

Abstract
Bacillus subtilis CsoR (Bsu CsoR) is a copper-sensing transcriptional repressor that regulates the expression of the copZA operon encoding a copper chaperone and a Cu efflux P-type ATPase, respectively. Bsu CsoR is a homolog of Mycobacterium tuberculosis CsoR (Mtb CsoR), representative of a large Cu(I)-sensing regulatory protein family. We show here that Bsu CsoR binds ≈1 mol equiv Cu(I) per monomer in vitro with an affinity ≥1021M-1. X-ray absorption spectroscopy shows Cu(I) adopts a trigonal S2N coordination like Mtb CsoR. Both apo and Cu(I)-bound Bsu CsoR are stable tetramers in the low micromolar monomer concentration range by sedimentation velocity and equilibrium ultracentrifugation. Apo-Bsu CsoR binds to a pseudo-palindromic 30 bp copZA operator-promoter DNA with a stoichiometry of two tetramers per DNA and stepwise affinities of K1apo=3.1(±0.8) × 107 M-1 and K2apo = 8.3 (±2.2) × 107 M-1 (0.4 M NaCl, 25 °C, pH 6.5). Cu(I) Bsu CsoR binds to the same DNA with greatly reduced affinities, K1Cu=2.9(±0.4) × 106 M-1 and K2Cu≤1.0 × 105 M-1 consistent with a copper-dependent derepression model. This Cu-dependent regulation is abrogated by a “second shell” Glu90-to-Ala substitution. Bsu CsoR binds Ni(II) with very high affinity but forms a non-native coordination geometry, as does Co(II) and likely Zn(II); none of these metals strongly regulate copZA operator DNA binding in vitro. The implications of these findings on the specificity of metal sensing sites in CsoR/RcnR proteins are discussed.
As an essential catalytic cofactor of metalloenzymes, copper (Cu) plays crucial roles in many important biological processes (1). The reversible oxidation of Cu(I) and reduction of Cu(II) makes Cu versatile in catalyzing a wide range of different chemical reactions in the cell. However, these redox properties also make Cu toxic with amounts of “free” Cu inside the cell thought to be strongly limiting. An undesirable pool of available Cu in the cytosol could mediate the production of reactive oxygen species (ROS), i.e. OH-., O2-., through the Fenton reaction, but may also be involved in amyloid formation and toxicity in mammals (2-4). Therefore, the total and bioavailable concentrations of Cu inside the cell are thought to be strictly regulated by a Cu homeostasis system, which includes Cu transporters, chaperones, scavengers and regulatory proteins.
Recent findings suggest that just like iron, copper and copper homeostasis related proteins can be required for full virulence of pathogenic bacteria. For example, a Cuefflux P-type ATPase CtpA in L. monocytogenes in liquid culture is a known virulence factor as is Cu-superoxide dismutase in Mycobacterium tuberculosis (Mtb) (5, 6). Furthermore, recent microarray studies carried out with Mtb at different Cu concentrations suggest a strong relationship between Cu toxicity and oxidative stress, the latter of which is one of the challenges Mtb encounters during its survival in macrophages (7). Another proteomics study carried out in L. lactis identified three oxidative stress related proteins as highly induced by Cu stress (8). Although precisely how Cu stress induces what appears to be an oxidative stress response is not firmly established, the reversible redox cycling of excess Cu in an anaerobic environment may well be at least partly causative; in any case, proteins involved in Cu homeostasis clearly play important roles in the defense mechanisms of the pathogens against oxidative stress (9, 10).
Bacillus subtilis CsoR (Bsu CsoR) has recently been found to be a Cu sensor that regulates the transcription of copZA operon, which encodes the Cu chaperone CopZ and the Cu-efflux P-type ATPase CopA (11). Bsu CsoR shares high sequence similarity with M. tuberculosis CsoR (Mtb CsoR), the first characterized member of what is predicted to be a large family of Cu regulatory proteins in bacteria (Figure 1) (12). It has been shown that Bsu CsoR specifically binds to a pseudo-palindromic DNA in the operator-promoter region of copZA operon and represses the transcription. Addition of Cu inhibits the binding and derepresses the transcription, allowing the expression of CopZ/CopA proteins to traffic and efflux the excess Cu out of the cell.
Figure 1.

Multiple sequence alignment of known CsoR/RcnR family Cu-sensing CsoR homologs from several mycobacteria, Bsu CsoR (11) and E. coli RcnR (13). Organisms and (locus tags) for the other entries are as follows: M. tuberculosis (Rv0967) (12); M. marinum (MM4874); M. ulcerans (MUL_0425); M. smegmatis (MSMEG_0230). Conserved metal binding residues are highlighted in red and proposed key residues for allosteric regulation of DNA binding are in blue. The unique C-terminal tails from CsoR homologs encoded by pathogenic mycobacteria are shaded in green. Secondary structure elements are labeled according to Mtb CsoR structure (12).
The Ni(II)/Co(II) sensor RcnR from E. coli is a distant ortholog of Mtb CsoR (Figure 1). RcnR regulates the expression of a Ni-efflux system in E. coli (13). Recent studies show that RcnR binds Ni(II)/Co(II) with a higher coordination number (n=5 or 6) relative to the trigonal planar Cu(I) site (n=3) in Mtb CsoR and is proposed to utilize a subset of CsoR Cu ligands as Ni(II)/Co(II) ligands in RcnR (12, 14). This observation, coupled with other CsoR/RcnR family proteins for which we have no biological and structural insight, opens up opportunities to understand the mechanisms of ligand (metal) selectivity at the molecular level of other CsoR orthologs, while expanding the range of known or potential functions in this new family of proteins (14).
Here, using a series of biochemical and biophysical experiments, we show that Bsu CsoR binds 1 mol equiv Cu(I) per monomer with very high affinity (≥1021M-1) in vitro. X-ray absorption spectroscopy (XAS) shows Cu(I) adopts, as expected, an S2N coordination geometry similar to Mtb CsoR (12). Both apo and Cu(I)-bound Bsu CsoRs are non-dissociable tetramers in the low micromolar monomer concentration range. Size exclusion chromatography reveals apo-Bsu CsoR binds a 30 bp copZA operator DNA with a stoichiometry of 8 monomers, or two tetramers, to one two-fold symmetric, pseudo-palindromic DNA sequence. The stepwise DNA binding affinities were further determined by fluorescence anisotropy for both apo-Bsu CsoR and Cu(I)-bound Bsu CsoR. The Cu(I)-dependent regulation of DNA binding is abrogated in an E90A mutant. Interestingly, Bsu CsoR is also capable of binding other divalent metal ions including Co(II), Zn(II) and Ni(II), and in fact binds Ni(II) and Zn(II) with 108-109 M-1 affinities at equilibrium. However, each of these metals adopts a non-native (non-trigonal) coordination geometry and each fails to strongly negatively regulate operator DNA binding in vitro.
Methods
Plasmid construction, protein expression and purification
The Bsu CsoR-pET16b expression plasmid was a generous gift of Dr. John D. Helmann (Cornell University). Amino acid substitutions were introduced to the plasmid by site-directed quick-change mutagenesis and the resulting plasmids were confirmed by DNA sequencing. Wild-type and E90A Bsu CsoRs were expressed and purified using similar procedures as described previously for Mtb CsoR (11, 12). Expression plasmids containing wild type or mutant Bsu CsoR were transformed into E. coli BL21(DE3) and grown in LB media until OD600 reached 0.6-0.8. 0.4 mM IPTG was then added and cells were grown for additional 2 h before harvesting by low speed centrifugation. Cell pellets were suspended in 200 mL Buffer A (25 mM MES, 2 mM DTT, 1 mM EDTA, pH 5.8) and lysed by sonication. The lysate was centrifuged and 0.15% (v/v) polyethyleneimine (PEI) was added to the supernatant to precipitate the nucleic acids. Bsu CsoR remained in the supernatant and was subjected to (NH4)2SO4 precipitation and the ammonium sulfate pellet was resuspended in Buffer A and dialyzed against Buffer A containing 0.05 M NaCl. The sample is then purified by SP Fast Flow, Superdex 200 size exclusion chromatography as described previously (12). The resultant proteins were pooled, concentrated and dialyzed against Buffer S (10 mM MES, 0.2 M NaCl, pH 6.5) in an anaerobic Vacuum Atmospheres glovebox. The purity of the final products was estimated by visualization of Coomassie-stained 18% Tris-glycine SDS-PAGE gels to be ≥90%. Protein concentration was determined by UV absorption using ε280nm=1615 M-1cm-1 and free thiols were determined by DTNB assay to be more than 95% of expected value (11, 12). Less than 0.1% copper was detected by atomic absorption spectroscopy in all purified protein samples.
Cu(I) binding and BCS competition monitored by UV-vis absorption spectrocopy
250 μL aliquots of 20 μM Bsu CsoR monomer with or without 50 μM bathocuprione disulfonate (BCS) in buffer S were prepared in an anaerobic glovebox. Different amounts of CuCl were added to each aliquot and UV-vis absorption spectrum was taken. Samples with BCS were equilibrated in the glovebox at room temperature (≈22 °C) for 4 hours before recording the spectrum to ensure that equilibrium was reached. Absorption at 240 nm and 483 nm were plotted against Cu concentration respectively. Cu(I) binding affinity was determined by fitting the BCS competition data by a simple competition model using Dynafit (15) with the overall affinity constant KCu(BCS)2 (β2) fixed at 1019.6 M-2. This value represents the corrected at pH 6.5 (used here) calculated from the reported value of β2=19.8, (pKa=5.7) measured at pH 8.0 (16).
Cu(I)-binding monitored by tyrosine fluorescence
1700 μL of 10 μM Bsu CsoR was loaded into an anaerobic cuvette fitted with an adjustable-volume Hamilton gastight syringe loaded with 100 μL of Cu(I) titrant prior to removal from the anaerobic glovebox. Emission spectra from 295 nm to 400 nm were taken at each ith addition of Cu(I) with λex=280 nm. The ratio of emission intensity at 307 nm after the ith addition (I) with the initial intensity (I0) were plotted against Cu(I) concentration after correcting for dilution (17). No significant photobleaching of the Tyr fluorescence was observed in these experiments.
Cu(I) X-ray absorption spectroscopy
Both wild-type and E90A Bsu CsoRs were mixed with 0.8 mol equiv of Cu(I) in 10 mM MES, 0.2 M NaCl, 30% (v/v) glycerol, pH 6.5 in an anaerobic environment to about 1 mM final protein concentration. Samples were loaded into a 5-well polycarbonate XAS cuvette and immediately frozen in liquid N2. XAS data were collected at Stanford Synchrotron Radiation Lightsource (SSRL) on beamline 9-3 with the SPEAR storage ring operating at 3.0 GeV. EXAFS data analysis was performed using EXAFSPAK software, using ab initio phase and amplitude functions computed with FEFF v7.2, according to standard procedures as described before (17). Given the minor contribution of Cu-N/O scattering to EXAFS that is dominated by Cu-S, models used for curve fitting contained a fixed Cu-N/O distance that is expected for 3-coordinated Cu(I) imidazole coordination (2.05Å).
Sedimentation velocity and equilibrium experiments
All analytical ultracentrifugation experiments were carried out using a Beckman model Optima XL-I analytical ultracentrifuge equipped with an An-60 Ti rotor in the Physical Biochemistry Instrumentation Facility at Indiana University. All samples were prepared in buffer S and loaded into centerpieces inside the anaerobic glovebox. 110 μL samples for equilibrium experiments were prepared at 8 μM, 14 μM and 20 μM monomer concentrations (about 0.3, 0.5 and 0.8 OD230 initially) and loaded into 6-channel Epon charcoal-filled centerpieces. Intensity scans at 230 nm were taken at speeds of 19,300, 30,600 and 38,700 rpm at 20 °C. All equilibrium data were fit globally to a single species model using Ultrascan as described (18). For sedimentation velocity experiments, 450 μL samples are loaded into a two-channel aluminum centerpiece with 1.2 cm path length. The rotor speed was 60,000 rpm at 20°C and intensity data at 230 nm were collected as a function of time. Sample concentrations were 8 μM and 20 μM Bsu CsoR monomer (≈0.3 and 0.8 OD230 initially). Data were analyzed using Ultrascan software interfaced with a genetic algorithm and Monte Carlo analysis package essentially as described (19-22).
Size exclusion chromatography
A 30 bp DNA derived from the copZA operator-promoter region (5′-TTGTAATACCCTACGGGGGTATGGTAGGAT-3′ and the complementary sequence) was used for all DNA binding experiments. 10 μM DNA was mixed with different concentrations of Bsu CsoR monomer up to 100 μM in buffer S with 2 mM DTT in room temperature. 100 μL of each mixture was loaded onto a Tricon Superdex 200 column (GE healthcare) on an Akta purifier. Elution profiles were obtained by monitoring the absorption at 240 nm, 260 nm and 280 nm simultaneously.
Fluorescence anisotropy
A 30 bp 5′-fluorescein labeled DNA with same sequence as above was used. The double stranded DNA was made by mixing the labeled strand with 1.1 mol equiv of the unlabeled complementary strand. The mixture was heated at 95 °C for 10 min and then slowly cooled to room temperature. Formation of double stranded DNA was further confirmed by native TBE gel electrophoresis. A typical anisotropy experiment was done with 4 nM DNA in 10 mM MES, 0.4 M NaCl, 2 mM DTT, pH 6.5 at 25°C unless noted otherwise. Anisotropy was monitored by exciting the fluorescein at 487 nm. With apo- or Cu(I)-bound Bsu CsoRs added, the average anisotropy of 5 measurements was reported for each addition. For Ni(II)- and Zn(II)-bound Bsu CsoRs, 1 mol equiv of metal ions were mixed with Bsu CsoR as titrant; an additional 5 μM Ni(II) or Zn(II) was present in the cuvette to ensure that only the metal-bound CsoR was present during these titrations. The resulting data were fitted to a stepwise model involving the binding of two non dissociable tetramers to one DNA using Dynafit assuming a linear change in anisotropy with fractional saturation of the DNA (15, 17). Since Cu(I)-bound Bsu CsoR does not reach saturation, the maximum anisotropy value was fixed at the same value as that obtained for apo-Bsu CsoR. The coupling free energy, ΔGc, is operationally defined by ΔG=-RTln(K1CuK2Cu/K2apoK2apo), where K1apo, K2apo, K1Cu and K2Cu are stepwise DNA binding constants for apo and Cu(I)-bound Bsu CsoRs respectively. This formalism for ΔGc was used since the saturating and presumably fully repressing complex invokes two bound tetramers bound per palindromic operator DNA segment, as well as the high inverse correlation between the magnitudes of K1 and K2.
Other metal binding experiments
Zn(II) binding was monitored by a chelator competition assay with mag-fura-2 (KZn=5.0 × 107 M-1 at pH 7.0) using UV-vis absorption spectroscopy as previously described (17, 18). The data were fit using a competitive binding model with Dynafit (15) to determine the Zn(II) binding affinity. Co(II) and Ni(II) binding experiments were carried out as described previously (17, 18) in buffer S. Ni(II) binding affinity was determined by a competition assay with EGTA (23). All concentrations of metal titrants were determined using atomic absorption spectroscopy.
Results
Bsu CsoR binds 1 monomer mol equiv Cu(I) with an affinity higher than that of BCS
It has been previously shown that Bsu CsoR regulates the expression of copZA operon by binding to the promoter region and that the addition of CuSO4 and DTT as reductant resulted in disruption of the DNA binding. It was therefore proposed that the DNA binding affinity of Bsu CsoR is regulated by Cu(I) binding. Here, we show that Bsu CsoR binds Cu(I) directly in vitro by both UV-vis and tyrosine fluorescence spectroscopies.
Addition of Cu(I) into an anaerobic solution of Bsu CsoR causes increased absorption in the ultraviolet region (ε240nm≈16,000 M-1cm-1); this reports on the formation of thiolate-copper coordination bonds and the spectrum is quite similar to that of Cu(I)-saturated Mtb CsoR (12, 17). This increase is saturable at about 1.0 mol equiv Cu(I) per monomer (Figure 2A). Significant quenching of tyrosine fluorescence, the importance of which is further discussed below (see Discussion), is observed upon Cu(I) binding as shown in Figure 2B. Maximum quenching is achieved upon addition of about 1.2 mol equiv Cu(I). Therefore, both UV-vis and tyrosine fluorescence suggest Bsu CsoR binds ≈1 Cu(I) ion per mol monomer.
Figure 2.

Bsu CsoR binds 1 mol equiv of Cu(I). (A) Apoprotein-subtracted molar absorptivity spectrum of Cu(I):Bsu CsoR mixture at 1:1 molar ratio with the binding isotherm shown in the inset. (B) Anaerobic titration of 10 μM apo-Bsu CsoR with Cu(I) as monitored by change in tyrosine fluorescence. Conditions: 10 mM MES, 0.2 M NaCl, pH 6.5, 25°C.
The Cu(I) binding affinity was estimated by a competition experiment using BCS, a Cu(I) specific competitor which can form a Cu(I)(BCS)2 complex that absorbs at 483 nm with a β2=19.8 (16). Figure 3 shows the change of the absorbance at 483 nm when Cu(I) is added to a mixture of 20 μM Bsu CsoR monomers and 50 μM BCS. No change of absorbance is observed until about 20 μM Cu(I) is added, suggesting the Cu(I) added initially is bound to Bsu CsoR but not BCS. After Bsu CsoR is saturated with 1 mol equiv Cu(I), additional Cu(I) forms a complex with BCS reported by the linear increase of absorbance at 483 nm, which is saturated at about 45 μM total Cu(I), reporting on the formation of ≈25 μM Cu(I)(BCS)2 complex. The fact that no Cu(I)(BCS)2 complex is formed until saturation of Bsu CsoR suggests that Bsu CsoR binds Cu(I) with much higher affinity than BCS. Due to the sharpness of the transition, this binding isotherm provides only a lower limit of the apparent binding affinity of KCu≥1021 M-1 when fit to a single site binding model [1:1 Cu(I) per protomer] under these solution conditions (see Methods, Table 2). Although CsoR is a tetramer, microscopic binding constants for individual sites in the tetramer, and thus any cooperativity of metal binding to these sites, cannot be resolved by this assay under these conditions given the stoichiometric nature of the complex formation.
Figure 3.
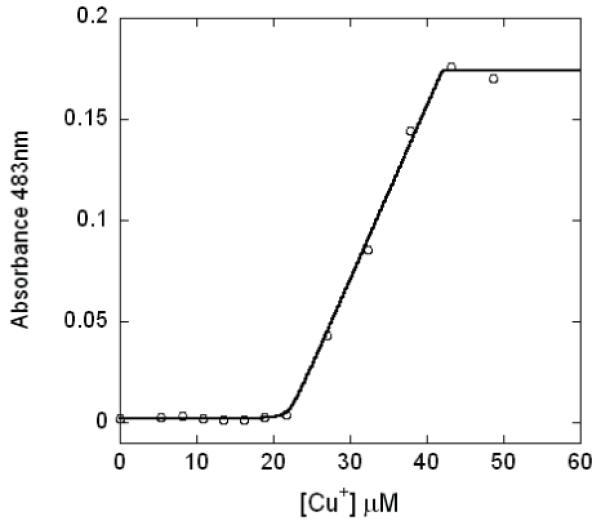
BCS competition experiment with wild-type Bsu CsoR. Different amounts of Cu(I) were mixed with 20 μM Bsu CsoR monomer and 50 μM BCS in buffer S with the absorption at 483 nm plotted against the total Cu(I) concentration. The solid curve represents the best-fit to a simple competition model (see text for details).
Table 2.
Summary of the metal and DNA binding affinities and allosteric coupling free energies for metalloderivatives of wild-type and E90A Bsu CsoR
| Bsu CsoR | metal | metal binding affinitya | DNA binding affinityb | Δ Gc | |
|---|---|---|---|---|---|
| (M-1) | K1 (M-1) | K2 (M-1) | (kcal/mol) | ||
| Wild-type | apo | - | 3.1 (±0.8) × 107 | 8.3 (±2.2) × 107 | - |
| Cu(I) | ≥ 1021 | 2.9 (±0.4) × 106 | ≤1.0 × 105 | ≥ +5.4 | |
| Co(II) | ≤ 105 | - | - | - | |
| Ni(II) | 3.6 (±0.3) × 109 | 5.7 (±1.0) × 106 | 3.1 (±0.5) × 107 | +1.6 (±0.3) | |
| Zn(II) | 1.6 (±0.1) × 108,c | 1.0 (±0.3) × 107 | 1.5 (±0.4) × 107 | +1.7 (±0.3) | |
| E90A | apo | - | 9.5 (±3.0) × 106 | 1.3 (±0.4) × 107 | - |
| Cu(I) | ≥ 1021 | 4.8 (±2.0) × 106 | 1.1 (±0.4) × 108 | -0.9 (±0.4) | |
The results of fitting to a simple 1:1 (metal:monomer) binding model.
Solution conditions: 10 mM MES, 0.4 M NaCl, 2 mM DTT, pH 6.5, 25 °C. We note that unique values of K1 and K2 are not readily resolved in this assay (see Methods)
A fit to two-site step-wise dimer binding models gives KZn1= 1.7 (±0.4) × 109 M-1; KZn2= 4.5 (±0.3) × 107 M-1 (see Supplementary Figure S1).
Cu(I) forms a trigonal S2N coordination site in Bsu CsoR
Copper K-edge X-ray absorption spectroscopy was used to determine the structure of Cu(I) complex formed at 0.8:1 Cu(I):Bsu CsoR monomer molar ratio (chosen to assure that all Cu(I) was bound to protein). The pre-edge peak at 8940 eV in the edge spectrum shown in Figure 4A is consistent with a 1s→4p excitation typical for 3-coordinate Cu(I) (24). The Cu K-edge extended X-ray absorption fine structure (EXAFS) spectrum as well as the Fourier transforms are shown in Figure 4B and 4C, respectively; structural parameters derived from EXAFS curve fitting are shown in Table 1. The data are best fit to a 3-coordinate model, with two Cu-S interactions at 2.20 Å and a single Cu-N/O interaction (fixed at 2.05 Å). This Cu-S distance is consistent with 3-coordinate Cu(I) and is similar to the Cu-S distance previously reported for Mtb CsoR. Significant outer-shell scattering observed between 3 and 4 Å is consistent with the third coordinating ligand being a nitrogen atom from a His residues, possibly from His70 which corresponds to known Cu(I) ligand His61 in Mtb CsoR (Figure 1) (12). These data are consistent with the idea that Cu(I) is coordinated by Cys45′, His70 and Cys74 in Bsu CsoR within a dimer, which are analogous to the Cu(I) ligands in Mtb CsoR dimer (Figure 1). Virtually identical spectra and curve-fitting results were obtained for E90A Bsu CsoR, suggesting no significant change in the first-shell coordination of Cu(I) in this mutant (see Discussion).
Figure 4.

X-ray absorption spectroscopy (XAS) of Cu(I)-bound Bsu CsoR. (A) Cu K-edge X-ray absorption edge spectra of Cu(I)-bound WT Bsu CsoR (solid) and E90A mutant (dashed). Copper K-edge EXAFS spectra and the Fourier transforms (k3 weighted, k=2-12 Å-1) for Cu(I)-bound WT Bsu CsoR (B) and the E90A mutant (C). In B and C, solid curves represent the experimental data and dashed curves represent best fits with parameters compiled in Table 1.
Table 1.
XAS fitting parametersa
| Sample(k range) Δk3χ | Fit | Shell | Ras (Å) | σas2 (Å2) | ΔE0 (eV) | f’b |
|---|---|---|---|---|---|---|
| WT Bsu CsoR | 1 | Cu-S2 | 2.20 | 0.0023 | -3.853 | 0.070 |
| (k = 2-12Å-1) | Cu-N1 | 2.05 c | 0.0016 | [-3.853]d | ||
| Δk3χ = 12.729 | Cu-C1 | [3.04] | [0.0032] | [-3.853] | ||
| Cu-C1 | [3.08] | [0.0033] | [-3.853] | |||
| Cu-N1 | [4.21] | [0.0020] | [-3.853] | |||
| Cu-C1 | [4.24] | [0.0020] | [-3.853] | |||
| E90A Bsu CsoR | 1 | Cu-S2 | 2.20 | 0.0024 | -4.322 | 0.076 |
| (k = 2-12Å-1) | Cu-N1 | 2.05 | 0.0016 | [-4.322] | ||
| Δk3χ = 14.157 | Cu-C1 | [3.04] | [0.0032] | [-4.322] | ||
| Cu-C1 | [3.08] | [0.0033] | [-4.322] | |||
| Cu-N1 | [4.21] | [0.0020] | [-4.322] | |||
| Cu-C1 | [4.24] | [0.0020] | [-4.322] |
Shell is the chemical unit defined for the multiple scattering calculation. Subscripts denote the number of scatterers per metal. Ras is the metal-scatterer distance. σas2 is a mean square deviation in Ras. ΔE0 is the shift in E0 for the theoretical scattering functions.
Underlined numbers were fixed at the indicated value (not optimized).
Numbers in square brackets were constrained to be either a multiple of the above value (σas2) or to maintain a constant difference from the above value (Ras, ΔE0).
Both apo and Cu(I)-bound Bsu CsoR are tetramers
A preliminary characterization using Superdex 200 size exclusion chromatography on both apo and Cu(I)-bound Bsu CsoR revealed a single species roughly corresponding to a homotetramer in both cases (data not shown). To better characterize the assembly state of Bsu CsoR in solution, analytical sedimentation equilibrium and velocity ultracentrifugation experiments were carried out with apo and Cu(I)-bound Bsu CsoRs in the low micromolar monomer concentration range.
The equilibrium scans were globally fit with a single ideal species model, with representative data and fits shown for the apo- and Cu(I)-bound Bsu CsoRs in Figure 5A and 5B, respectively. For apo and Cu(I)-bound Bsu CsoR, the single species molecular weights of 41.0±0.5 kDa and 39.8±0.6 kDa, respectively, were obtained. This suggests that the assembly state of both forms of CsoR is tetrameric under these solution conditions (expected molecular weight of 45.9 kDa) and a lower limit of the dimer-tetramerization equilibrium constant is 107 (M dimer)-1. This is in full agreement with results for the Ni(II)/Co(II) sensor E. coli RcnR (14), but distinct from what we reported for Mtb CsoR, where a significant dimer-tetramer equilibrium was observed (12). One possible explanation is that the dimer-tetramer equilibrium of Mtb CsoR is influenced by the 30 amino acid C-terminal tail found only in CsoR homologs from pathogenic mycobacteria (see Figure 1).
Figure 5.

Analytical ultracentrifugation of apo and Cu(I)-bound Bsu CsoRs. (A) and (B) Representative equilibrium data with a global fit to a single ideal species model and residuals for both apo and Cu(I)-bound Bsu CsoR at 0.3 AU230, respectively. The three data sets correspond to different rotor speeds: 1) 19,300 rpm; 2) 30,600 rpm; 3) 38,700 rpm. The solid curves show a global fit to a single ideal species model (residues in upper panel). (C) and (D) Distribution of sedimentation coefficient of apo and Cu(I)-bound Bsu CsoR, respectively. All parameters derived from sedimentation velocity fits are compiled in Table 3.
To further confirm the assembly state and obtain further insights into the hydrodynamic properties of the apo- and Cu(I)-bound tetramers, sedimentation velocity experiments were carried out under the same solution conditions. Consistent with the equilibrium experiments, a single boundary was observed for all samples; quantitative analysis of these distribution plots is consistent with a single species of sedimentation coefficient 3.03 S for apo-Bsu CsoR and 3.34 S for Cu(I)-bound Bsu CsoR (Figure 5C and 5D, respectively). The fitted parameters and predicted hydrodynamic characteristics are compiled in Table 3. These data taken collectively suggest that the dominant assembly state of each form of CsoR is that of a highly asymmetric tetramer with Cu(I) binding inducing a small but measurable change in the hydrodynamic properties of the molecule (see Discussion).
Table 3.
Summary of fitted parameters derived from the sedimentation velocity experiments with Bsu CsoR
| CsoR | s × 1013 (sec) | D × 107 (cm2/sec) | RMSD | f/f0a |
|---|---|---|---|---|
| apo (0.3 OD230) | 3.05 | 6.84 | 0.0067 | 1.50 |
| apo (0.8 OD230) | 3.01 | 7.08 | 0.011 | 1.52 |
| Cu(I)-bound (0.3 OD230) | 3.34 | 6.07 | 0.0082 | 1.37 |
| Cu(I)-bound (0.8 OD230) | 3.34 | 6.10 | 0.0095 | 1.37 |
Frictional coefficients calculated from s and D upon fixing the molecular weight of the tetramer to 45.9 kDa.
Apo-Bsu CsoR binds operator DNA with 2:1 tetramer:DNA stoichiometry
It was previously shown that Bsu CsoR is capable of binding to an operator in the promoter region of copZA operon, although the stoichiometry and affinity were not investigated in detail (11). Size exclusion chromatography was first used to determine the DNA binding stoichiometry (Figure 6A). Addition of 40 μM apo-Bsu CsoR monomer to 10 μM DNA gives rise to a new peak with an elution volume of 13.7 mL, assigned to a Bsu CsoR-DNA complex, with a significant fraction of DNA. Addition of 80 μM apo-Bsu CsoR monomer to 10 μM DNA reveals that only the 13.7 mL peak is observed with no evidence of free DNA, suggesting that all the protein and DNA added form the complex. Further addition of protein does not affect the protein-DNA complex peak at 13.7 mL, with only free protein peak eluting at 15.9 mL (data not shown). This suggests that the Bsu CsoR-DNA interaction saturates at 8:1 monomer:DNA ratio corresponding to two tetramers per DNA. Similar experiments carried out with Cu(I)-bound Bsu CsoR reveals the elution of only free DNA and free protein and no protein-DNA complex peak (data not shown), which is consistent with the fact that Cu(I) binding to Bsu CsoR significantly decreases its DNA binding affinity.
Figure 6.
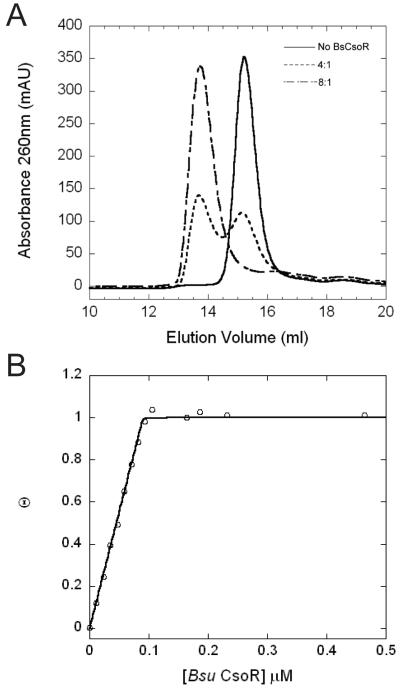
Bsu CsoR-copZA operator DNA binding stoichiometry. (A) Elution profile obtained with different Bsu CsoR:DNA ratios from a Superdex 200 column as monitored by absorption at 260nm. Conditions: 10 mM MES, 0.2 M NaCl, 2 mM DTT, pH 6.5. (B) Normalized DNA binding isotherm based on fluorescence anisotropy with 10 nM DNA in 10 mM MES, pH6.5, 0.2 M NaCl, 2 mM DTT, 25°C.
Fluorescence anisotropy experiments carried out in buffer S (0.2 M NaCl) with 2 mM DTT with 10 nM 30 bp DNA further confirm the stoichiometry. Under these conditions, apo-Bsu CsoR binds the fluorescein-labeled 30 bp DNA with very high affinity (≥109 M-1) as revealed by the stoichiometric binding curve (Figure 6B). The binding isotherm increases linearly and saturates at about 8 Bsu CsoR monomers to 1 DNA, consistent with the stoichiometry determined by size exclusion chromatography. Since free Bsu CsoR is a stable tetramer, these data are consistent with a stoichiometry of two tetramers binding to one two-fold symmetric DNA.
DNA binding affinity of apo and Cu(I)-bound Bsu CsoR
The apparent DNA binding affinity of apo-Bsu CsoR was estimated to be 2×107 M-1 by electrophoretic mobility shift assay (EMSA) in 20 mM Tris, 50 mM NaCl, 1 mM DTT at pH 8.0 (11). To determine the binding affinity quantitatively, we performed the fluorescence anisotropy experiments at 0.4 M NaCl in Buffer S. Figure 7A shows a typical normalized Bsu CsoR-DNA binding curve monitored by fluorescence anisotropy. Since only a lower limit of tetramerization constant of 107 M-1 was determined by analytical ultracentrifugation, these DNA binding data were fit using a stepwise binding model of two non-dissociable tetramers to one DNA. Apo-Bsu CsoR binds to the DNA with K1apo=3.1(±0.8) × 107 M-1 and K2apo=8.3 (±2.2) × 107 M-7, while Cu(I)-bound Bsu CsoR binds the same DNA with K1Cu=2.9(±0.4) × 106 M-1 and K2Cu≤1.0 × 105 M-1. This gives an operationally defined coupling free energy (ΔGc) ≥ 5.4 kcal/mol (Table 2). The second binding event was not observed for Cu(I)-bound protein under the experimental conditions, therefore only an upper limit for K2Cu is reported here.
Figure 7.

Normalized fluorescence anisotropy-based DNA binding isotherms of WT Bsu CsoR (A) and E90A (B) acquired in the absence (○) and presence (□) of Cu(I). Curves represent the best fit using a stepwise two tetramer DNA binding model with the fitting parameters given in Table 2.
DNA binding by E90A CsoR is not regulated by Cu(I) binding
How Cu(I) binding changes the conformation and/or assembly state of CsoR in a way that results in an allosterically inhibited conformation remains unclear; however, previous preliminary observations suggested that a highly conserved residue in the α3 helix, Glu81 in Mtb CsoR, may be crucial in driving this switch (Figure 1) (12). Therefore, to test whether Bsu CsoR shares an allosteric mechanism that is similar to that suggested for Mtb CsoR, the equivalent residue in Bsu CsoR, Glu90, was substituted with an Ala. As shown in Figure 7B, Cu(I)-bound E90A binds the 30 bp DNA with a high affinity that is similar in magnitude to that of apo-E90A CsoR, suggesting that Cu(I)-binding does not negatively regulate the DNA binding affinity of this mutant. Since the Cu(I)-binding affinity (data not shown) and the coordination environment of E90A CsoR (Figure 4) is indistinguishable from that wild-type CsoR, these data suggest that E90 plays an important role in mediating Cu(I)-dependent negative regulation of DNA binding.
The Binding of Zn(II), Co(II) and Ni(II) to Bsu CsoR
Although studies in Mtb and in M. smegmatis reveal that Cu(I) is the primary inducer of csoR-dependent gene expression, it was of interest to determine the specificity of Cu(I) binding and the degree to which other metals could allosterically regulate copZA operator-promoter binding. A Zn(II) titration using mag-fura-2 as a competition chelator shows that Bsu CsoR binds Zn(II) with KZn=1.6 (±0.1) × 108 M-1 (see Supplemental Figure S1A; Table 2). To provide insight into the coordination geometry of Zn(II), Co(II) was used as a structural surrogate for Zn(II) (17, 18). Not surprisingly, Co(II) binds to Bsu CsoR with an affinity far lower than that of Zn(II), with KCo ≤105 M-1 under the same conditions. As shown in Figure 8A, Co(II) bound Bsu CsoR shows strong ligand-to-metal charge transfer (LMCT) at 290 nm with an ε=1500 M-1cm-1 and at 335 nm with an ε=800 M-1cm-1. The d-d transition envelope centered at ≈600 nm gives an ε≈300 M-1cm-1. These data taken together are consistent with a tetrahedral or distorted tetrahedral Co(II) complex with one or two of the Cys residues as donor atoms to the Co(II). Since Co(II) is bound tetrahedrally, Zn(II) may well bind with the same coordination geometry, although this was not directly determined here.
Figure 8.
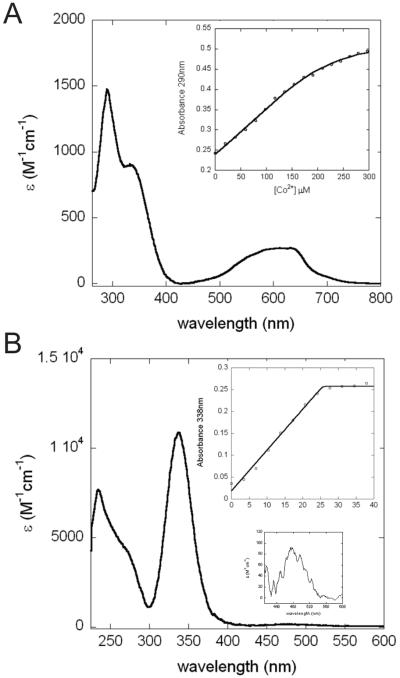
Co(II) and Ni(II) binding to Bsu CsoR. Apoprotein-subtracted molar absorptivity spectra of Co(II):Bsu CsoR mixture at 2:1 molar ratio (A) or Ni(II):Bsu CsoR mixture at 1:1 molar ratio (B). The binding isotherms shown in the insets are fitted with a simple 1:1 binding model with parameters collected in Table 2.
When apo-Bsu CsoR binds Ni(II), the UV-vis absorption spectrum shows a feature at ≈480 nm with a molar intensity of ε=100 M-1cm-1(Figure 8B and inset). This feature is consistent with a square planar or distorted square planar coordination geometry as observed previously for E. coli NikR (23) and nickel-substituted mutant retroviral-type zinc-finger peptides (25). The intense charge transfer transitions in the near ultraviolet region suggest that Cys residues are involved in coordinating Ni(II). The binding isotherm as shown in the inset of Figure 8B shows a linear increase up to about 1.2 mol equiv Ni(II) followed by a sharp transition to a plateau, revealing that Bsu CsoR binds ≈1 mol equiv Ni(II) with a binding affinity ≥107 M-1 (i.e., stoichiometrically). The binding affinity estimated by an EGTA competition experiment was determined to be KNi=3.6(±0.3) × 109 M-1 (see Table 2, Supplemental Figure S1B).
These metal binding experiments clearly show that Bsu CsoR is also capable of binding other divalent metal ions with widely different affinities and coordination geometries. To test whether these metal ions are significant allosteric negative effectors of DNA binding, fluorescence anisotropy experiments analogous to that shown in Figure 7 for Cu(I) were carried out with excess metal ion to ensure that the metal-bound form of the protein is the predominant species in solution. The fitted parameters are compiled in Table 2. Both Zn(II) and Ni(II) complexed Bsu CsoR bind to the 30 bp copZA operator DNA with affinity close to that of apo-Bsu CsoR, each resulting in only a small positive coupling free energy (Supplemental Figure S2). This is consistent with previous observations that divalent metal ions are poor regulators of DNA binding of Mtb CsoR both in vitro and in vivo, despite their high equilibrium affinities for these metals (12).
Discussion
Bsu CsoR regulates the transcription of copZA operon, which encodes two important components of the Cu homeostasis system, the Cu-chaperone CopZ and the Cu-effluxer CopA. It is widely accepted that in both eukaryotes and prokaryotes, the intracellular trafficking of copper ions is dependent on metallochaperones which reinforces the idea that there is little or no “free” or bioavailable copper ions in the cell (26-28). It is still unclear how Bsu CsoR, as a Cu-sensor, obtains its copper ion in the cell, although one strong possibility is from CopZ. Such a CopZ-dependent transfer of Cu to Bsu CsoR is analogous to the well-studied mechanism in E. hirae, where the Cu-sensor CopY acquires Cu from the Cu-chaperone CopZ (29). Under uninduced conditions, a background level of CopZ in the cell functions as a copper chelator or buffer, perhaps delivering copper to target proteins. The ratio between apo and Cu-bound CopZ may be thermodynamically and kinetically maintained in a certain window by various cellular protein-protein interactions and Cu-transfer reactions. Upon Cu stress, Cu-bound CopZ may be quickly formed, therefore making it possible to transfer Cu to Bsu CsoR, leading to derepression of the copZA operon; this, in turn, results in increased expression of apo-CopZ and CopA required to efflux excess Cu out of cytosol, bringing this ratio back into a normal or unstressed range. Cu(I)-bound Bsu CsoR may then be degraded or potentially transfer Cu to other target proteins, e.g. CopA itself, via a ligand exchange reaction (30).
Bsu CsoR and Mtb CsoR share very high amino acid sequence identity. In particular, all three proposed Cu ligands are conserved as are two proposed “second shell” residues, corresponding to Tyr35 (Tyr44 in Bsu) and Glu81 (Glu90 in Bsu) in Mtb CsoR. The major difference between the two CsoRs is that Mtb CsoR contains a unique ≈30 amino acid C-terminal tail which is missing in the previously solved crystal structure of Cu(I)-CsoR from Mtb (Figure 1) (12). Aside from this, these two CsoRs possess very similar biochemical and biophysical properties. Both coordinate 1 monomer mol equiv of Cu(I) with very high affinity to form a trigonal planar S2N coordination geometry with very similar Cu-S distances. It is also the case that Bsu CsoR is a stable tetramer in the low micromolar (monomer) concentration range, while a dimer-tetramer equilibrium was observed in previous analytical ultracentrifugation studies carried out on full-length Mtb CsoR (12). Adjacent C2-symmetric dimers in the crystal structure of Cu(I)-bound Mtb CsoR pack against one another to form a tetramer with D2 rotational symmetry. In this configuration of the tetramer, the C-terminal tail of Mtb CsoR would be positioned at the dimer-dimer interface; it therefore seems possible that the flexible C-terminal tail may destabilize the Mtb CsoR tetramer toward dimer. This has yet to be investigated systematically.
We find no significant differences in assembly states of the apo and Cu(I)-bound forms of Bsu CsoR, which are both tetrameric under the conditions investigated. Therefore, only a lower limit of the tetramerization constant (Ktet) of about 107 (M dimer)-1 could be estimated from these data. However, differences in Ktet in the nanomolar concentration range cannot be ruled out by these data; if this is the case, they may be partly responsible for the different DNA binding affinities reported here for apo- and Cu(I)-CsoR. On the other hand, the sedimentation velocity experiments show a small increase in sedimentation coefficient in Cu(I)-bound Bsu CsoR, suggesting a conformation with a smaller frictional coefficient, thus more spherical relative to the apoprotein (Table 3). However, this change may not play a primary role in allosteric regulation of DNA binding because a similar change in sedimentation coefficient appears to characterize the E90A mutant as well (data not shown).
Understanding what happens when a particular metal sensor binds the “wrong” metal is just as important as understanding how the cognate metal ion drives regulation of gene expression (31). We show here that the metal binding site of Bsu CsoR can adopt a range of distinct coordination numbers and geometries upon binding different metal ions. Co(II), and by inference Zn(II), adopts a tetrahedral or distorted tetrahedral geometry while Ni(II) appears to form a square planar or distorted square planar geometry. Each complex incorporates one or both Cys residues in Bsu CsoR; in fact, it is formally possible that each employs all three Cu(I) ligands while adding a fourth ligand, perhaps from the N-terminal region like in RcnR (14), or from solvent. In any case, the Co(II) and Ni(II) coordination geometries are clearly distinct from that of RcnR, where each metal adopts an octahedral or pseudo-octahedral complex (14). Strikingly, while the binding of Cu(I) stabilizes an allosterically inhibited conformation of Bsu CsoR, neither Zn(II) nor Ni(II) is capable of strongly regulating the DNA binding, despite their high equilibrium affinities (albeit ≥10 orders of magnitude smaller than KCu) (Table 1). In fact, KNi for CsoR may well be comparable to KNi for the bona fide Ni/Co sensor E. coli RcnR; in contrast, KCo is at least 104-smaller for Bsu CsoR (KZn has not been reported for RcnR) (14). These features are consistent with the emerging theme that formation of the “native” coordination geometry is most closely linked to biological metalloregulation, rather than absolute metal binding affinity (18, 32, 33). This, in turn, suggests that specific features of trigonal planar Cu(I) coordination complex in CsoR may organize a “second coordination shell” of interactions used to drive defined conformational changes that are linked to Cu(I)-mediated derepression of gene expression (12, 17).
One strong candidate for propagating this structural change in Cu(I)-CsoRs is the Nε face of His70 (His61 in Mtb CsoR). The crystal structure of Mtb CsoR reveals that this face of the His61 imidazole ring is in close proximity to the side chains of both Glu81 and Tyr35′ in Mtb CsoR (12) which correspond to Glu90 and Tyr44′ in Bsu CsoR. Glu90 is a highly conserved residue in the α3 helix of all Cu(I)-specific CsoRs (Figure 1), and we show here that an Ala substitution abolishes negative regulation of DNA binding without significantly interfering with the Cu(I) affinity or coordination geometry. We note that the significant quenching of the steady-state tyrosine fluorescence upon Cu(I) binding is consistent with a tyrosinate-like species, that might form as a result of hydrogen bonding of its hydroxyl proton with an as yet unidentified acceptor, an excellent candidate for which is Glu90 (34). Interestingly, both of these conserved residues have also been shown to be crucial for allosteric regulation in Mtb CsoR (Ma, Z. and Giedroc, D. P., unpublished observations).
One model for negative allosteric regulation of DNA binding of CsoR by Cu(I) posits that the α3 helix is only loosely associated with the α2′ helix of four-helical dimer bundle (α1-α2-α1′-α2′) within the dimer and/or tetramer in the absence of Cu(I), and may in fact physically interact with the adjacent tetramer when bound to the DNA, thereby stabilizing an octameric assembly state on the DNA. Cu(I) binding to this species may reorient and strongly stabilize the intramolecular α3-α2′ interface within a tetramer, leading to a conformation that is not optimized for high affinity DNA binding and/or forms a limiting 1:1 tetramer:DNA complex that is nonfunctional for repression. Uninducing metal ions, e.g., Ni(II) and Zn(II), while detectably regulatory (see Table 2) do not block formation of the repressing 2:1 tetramer:DNA complex and are therefore nonfunctional; this model is consistent with the DNA binding characteristics of each metalloderivative of Bsu CsoR reported here. Further biochemical and structural support for this proposed mechanism of regulation is required.
Supplementary Material
Acknowledgements
We thank Dr. John Helmann, Cornell University, for the gift of the overexpression plasmid for Bsu CsoR and Dr. Todd Stone, Indiana University, for help in analyzing the analytical ultracentrifugation experiments. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program.
This work was supported by grants from the National Institutes of Health to D. P. G. (GM042569) and R. A. S. (GM042025).
Abbreviations used are as follows
- CsoR
copper-sensitive operon repressor
- DTNB
5, 5′-dithiobis-(2-nitrobenzoic acid)
- mag-fura-2
2-[6-[bis(carboxymethyl)amino]-5-(carboxymethoxy)-2-benzofuranyl]-5-oxazolecarboxylic acid
REFERENCES
- 1.Davis AV, O’Halloran TV. A place for thioether chemistry in cellular copper ion recognition and trafficking. Nat Chem Biol. 2008;4:148–151. doi: 10.1038/nchembio0308-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Solioz M, Stoyanov JV. Copper homeostasis in Enterococcus hirae. FEMS Microbiol Rev. 2003;27:183–195. doi: 10.1016/S0168-6445(03)00053-6. [DOI] [PubMed] [Google Scholar]
- 3.Dong J, Atwood CS, Anderson VE, Siedlak SL, Smith MA, Perry G, Carey PR. Metal binding and oxidation of amyloid-beta within isolated senile plaque cores: Raman microscopic evidence. Biochemistry. 2003;42:2768–2773. doi: 10.1021/bi0272151. [DOI] [PubMed] [Google Scholar]
- 4.Meloni G, Sonois V, Delaine T, Guilloreau L, Gillet A, Teissie J, Faller P, Vasak M. Metal swap between Zn7-metallothionein-3 and amyloid-[beta]-Cu protects against amyloid-[beta] toxicity. Nat Chem Biol. 2008;4:366–372. doi: 10.1038/nchembio.89. [DOI] [PubMed] [Google Scholar]
- 5.Francis MS, Thomas CJ. Mutants in the CtpA copper transporting P-type ATPase reduce virulence of Listeria monocytogenes. Microb Pathog. 1997;22:67–78. doi: 10.1006/mpat.1996.0092. [DOI] [PubMed] [Google Scholar]
- 6.Spagnolo L, Toro I, D’Orazio M, O’Neill P, Pedersen JZ, Carugo O, Rotilio G, Battistoni A, Djinovic-Carugo K. Unique features of the sodC-encoded superoxide dismutase from Mycobacterium tuberculosis, a fully functional copper-containing enzyme lacking zinc in the active site. J Biol Chem. 2004;279:33447–33455. doi: 10.1074/jbc.M404699200. [DOI] [PubMed] [Google Scholar]
- 7.Ward SK, Hoye EA, Talaat AM. The Global Responses of Mycobacterium tuberculosis to Physiological Levels of Copper. J. Bacteriol. 2008;190:2939–2946. doi: 10.1128/JB.01847-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Magnani D, Barre O, Gerber SD, Solioz M. Characterization of the CopR Regulon of Lactococcus lactis IL1403. J. Bacteriol. 2008;190:536–545. doi: 10.1128/JB.01481-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robinson NJ. A bacterial copper metallothionein. Nat Chem Biol. 2008;4:582–583. doi: 10.1038/nchembio1008-582. [DOI] [PubMed] [Google Scholar]
- 10.Gold B, Deng H, Bryk R, Vargas D, Eliezer D, Roberts J, Jiang X, Nathan C. Identification of a copper-binding metallothionein in pathogenic mycobacteria. Nat Chem Biol. 2008;4:609–616. doi: 10.1038/nchembio.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smaldone GT, Helmann JD. CsoR regulates the copper efflux operon copZA in Bacillus subtilis. Microbiology. 2007;153:4123–4128. doi: 10.1099/mic.0.2007/011742-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu T, Ramesh A, Ma Z, Ward SK, Zhang L, George GN, Talaat AM, Sacchettini JC, Giedroc DP. CsoR is a novel Mycobacterium tuberculosis copper-sensing transcriptional regulator. Nat Chem Biol. 2007;3:60–68. doi: 10.1038/nchembio844. [DOI] [PubMed] [Google Scholar]
- 13.Iwig JS, Rowe JL, Chivers PT. Nickel homeostasis in Escherichia coli - the rcnR-rcnA efflux pathway and its linkage to NikR function. Mol Microbiol. 2006;62:252–262. doi: 10.1111/j.1365-2958.2006.05369.x. [DOI] [PubMed] [Google Scholar]
- 14.Iwig JS, Leitch S, Herbst RW, Maroney MJ, Chivers PT. Ni(II) and Co(II) sensing by Escherichia coli RcnR. J Am Chem Soc. 2008;130:7592–7606. doi: 10.1021/ja710067d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuzmic P. Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal Biochem. 1996;237:260–273. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- 16.Xiao Z, Loughlin F, George GN, Howlett GJ, Wedd AG. C-terminal domain of the membrane copper transporter Ctr1 from Saccharomyces cerevisiae binds four Cu(I) ions as a cuprous-thiolate polynuclear cluster: sub-femtomolar Cu(I) affinity of three proteins involved in copper trafficking. J Am Chem Soc. 2004;126:3081–3090. doi: 10.1021/ja0390350. [DOI] [PubMed] [Google Scholar]
- 17.Liu T, Chen X, Ma Z, Shokes J, Hemmingsen L, Scott RA, Giedroc DP. A CuI-Sensing ArsR Family Metal Sensor Protein with a Relaxed Metal Selectivity Profile. Biochemistry. 2008;47:10564–10575. doi: 10.1021/bi801313y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pennella MA, Shokes JE, Cosper NJ, Scott RA, Giedroc DP. Structural elements of metal selectivity in metal sensor proteins. Proc Natl Acad Sci U S A. 2003;100:3713–3718. doi: 10.1073/pnas.0636943100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brookes E, Demeler B. Parallel computational techniques for the analysis of sedimentation velocity experiments in UltraScan. Colloid & Polymer Science. 2008;286:139–148. [Google Scholar]
- 20.Demeler B, Brookes E. Monte Carlo analysis of sedimentation experiments. Colloid & Polymer Science. 2008;286:129–137. [Google Scholar]
- 21.Brookes E, Demeler B. Analytical Ultracentrifugation. VIII. 2006. Genetic Algorithm Optimization for Obtaining Accurate Molecular Weight Distributions from Sedimentation Velocity Experiments; pp. 33–40. [Google Scholar]
- 22.Tan X, Kagiampakis I, Surovtsev IV, Demeler B, Lindahl PA. Nickel-Dependent Oligomerization of the Alpha Subunit of Acetyl-Coenzyme A Synthase/Carbon Monoxide Dehydrogenase. Biochemistry. 2007;46:11606–11613. doi: 10.1021/bi7014663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang SC, Dias AV, Bloom SL, Zamble DB. Selectivity of metal binding and metal-induced stability of Escherichia coli NikR. Biochemistry. 2004;43:10018–10028. doi: 10.1021/bi049405c. [DOI] [PubMed] [Google Scholar]
- 24.Kau LS, Spira-Solomon DJ, Penner-Hahn JE, Hodgson KO, Solomon EI. X-ray absorption edge determination of the oxidation-state and coordination-number of copper - application to the type-3 site in rhus-vernicifera laccase and its reaction with oxygen. J. Am. Chem. Soc. 1987;109:6433–6442. [Google Scholar]
- 25.Chen X, Chu M, Giedroc DP. Spectroscopic characterization of Co(II)-, Ni(II)-, and Cd(II)-substituted wild-type and non-native retroviral-type zinc finger peptides. J Biol Inorg Chem. 2000;5:93–101. doi: 10.1007/s007750050012. [DOI] [PubMed] [Google Scholar]
- 26.Rae TD, Schmidt PJ, Pufahl RA, Culotta VC, O’Halloran TV. Undetectable intracellular free copper: the requirement of a copper chaperone for superoxide dismutase. Science. 1999;284:805–808. doi: 10.1126/science.284.5415.805. [DOI] [PubMed] [Google Scholar]
- 27.Rosenzweig AC, O’Halloran TV. Structure and chemistry of the copper chaperone proteins. Curr Opin Chem Biol. 2000;4:140–147. doi: 10.1016/s1367-5931(99)00066-6. [DOI] [PubMed] [Google Scholar]
- 28.Rosenzweig AC. Copper delivery by metallochaperone proteins. Acc Chem Res. 2001;34:119–128. doi: 10.1021/ar000012p. [DOI] [PubMed] [Google Scholar]
- 29.Cobine PA, George GN, Jones CE, Wickramasinghe WA, Solioz M, Dameron CT. Copper transfer from the Cu(I) chaperone, CopZ, to the repressor, Zn(II)CopY: metal coordination environments and protein interactions. Biochemistry. 2002;41:5822–5829. doi: 10.1021/bi025515c. [DOI] [PubMed] [Google Scholar]
- 30.Banci L, Bertini I, Cantini F, Felli IC, Gonnelli L, Hadjiliadis N, Pierattelli R, Rosato A, Voulgaris P. The Atx1-Ccc2 complex is a metal-mediated protein-protein interaction. Nat Chem Biol. 2006;2:367–368. doi: 10.1038/nchembio797. [DOI] [PubMed] [Google Scholar]
- 31.Waldron KJ, Robinson NJ. How do bacterial cells ensure that metalloproteins get the correct metal? Nat Rev Micro. 2009;7:25–35. doi: 10.1038/nrmicro2057. [DOI] [PubMed] [Google Scholar]
- 32.Pennella MA, Arunkumar AI, Giedroc DP. Individual metal ligands play distinct functional roles in the zinc sensor Staphylococcus aureus CzrA. J Mol Biol. 2006;356:1124–1136. doi: 10.1016/j.jmb.2005.12.019. [DOI] [PubMed] [Google Scholar]
- 33.Giedroc DP, Arunkumar AI. Metal sensor proteins: nature’s metalloregulated allosteric switches. Dalton Trans. 2007:3107–3120. doi: 10.1039/b706769k. [DOI] [PubMed] [Google Scholar]
- 34.Dietze EC, Wang RW, Lu AYH, Atkins WM. Ligand Effects on the Fluorescence Properties of Tyrosine-9 in Alpha 1-1 Glutathione S-Transferase. Biochemistry. 1996;35:6745–6753. doi: 10.1021/bi9530346. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.