected (Van Raamsdonk et al., 2007). The deficits seen for the YAC128 in the present set of experiments are less pronounced than those in other publications, however, with the YAC128B offering a similar yet slightly less variable phenotype on a C57Bl/6 background. The milder phenotype of the YAC128, as compared to what others have observed (Van Raamsdonk et al., 2005), maybe due to the way outliers or abnormal behavior is handled. For example, we removed animals that froze in the open field and light–dark tests, whereas Slow et al. (2003) removed animals that circled in their open field. Other specific experimental details used in these experiments, housing enriched or non-enriched environments or other husbandry details can also have effects on the behavioral deficits measured. It is possible that the specific testing paradigm with enriched conditions used in this study resulted in a particularly more attenuated phenotype of the YAC128 mice on the FVB/N strain. The milder deficits observed with the FVB/N strain suggest that it is not more suitable than the C57Bl/6 for drug testing using behavioral outcome measures.
Increased body weight in the BACHD, YAC128 and YAC128B models, is a possible confounding factor, especially for tests of motor function. We show evidence, however, that the rotarod deficit can be found over and above body-weight differences in the BACHD males, as did the original report (Gray et al., 2008) and YAC128B mice, although the analysis was not possible in BACH HD females. The progression of behavioral deficits in BACHD mice between 2 and 6 months of age parallels the progression of electrophysiological deficits in cortical pyramidal neurons, cortical interneurons, and striatal medium spiny neurons (Gray et al., 2008; Spampanato et al., 2008) suggesting that the abnormal cortical striatal neuronal circuit may account for some of the behavioral deficits in BACHD mice.
As body-weight increase is likely to result from the addition of full-length mutant protein onto the load of normal protein from the endogenous HD genes it is not surprising that the three transgenic lines showed increased body weight (Van Raamsdonk et al., 2006a). Consistently, the HdhQ111 knock-in model, which expresses mutant huntingtin from the endogenous mouse gene, showed reduced body weight. We were unable show independence between body weight and rotarod performance in YAC128 mice, and thus care is still needed when interpreting functional endpoints that may be influenced by body weight (i.e. rotarod, swimming tasks).
In summary, our data support the use of the R6/2 and BACHD models for drug testing using behavioral outcome measures, amid the lines compared in this work. The results from the HdhQ111 motor phenotyping were not robust enough to justify using this line for testing compounds, although the literature suggests that the histopathology of this model is robust. The results from the YAC128 line are encouraging, although less robust than expected. This may be explained in part due to baseline behavioral differences we observed between the WT FVB/NCrl mice used as controls for the YAC128 mice and the WT FVB/NJ mice used as controls for the BACHD mice under the conditions used in this study. Other lines exist, such as the Q150 model, with strong phenotypes for which a thorough comparison head-to-head can be used to judge their utility in behavioral or neuropathology preclinical testing, or in basic research to understand the pathophysiological mechanisms of HD (Woodman et al., 2007).
No model can be proved superior to others on all aspects, in particular, the homology of the genetic insult and neuropathology between the mouse model and HD, and the robustness of the behavioral dysfunction phenotype. However, although the cost of screening potential therapeutic compounds in a model with a slower progression is considerable, the need for another model to complement to drug screening in the R6/2 mice can hardly be disputed.
Acknowledgments
We are grateful to Drs. Michael Hayden and Blair Leavitt for YAC128 mice, Dr. Jeff Schneider for the statistical support, Dr. Marie Françoise Chesselet, for numerous discussions and suggestions. Our thanks to Jennifer Goodman, Albert Jimenez, Jennie Murphy, Natalie Morgan, Morgen Peck, Jennifer Galarza, Sidra Khalid Melinda Ruiz and Meredith Taylor for help with the breeding, behavioral experiments and operations. Dr. X. William Yang was supported by an NINDS/NIH (R01 NS049501) grant and grants from CHDI and HDF.
References
- ALS Association http://www.alsa.org/
- Argmann CA, Auwerx J. Minimizing variation due to genotype and environment. Curr Protoc Mol Biol. 2006 doi: 10.1002/0471142727.mb29a02s73. Chapter 29, Unit 29A 2. [DOI] [PubMed] [Google Scholar]
- Bowes C, et al. Retinal degeneration in the rd mouse is caused by a defect in the beta subunit of rod cGMP-phosphodiesterase. Nature. 1990;347:677–680. doi: 10.1038/347677a0. [DOI] [PubMed] [Google Scholar]
- Carter RJ, et al. Characterization of progressive motor deficits in mice transgenic for the human Huntington’s disease mutation. J. Neurosci. 1999;19:3248–3257. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlai R. Gene-targeting studies of mammalian behavior: is it the mutation or the background genotype? Trends Neurosci. 1996;19:177–181. doi: 10.1016/s0166-2236(96)20020-7. [DOI] [PubMed] [Google Scholar]
- Glass M, et al. Delayed onset of Huntington’s disease in mice in an enriched environment correlates with delayed loss of cannabinoid CB1 receptors. Neuroscience. 2004;123:207–212. doi: 10.1016/s0306-4522(03)00595-5. [DOI] [PubMed] [Google Scholar]
- Gray M, et al. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J. Neurosci. 2008;28:6182–6195. doi: 10.1523/JNEUROSCI.0857-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusella JF, et al. A polymorphic DNA marker genetically linked to Huntington’s disease. Nature. 1983;306:234–238. doi: 10.1038/306234a0. [DOI] [PubMed] [Google Scholar]
- Hickey MA, et al. Early behavioral deficits in R6/2 mice suitable for use in preclinical drug testing. Neurobiol. Dis. 2005;20:1–11. doi: 10.1016/j.nbd.2005.01.024. [DOI] [PubMed] [Google Scholar]
- Hockly E, et al. Environmental enrichment slows disease progression in R6/2 Huntington’s disease mice. Ann. Neurol. 2002;51:235–242. doi: 10.1002/ana.10094. [DOI] [PubMed] [Google Scholar]
- Hockly E, et al. Standardization and statistical approaches to therapeutic trials in the R6/2 mouse. Brain Res. Bull. 2003;61:469–479. doi: 10.1016/s0361-9230(03)00185-0. [DOI] [PubMed] [Google Scholar]
- Koller WC, Trimble J. The gait abnormality of Huntington’s disease. Neurology. 1985;35:1450–1454. doi: 10.1212/wnl.35.10.1450. [DOI] [PubMed] [Google Scholar]
- Kopp C, et al. Comparative study of emotional behaviour in three inbred strains of mice. Behav. Proc. 1999;47:161–174. doi: 10.1016/s0376-6357(99)00057-1. [DOI] [PubMed] [Google Scholar]
- Kuhn A, et al. Mutant huntingtin’s effects on striatal gene expression in mice recapitulate changes observed in human Huntington’s disease brain and do not differ with mutant huntingtin length or wild-type huntingtin dosage. Hum. Mol. Genet. 2007;16:1845–1861. doi: 10.1093/hmg/ddm133. [DOI] [PubMed] [Google Scholar]
- Lazic SE, et al. Neurogenesis in the R6/1 transgenic mouse model of Huntington’s disease: effects of environmental enrichment. Eur. J. Neurosci. 2006;23:1829–1838. doi: 10.1111/j.1460-9568.2006.04715.x. [DOI] [PubMed] [Google Scholar]
- Lin CH, et al. Neurological abnormalities in a knock-in mouse model of Huntington’s disease. Hum. Mol. Genet. 2001;10:137–144. doi: 10.1093/hmg/10.2.137. [DOI] [PubMed] [Google Scholar]
- Lione LA, et al. Selective discrimination learning impairments in mice expressing the human Huntington’s disease mutation. J. Neurosci. 1999;19:10428–10437. doi: 10.1523/JNEUROSCI.19-23-10428.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloret A, et al. Genetic background modifies nuclear mutant huntingtin accumulation and HD CAG repeat instability in Huntington’s disease knock-in mice. Hum. Mol. Genet. 2006;15:2015–2024. doi: 10.1093/hmg/ddl125. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- Menalled L, Chesselet M. Mouse models of Huntington’s disease. Trends Pharmacol. Sci. 2002;23:32–39. doi: 10.1016/s0165-6147(00)01884-8. [DOI] [PubMed] [Google Scholar]
- Menalled LB, et al. Early motor dysfunction and striosomal distribution of huntingtin microaggregates in Huntington’s disease knock-in mice. J. Neurosci. 2002;22:8266–8276. doi: 10.1523/JNEUROSCI.22-18-08266.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton AJ, et al. Progressive formation of inclusions in the striatum and hippocampus of mice transgenic for the human Huntington’s disease mutation. J. Neurocytol. 2000;29:679–702. doi: 10.1023/a:1010887421592. [DOI] [PubMed] [Google Scholar]
- Morton AJ, et al. Disintegration of the sleep–wake cycle and circadian timing in Huntington’s disease. J. Neurosci. 2005;25:157–163. doi: 10.1523/JNEUROSCI.3842-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton AJ, et al. Paradoxical delay in the onset of disease caused by super-long CAG repeat expansions in R6/2 mice. Neurobiol. Dis. 2009;33:331–341. doi: 10.1016/j.nbd.2008.11.015. [DOI] [PubMed] [Google Scholar]
- Nadeau JH. Modifier genes and protective alleles in humans and mice. Curr. Opin. Genet. Dev. 2003;13:290–295. doi: 10.1016/s0959-437x(03)00061-3. [DOI] [PubMed] [Google Scholar]
- Nance MA, et al. Analysis of a very large trinucleotide repeat in a patient with juvenile Huntington’s disease. Neurology. 1999;52:392–394. doi: 10.1212/wnl.52.2.392. [DOI] [PubMed] [Google Scholar]
- Paylor R, Crawley J. Inbred strain differences in prepulse inhibition of the mouse startle response. Psychopharmacology. 1997;132:169–180. doi: 10.1007/s002130050333. [DOI] [PubMed] [Google Scholar]
- Sathasivam K, et al. Identification of an HD patient with a (CAG)180 repeat expansion and the propagation of highly expanded CAG repeats in lambda phage. Hum. Genet. 1997;99:692–695. doi: 10.1007/s004390050432. [DOI] [PubMed] [Google Scholar]
- Schilling G, et al. Environmental, pharmacological, and genetic modulation of the HD phenotype in transgenic mice. Exp. Neurol. 2004;187:137–149. doi: 10.1016/j.expneurol.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Singer J. Using SAS PROC MIXED to fit multilevel models, hierarchical models, and individual growth models. J. Educ. Behav. Stat. 1998;23:323. [Google Scholar]
- Slow EJ, et al. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum. Mol. Genet. 2003;12:1555–1567. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- Slow EJ, et al. Absence of behavioral abnormalities and neurodegeneration in vivo despite widespread neuronal huntingtin inclusions. Proc. Natl. Acad. Sci. U. S. A. 2005;102:11402–11407. doi: 10.1073/pnas.0503634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMA Foundation http://www.smafoundation.org.
- Spampanato J, et al. Progressive synaptic pathology of motor cortical neurons in a BAC transgenic mouse model of Huntington’s disease. Neuroscience. 2008;157:606–620. doi: 10.1016/j.neuroscience.2008.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires TL, et al. Environmental enrichment rescues protein deficits in a mouse model of Huntington’s disease, indicating a possible disease mechanism. J. Neurosci. 2004;24:2270–2276. doi: 10.1523/JNEUROSCI.1658-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, et al. Impaired prepulse inhibition of acoustic and tactile startle response in patients with Huntington’s disease. J. Neurol. Neurosurg. Psychiatry. 1995;58:192–200. doi: 10.1136/jnnp.58.2.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telenius H, et al. Molecular analysis of juvenile Huntington disease: the major influence on (CAG)n repeat length is the sex of the affected parent. Hum. Mol. Genet. 1993;2:1535–1540. doi: 10.1093/hmg/2.10.1535. [DOI] [PubMed] [Google Scholar]
- The Huntington’s Disease Collaborative Research Group A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- TREAT-NMD http://www.treat-nmd.eu/.
- van Dellen A, et al. Delaying the onset of Huntington’s in mice. Nature. 2000;404:721–722. doi: 10.1038/35008142. [DOI] [PubMed] [Google Scholar]
- van Dellen A, et al. Wheel running from a juvenile age delays onset of specific motor deficits but does not alter protein aggregate density in a mouse model of Huntington’s disease. BMC Neurosci. 2008;9:34. doi: 10.1186/1471-2202-9-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Raamsdonk JM, et al. Cognitive dysfunction precedes neuropathology and motor abnormalities in the YAC128 mouse model of Huntington’s disease. J. Neurosci. 2005;25:4169–4180. doi: 10.1523/JNEUROSCI.0590-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Raamsdonk JM, et al. Body weight is modulated by levels of full-length Huntingtin. Hum. Mol. Genet. 2006a;15:1513. doi: 10.1093/hmg/ddl072. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk JM, et al. Wild-type huntingtin ameliorates striatal neuronal atrophy but does not prevent other abnormalities in the YAC128 mouse model of Huntington disease. BMC. Neurosci. 2006b;7:80. doi: 10.1186/1471-2202-7-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Raamsdonk JM, et al. Phenotypic abnormalities in the YAC128 mouse model of Huntington disease are penetrant on multiple genetic backgrounds and modulated by strain. Neurobiol. Disease. 2007;26:189–200. doi: 10.1016/j.nbd.2006.12.010. [DOI] [PubMed] [Google Scholar]
- Woodman B, et al. The Hdh(Q150/Q150) knock-in mouse model of HD and the R6/2 exon 1 model develop comparable and widespread molecular phenotypes. Brain Res. Bull. 2007;72:83–97. doi: 10.1016/j.brainresbull.2006.11.004. [DOI] [PubMed] [Google Scholar]