

Abstract
Life-long addition and elimination of neurons within the adult olfactory epithelium and olfactory bulb allows for adaptive structural responses to sensory experience, learning, and recovery after injury. The interdependence of the two structures is highlighted by the shortened life span of sensory neurons deprived of bulb contact, and has prompted the hypothesis that trophic cues from the bulb contribute to their survival. The specific identity and source of these signals remain unknown. To investigate the potential role of target neurons in this support, we employed a neurotoxic lesion to selectively remove them while preserving the remaining nerve projection pathway, and examined the dynamics of sensory neuron proliferation and survival. Pulse-labeling of progenitors with bromodeoxyuridine showed that, as with surgical bulb removal, increased apoptosis in the epithelium triggered accelerated production of new neurons after chemical depletion of target cells. Rather than undergoing premature death, a large subpopulation of these neurons survived long term. The combination of increased proliferation and extended survival resulted in essentially normal numbers of new sensory neurons surviving for as long as 5 weeks, with an accompanying restoration of olfactory marker protein expression. Changes in neurotrophic factor expression levels as measured by quantitative polymerase chain reaction (Q-PCR), and in bulb cell populations, including the addition of new neurons generated in the subventricular zone, were observed in the injured bulb. These data indicate that olfactory sensory neurons can adapt to reductions in their normal target field by obtaining sufficient support from remaining or alternative cell sources to survive and maintain their projections.
Keywords: olfactory bulb, neurotrophic factor, apoptosis, regeneration, subventricular zone
INTRODUCTION
Sensory neurons of the adult olfactory epithelium (OE) normally undergo continuous turnover throughout life. Most have an average lifespan of only 30-60 days due to their vulnerable, exposed location in the nasal cavity, at which time they undergo caspase-mediated programmed cell death (Farbman, 1990; Cowan and Roskams, 2002). New neurons are generated from multipotent stem cells located in the basal compartment of the epithelium, along a defined lineage (Calof et al., 1998; Schwob, 2002; Beites et al., 2005). A number of critical regulatory signals that control normal epithelial neurogenesis have been identified, and these act locally on cells at specific stages within the lineage to control the size of the neuronal population (Calof et al., 1998; Schwob, 2002; Wu et al., 2003; Kawauchi et al., 2004). Damage that produces widespread death of olfactory sensory neurons (OSNs) leads to a compensatory increase in neurogenesis as the balance of these same regulatory pathways shifts in an attempt to replenish the population and maintain olfactory function (Bauer et al., 2003; Leung et al., 2007; Iwai et al., 2008).
Ongoing neuronal replacement and afferent turnover within the olfactory pathway leads to preservation of many developmental features in the mature system. Adult-generated sensory neurons elaborate axons that grow out toward the olfactory bulb in search of synaptic targets. Immature neurons that successfully establish synapses with bulb neurons survive and mature, while others, presumably less successful in this endeavor, undergo apoptosis. At any given time, the population of sensory neurons undergoing normal apoptosis in the adult OE includes both mature neurons that have reached the end of their lifespan, and this subpopulation of immature neurons (Holcomb et al., 1995; Mahalik, 1996; Cowan and Roskams, 2002). The generation of immature neurons in excess of the numbers that will ultimately mature and survive is reminiscent of developmental apoptosis characteristic of neuronal populations that compete for limited levels of target-derived trophic factors (Burek and Oppenheim, 1996), suggesting that the adult olfactory bulb provides retrograde trophic support that works in combination with local mechanisms in the OE to regulate sensory neuron lifespan.
Experimental evidence that the bulb provides trophic support for OSNs comes from in vivo manipulations that interrupt connections with the epithelium. OSNs undergo rapid apoptosis when their axons are severed either by nerve transection or by surgical removal of the bulb (bulbectomy), which also severs these axons (Holcomb et al., 1995; Schwob, 2002; Cowan and Roskams, 2002; Shetty et al., 2005). Subsequently, compensatory neurogenesis takes place, and in nerve-transected animals, the OE is restored over time as the bulb is reinnervated (Costanzo, 1984, Christensen et al., 2001). However, bulbectomy leads to the formation of scar tissue that prevents new axons from reaching the CNS, and the OE remains permanently atrophied under these conditions (Costanzo, 1984; Schwob et al., 1992). Apoptosis in the chronically disconnected OE remains elevated, as does progenitor proliferation and the number of immature neurons expressing growth associated protein-43 (GAP-43; Schwob et al., 1992; Holcomb et al., 1995). Neuronal lifespan is considerably shortened, with almost 90% of new cells dying within two weeks of their birth, and permanent depletion of the mature OSN population (Schwob et al., 1992; Carr and Farbman, 1993). Even partial loss of neuronal targets, achieved by axotomizing bulb mitral cells, stimulates cell death and proliferation in the OE (Weiler and Farbman, 1999). The possibility that trophic mechanisms limit the survival of targetless OSNs is supported by bulbectomy studies of transgenic mice with altered expression of pro- or anti-apoptotic proteins, or caspases (Cowan et al., 2001; Robinson et al., 2003; Hayward et al., 2004). For example, OSNs born after bulbectomy live longer in mice in which Bcl-2, an anti-apoptotic factor that allows some neurons to withstand trophic factor deprivation (Allsopp et al., 1993) is constitutively expressed by mature OSNs. In comparison to non-transgenic mice, numbers of mature cells in the targetless OE recover to nearly normal levels over time (Hayward et al., 2004).
The identity of the putative target-derived signal is not known, but previous studies have detailed the different types of neurotrophic factors synthesized in the mature bulb (reviewed by Mackay-Sim and Chuah, 2000). Neuronal populations contacted by sensory axons express a variety of factors including members of the nerve growth factor (NGF), insulin-like growth factor (IGF), transforming growth factor (TGF), platelet-derived growth factor (PDGF) and fibroblast growth factor (FGF) families. Olfactory ensheathing glia (OEG) within the olfactory nerve and bulb nerve layer are also a source of growth factors, including ciliary neurotrophic factor (CNTF), insulin-like growth factor-1 (IGF-1) and fibroblast growth factor-2 (FGF2) (MacKay-Sim and Chuah, 2000). Survival-promoting effects on cultured OSNs have been demonstrated for individual substances within several of these classes of factors, including PDGFs (Newman et al., 2000) and neurotrophins (Holcomb et al., 1995) and co-culture of OSNs with bulb cells similarly promotes their maturation and survival (Grill and Pixley, 1997).
To assess the specific contribution of target neurons to sensory neuron survival mechanisms, we eliminated olfactory bulb neurons in vivo using N-methyl-D-aspartate (NMDA) infusion, a strategy used by Sofroniew and colleagues (1990) to examine the trophic dependence of adult cholinergic forebrain neurons on hippocampal targets, and compared NMDA lesion effects with bulbectomy. NMDA rapidly kills bulb neurons, but leaves the epithelium, the olfactory nerve pathway, and the bulb’s OEG population intact. We hypothesized that dependence on target neurons would leave OSNs vulnerable to bulb NMDA lesion, and reduce their long-term survival in much the same way as bulbectomy. Our analyses included light and electron microscopic examination of degenerative changes in deprived OSNs and their axons within the neuron-depleted bulb, quantification of apoptosis, and measurement of OSN life-span using pulse labeling of progenitors with 5-bromo-2′-deoxyuridine (BrdU). To compare trophic factor expression levels in normal and lesioned target tissue, we aditionally carried out quantitative PCR (Q-PCR). We report here that sensory neurons demonstrated surprising resistance to target field depletion, and that changes in the cellular composition of the lesioned bulb were accompanied by alterations in the expression of specific neurotrophic factors.
MATERIALS AND METHODS
Surgeries and histology
All surgeries were carried out using Adult male Wistar rats (185-225g) from Charles River Laboratories. Procedures were performed in accordance with National Institutes of Health guidelines and were approved by the Florida Atlantic University Institutional Animal Care and Use Committee. Rats were anesthetized with ketamine hydrochloride/xylazine (150 mg/kg and 75mg/kg; i.p.), and placed in a stereotaxic apparatus. N-methyl-D-aspartic acid (NMDA; Tocris, 12 mg/ml, pH 7.0, 1.5 μl), or an equivalent volume of sterile saline, was injected at two locations in the right olfactory bulb using a Hamilton syringe with a 25 gauge needle. The first injection was made 6.5-7.5 mm anterior to Bregma and the second was placed 1.1 mm anterior to this. Both were positioned 1 mm lateral (right) of the midsagittal suture, and 2 mm ventral to the bulb surface. Infusions were made over 10 min, and the needle remained in place for an additional 10 min. Rats were euthanized with sodium pentobarbital (150 mg/kg, i.p.) at 24, 32, 48, 72 hours, 6 days, 2, 3, 5, or 8 weeks after surgery (n=4-7 per time point). Animals were perfused with phosphate buffered saline (PBS) followed by 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.3). Nasal cavities were postfixed, decalcified in 0.37 M ethylenediamine tetraacetic acid (pH 7.5), and cryoprotected in 20% sucrose. Tissue was embedded in OCT compound (Sakura Finetek, Torrance, CA), and frozen in isopentane (-55°C). To evaluate lesions, sections through the bulbs and piriform cortex (20-25 μm) were stained with cresyl violet, FluoroJade (Histo-Chem Inc, Jefferson, AR; Schmued et al., 1997), and with the antibodies listed in Table 1. Controls included omission of primary antibodies from the immunostaining procedures.
Table 1.
Summary of primary antibodies used: *Details of antibody characterization shown below.
| Directed against: | Immunogen: | Source and species: | Dilution |
|---|---|---|---|
| olfactory marker protein (OMP) |
purified goat OMP | Wako Chemicals (Richmond, VA), goat polyclonal, #544-10001 |
1:7000 |
| growth-associated protein-43 (GAP-43) |
purified protein from feline brain |
Millipore/Chemicon (Temecula, CA), rabbit polyclonal, #AB5312 |
1:500 |
| NeuN | purified mouse brain nuclei |
Millipore (Temecula, CA) mouse monoclonal, # MAB377 |
1:200 |
| glial fibrillary acidic protein (GFAP) |
purified bovine GFAP | Dako USA (Carpinteria, CA) rabbit polyclonal, #Z0334 |
1:500 |
| tyrosine hyroxylase (TH) |
purified TH from rat pheochromocytoma |
Millipore (Temecula, CA) rabbit polyclonal, #AB152 |
1:500 |
| Fractin | C-term inal end of 244 AA beta-actin fragment produced by caspase-3 cleavage |
Millipore (Temecula, CA) rabbit polyclonal, #AB3150 |
1:2000 |
| bromodeoxy- uridine (BrdU) |
bromodeoxyuridine | Accurate Chemical (Westbury, NY) rat monoclonal, #OBT0030 |
1:50 |
| Ki-67 | the synthetic peptide TPKEKAQALEDLAG FKELFQT |
Drs. SJ Watson and C Isgor rabbit polyclonal |
1:10,000 |
The OMP antibody was originally generated by Dr. F. Margolis and recognizes a single band of 16.5 kDa on Western blots of protein isolated from mouse olfactory epithelium (Keller and Margolis, 1976). It specifically labels mature rodent OSNs, as charaterized by Keller and Margolis (1975). Within the OE, growth-associated protein-43 (GAP-43) is specifically expressed by immature OSNs elaborating axons, as described in detail by Biffo et al. (1990), and the GAP-43 antibody used here detects a single band of ∼50 kDa on Western blots of protein isolated from developing visual cortex. The GFAP antibody recognizes the well-known intermediate filament protein expressed by astrocytes, and detects a band of ∼51 kDa of Western blots of rodent brain extracts. Mouse monoclonal antibody to NeuN recognizes 2-3 bands (46-48 kDa) on blots of nuclear protein isolated from mouse brain, and labels the nucleus of most brain neurons, but not mitral cells. Tyrosine hydroxylase (TH) is expressed by subpopulations of bulb tufted and periglomerular cells and the antibody detects a single band of ∼60 kDa on brain tissue blots. Fractin, the N-terminal fragment of actin, is generated by cleavage of beta-actin by activated caspase-3. The antibody detects this 32 kDa fragment, but not full-length beta-actin, and labels apoptotic cells, including neurons and their processes (Suurmeijer et al., 1999).
Three lesioned rats also received bilateral bulbar injections of biotinylated dextran 2 weeks after NMDA, at the same right side coordinates (4 μl each site, 100 mg/ml, MW 10,000, Invitrogen, Carlsbad, CA; left coordinates=7.0 mm and 8.1 mm anterior to Bregma, 1.2 mm left of midline, and 2.0 mm ventral from the tissue surface). These rats were euthanized 10 days later and sections through the anterior piriform cortex were stained using avidin-biotin-HRP (1:1000, Elite ABC kit, Vector Laboratories, Burlingame, CA) followed by chromagen development with IMPACT-DAB peroxidase substrate (Vector Labs). An additional cohort of rats (n=46), anesthetized as above, underwent right olfactory bulbectomy via surgical aspiration, followed by gelfoam packing of the wound cavity, or sham surgery. These rats were euthanized and perfused at the same survival intervals up to three weeks (n=4-5 NMDA and 2 shams per time point). OE tissue was processed for TUNEL and in situ hybridization. Photomicrographs were collected using an Olympus AX70 microscope, with images adjusted for brightness and contrast using Adobe Photoshop.
Terminal transferase mediated-UTP-nick end labeling of fragmented DNA (TUNEL)
Serial coronal cryosections through the nasal cavities (20 μm) incubated as described (Ardiles et al., 2007) for 2 hrs at 37°C, using 200U/ml of terminal transferase (New England Biolabs, Ipswich, MA) and 0.5 μl/ml digoxigenin-11-dUTP (Roche Applied Science). Controls included omission of terminal transferase from the TUNEL reaction, and treatment of some sections with DNAse (1000U/ml) for 25 min at 37°C, prior to TUNEL labeling. Immunodetection of DIG employed sequential incubation in mouse antibody to DIG, which recognizes both free and bound DIG (#11 333 062 910; Roche Applied Science), followed by donkey anti-mouse IgG conjugated to AlexaFluor 555 (1:1000, Invitrogen, Carlsbad, CA). TUNEL-labeled cells were counted on each side of the epithelium in eight representative sections per animal, sampled at equivalent levels of the nasal cavity (based on the presence of turbinates), spaced at ∼280 μm intervals. Counts in tissue from NMDA- and saline-treated rats were made throughout the full extent of the epithelium at 100× objective magnification using an Olympus AX70 fluorescence microscope. For time course comparisons, cell counts for bulbectomized rats were made along 2 mm of the left and right OE lining the nasal septum, from the dorsal recess extending ventrally. Only densely labeled intact nuclei were counted. Group means (+/- standard error of the mean, SEM) were generated from cell counts per mm (bulbectomy) or per section (NMDA) from the left and right OE for each animal (n=4-6) to characterize patterns of apoptosis as a function of survival time for each type of lesion. Counts were corrected using the Abercrombie method (Abercrombie, 1946). TUNEL+ nuclei were measured microscopically from one section within 240 μm of the left and the right septum of rats killed at 24 hrs post-bulbectomy (n=4). Nuclei were assumed to be spherical and the mean diameter of labeled nuclei was 4.05 +/- 0.4 μm. Section thickness was 20 μm, and the calculated correction factor was 0.83. For double-labeling, sections were processed for TUNEL as described, with goat antibody to OMP (1:7000) included in the DIG antibody incubation, followed by detection with AlexaFluor 488-labeled secondary IgG (1:1000, Invitrogen). Confocal images were collected using a BioRad Radiance 2100 laser scanning confocal microscope.
In situ hybridization, densitometry, and epithelial depth
Sections through the OE were hybridized with 35S-labeled OMP or GAP-43 cRNA and processed for film autoradiography and quantitative densitometry as described (Ardiles et al., 2007). Sections of olfactory bulbs from injected rats were hybridized with 35S-labeled neuropeptide Y (NPY) cRNA to evaluate survival of olfactory ensheathing glia. The 218 base OMP riboprobe was complementary to positions 692-900 of the GenBank sequence NM-012616, and the GAP-43 cRNA was complementary to positions 334-897 of GenBank sequence M16228. The NPY cRNA comprised 551 bases complementary to the full coding sequence of the mature peptide (GenBank sequence M20373), and recognizes preproNPY mRNA (∼800 bp) in Northern blots of rat brain tissue (Higuchi et al., 1988). Labeling specificity was verified by hybridization of sense RNA sequences and changes in hybridization density ipsilateral to lesion were expressed as percent of contralateral measures. We did not observe specific labeling patterns when sections were incubated with the control, radiolabeled sense RNA sequences. Sections processed for in situ hybridization were counterstained with neutral red and examined at 60X objective magnification to measure the thickness of the olfactory epithelium from the basal lamina to the lumenal surface using an eyepiece micrometer. Measurements were made at 8-12 equivalent locations along the left and right sides of the septum from eight serial sections per animal and lesion effects expressed as percentage thickness of the untreated side.
Ki-67 Immunohistochemistry
Saline- and NMDA-treated rats (n=4 each) were overdosed with sodium pentobarbital (150 mg/kg; i.p.) at 3 weeks post-lesion, decapitated, and fresh nasal tissue collected. OE sections (14 μm) were processed for Ki-67 immunostaining using a rabbit antibody to Ki-67 as described (Table 1, Ardiles et al., 2007). Ki-67 is a nuclear protein expressed by actively proliferating cells, and is not expressed by quiescent (G0) or postmitotic cells. The antibody was generated against a synthetic peptide corresponding to amino acids 1213-1233 of the GenBank sequence CAH73169. It recognizes a band of ∼375 kDa on Western blots and labels proliferating cells in the adult rodent brain (Isgor and Watson, 2005). Selected sections were counterstained with methyl green. Counts of labeled nuclei in equivalent portions of the left and right OE were made at 100X magnification along 2 mm of the nasal septum, from the dorsal recess extending ventrally. All counts were restricted to the deepest half of the OE, and were corrected using the Abercrombie method. The mean diameter of Ki-67+ nuclei was measured within 240 μm of both the left and right septal OE in one section per saline-treated rat (n=4), and was found to be 4.51 +/-0.5 μm. Eight sections per rat, spaced at ∼280 μm intervals, were quantified to generate mean numbers of labeled cells per mm, per animal, and these numbers were corrected using a factor of 0.76 (Abercrombie, 1946). Group means were calculated from these values and Student’s t-test was used for paired comparisons.
Bromodeoxyuridine labeling
Three weeks after bulb injections, rats received 200 mg/kg BrdU (Roche Applied Science) by multiple i.p. injections (10mg/ml in 0.007 N NaOH made in sterile saline) administered over 3 hrs. Animals were euthanized at 4 days, 2 weeks or 5 weeks after BrdU treatment (n=4 NMDA- and 2 saline-injected per time point). Euthanasia, perfusion, and sectioning were carried out as described for TUNEL. Sections through the OE (20 μm) rinsed in Tris-buffered saline (TBS, pH 7.5), followed by incubation in 0.6% H2O2 in TBS for 10 min. Tissue was treated with 50% formamide in salt sodium citrate buffer for 2 hrs at 65°C, followed by incubation in 2N HCl for 30 min at 37°C. Sections were exposed to 0.1% trypsin and then treated with 0.1 M sodium borate (pH 8.5). Tissue blocked in 5% normal serum in TBS containing 0.3% Triton-X-100, prior to incubation for 48 hrs in rat anti-BrdU antibody (1:50; Accurate Chemical, Westbury, NY) in TBS containing 5% NRS. This monoclonal BrdU antibody detects free and DNA-bound bromodeoxyuridine. Sections incubated in biotinylated rabbit anti-rat IgG (1:100; Vector Laboratories), followed by avidin-biotin-HRP treatment (Vector Elite ABC kit). Chromagen was developed using IMPACT-DAB peroxidase substrate (Vector), with methyl green used to counterstain selected sections. Counts of BrdU-labeled nuclei at 4 days were made along 2 mm of both sides of the OE lining the septum, from 8 spaced sections. Counts at 2-5 weeks were made throughout the entire left and right OE from each of 8 serial sections at 100X (matched locations based on turbinates). Stained sustentacular cells near the surface of the epithelium were not included, and the full length of the OE in sampled sections was measured using a Mac G4 computer-based imaging system and NIH Image J software. The size of stained nuclei within matched 240 μm sample regions in the left and right septal OE of NMDA-treated rats (5 wk survival, n=4) was measured microscopically and nuclei were assumed to be spherical. Mean diameter was 6.03 +/- 0.5 μm, and a correction factor of 0.77 was applied to cell counts (Abercrombie, 1946). For double-labeling, tissue was processed as above, and goat anti-OMP was added to the anti-BrdU incubation, followed by AlexaFluor-488 and -594 secondary IgG incubations (1:1000; Invitrogen). Counts of BrdU-labeled mature neurons were made at 60-100X throughout the entire OE in 8 serial sections per animal at 5 weeks after BrdU, and confocal imaging was carried out as described for TUNEL-double labeling. Sections through the olfactory bulbs of rats treated with BrdU were also examined for the presence of double-labeled cells 3 and 5 weeks later, in combination with immunolocalization of tyrosine hydroxylase (TH; 1:500; Chemicon) and NeuN (1:200; Chemicon).
Electron microscopy
Three weeks after surgery, rats treated with NMDA (n=4) or saline (n=2) were euthanized with sodium pentobarbital (150 mg/kg), and perfused intracardially with PBS followed by 4% paraformaldehyde and 1% gluteraldehyde in 0.1 M phosphate buffer (pH 7.3). Brains remained within the cranium for 48 hours at 4°C, prior to dissection and immersion in the same fixative. Sections through the olfactory bulbs were cut at 50 μm with a vibratome, postfixed an additional 48 hrs, rinsed in PBS, and were then transferred to 1% osmium tetroxide, followed by ethanol dehydration and propylene oxide immersion. Blocks containing embedded bulb tissue were trimmed under a dissecting microscope, and a series of ultra-thin sections (60-80nm) was cut using a Reichert-Jung ultramicrotome. Sequential sections were collected onto mesh grids and stained with uranyl acetate and lead citrate. Tissue was examined with a Phillips CM-10 transmission electron microscope and images were collected using a Gatan digital camera.
Semi-Quantitative RT-PCR
At 3 weeks post-lesion, NMDA- and saline-injected rats (n=4 per group) were overdosed with sodium pentobarbital, decapitated, and the olfactory bulbs dissected and rapidly frozen in isopentane (-55°C). Total RNA was purified from individual bulbs using the RNAEasy Plus kit (Qiagen, Valencia, CA) and cDNA was generated using the AffinityScript cDNA kit and random primers (Agilent/Stratagene, La Jolla, CA). Q-PCR reactions used Taqman probe/primer sets (Applied Biosystems Inc, Foster City, CA) specific for rat trophic factor sequences. We tested 4 sequences as potential “housekeeping” genes. Expression of 18S RNA was highly variable. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and hypoxanthine-guanine phosphoribosyltransferase (HPRT) expression varied systematically with lesion treatment. In our hands, expression of beta-actin proved stable across bulb samples and was used for normalization. Standard curves were established, and all amplification efficiencies ranged from 94-105%. Q-PCR was carried out using Stratagene’s Brilliant ll QPCR master mix. Single-plex assays were performed on the MX3005P Q-PCR system, with a normalizing assay included for each sample. Beta-actin assays were run with 3ng of cDNA, while assays for lower-incidence trophic factor sequences were run with 15-30ng cDNA. Cycling was carried out at 95°C for 10 min, followed by 40 cycles of 95°C for 20 sec and 60°C for 1 min. Comparative quantification was calculated using MXPro software according to the 2-ΔΔCt method, with cDNA from one left control bulb (saline rat, triplicate) assigned as calibrator in each assay.
RESULTS
Elimination of olfactory bulb neurons
The effects of adult bulbectomy on OSN lifespan have been well characterized. We sought to compare these effects with those produced by a neurotoxic lesion that spared the sensory nerve from damage, and allowed regenerating axons to connect with the remaining bulb tissue. Extent of lesion and time-course of neuron death was evaluated by FluoroJade, Nissl staining, and immunoreactivity for neuronal and glial markers as described (Ardiles et al., 2007). As shown in figure 1, widespread cell death was evident within 24 hrs of NMDA infusion in all neuronal laminae. By this time, virtually all mitral cells and most tufted cells had already disappeared, with the mitral cell layer identifiable as a series of holes where the neurons had been (Fig. 1B). The initial wave of neuronal death was largely complete by 3 days, and collapse of the external plexiform (EPL) and granule cell layers (GCL) caused bulb shrinkage by 6 days (Fig. 1D). Tissue around the lesion core showed evidence of invasion by immune cells, including probable macrophages, and immunoreactivity (IR) for GFAP and OX-42 demonstrated rapid accumulation of reactive glia (Fig. 2). NeuN-IR verified neuronal loss throughout the bulb by 6 days. However, some NeuN+ nuclei remained deep in the remnant tissue, near the developing lesion cavity (Figs. 1E-F). Small numbers of NeuN+ cells also persisted in the glomerular region, possibly due to their distance from the injection site. Loss of output neurons was indicated by the absence of dextran transport from the lesioned bulb to ipsilateral piriform cortex, and layer IIa piriform neurons degenerated after lesion, as they do when deafferented by bulbectomy (Figs. 1G-H; Capurso et al., 1997). Following the initial wave of neuronal death, cavitation developed, with reactive astrocytes walling off the lesion cavity by 2 weeks, and further shrinkage of the remnant by 3 weeks (Fig. 1I). With the injection sites employed, the accessory olfactory bulb and the frontal pole of neocortex were consistently damaged, however a variable amount sparing occurred in the most caudal, medial bulb and in some rats, the rostral tip.
Figure 1.

A. Cresyl-violet stained section showing lamination of the normal olfactory bulb. B-D. Sections through the NMDA-lesioned bulb. B and C show FluoroJade and Nissl staining at 24 hrs post-injection, respectively. Most mitral cells are gone, and remaining degenerating neurons and dendrites are brightly labeled with FluoroJade. The inset in C shows higher magnification of degenerating cells (arrow) in the granule cell layer (gcl). By 6 days (D), laminar organization is gone, the external plexiform layer (epl) has collapsed, and glomeruli (asterisks) appear shrunken, with ill-defined borders. The dense accumulation of cells below glomeruli is the result of reactive gliosis. E-F. Comparison of NeuN-IR in normal (E) and lesioned bulbs (F) confirms that most neurons have disappeared within 6 days of NMDA treatment. However, small numbers of stained nuclei are found in portions of the glomerular region (arrows in F), and deep in the remnant, near the lesion cavity. G-H. Anterograde transport of biotinylated dextran from the intact (G) and lesioned bulb (H) to the anterior pirifrom cortex (pir). Removal of output neurons eliminates dextran transport on the NMDA-treated side. I. FluoroJade staining in the ipsilateral piriform cortex 48 hrs after lesion shows dying neurons (arrow) in layer II. J. Loss of neurons causes dramatic shrinkage of the treated bulb (right) by 3 weeks, however NPY-expressing olfactory ensheathing glia in the olfactory nerve layer (onl) survive. mcl, mitral cell layer. LOT, lateral olfactory tract. Bar in I= 100 μm in A-F, 1150 μm in G-H, 720 μm in I, 1250 μm in J.
Figure 2.

A-D. Glia in the lesioned olfactory bulb. A. Horizontal section showing part of the NMDA-treated bulb at 2 weeks. A GFAP+ glial scar has formed around the central lesion cavity. The distribution of normal astrocytes in the bulb (B) is replaced by widespread reactive astrogliosis (C) in response to neuronal degeneration. Small glomerular structures seen at 2 weeks (asterisks) are surrounded by GFAP+ processes. D. OX-42+ reactive microglia are also found throughout the injured bulb. E-I. TUNEL labeling in the normal olfactory epithelium (E), 32 hrs after bulbectomy (F), and 2 weeks after NMDA lesion (G). H-I. Double-labeling for TUNEL and OMP at 2 weeks (H) and 8 weeks (I) after NMDA. Arrows indicate labeled nuclei. J. Bar graph comparing temporal patterns of apoptosis in the target-deprived OE after bulbectomy and NMDA lesion. Significant increases (p<0.01) in TUNEL labeling occur rapidly and persist at all time points after bulbectomy compared to sham-lesions (not indicated on graph). The incidence of apoptotic cells increases gradually after NMDA and remains low in magnitude compared to bulbectomy. *p<0.05; **p<0.01, Student’s t test, ipsilateral vs pooled left + right saline. K-L. Immunostaining for OMP and GAP-43 in the contralateral (K) and ipsilateral OE (L) 3 weeks after NMDA. The mature OSN population is reduced while the immature population is increased in this portion of the deprived OE. Bar in L=375 μm in A, 150 μm in B-C, 90 μm in D, 32 μm in E-G, 15 μm in H-I, and 40 μm in K-L. Magenta-green version of this figure available as supplementary figure 1.
The presence of NeuN+ cells deep within the lesioned bulb suggested that new neurons were reaching the remnant via the SVZ/rostral migratory stream (RMS; Alvarez-Buylla et al., 2000), and this was verified by BrdU localization in sections from rats treated with BrdU 3 weeks after NMDA and euthanized 2 or 5 weeks later (see below). Persistent NPY mRNA expression demonstrated that OEG in the olfactory nerve layer (ONL) survived NMDA treatment (Fig. 1I), and the nerve layer remained grossly intact at all survival intervals.
Apoptosis in the target-deprived OE
Cell counts of TUNEL+ nuclei in the OE ipsilateral to NMDA injection demonstrated gradual, delayed increases in apoptosis after lesion (Fig. 2). This limited response, noted previously in mice (Carson et al., 2005; Ardiles et al., 2007), was surprising given the extent of target neuron loss. The rapid clearance of apoptotic cells and the limited time during which fragmented DNA can be detected using the TUNEL technique, prompted us to examine a large number of post-NMDA lesion time points in parallel with tissue from bulbectomized rats to ensure we did not overlook a vulnerable phase. An increase in TUNEL labeling with NMDA occurred at 6 days, and counts of positive nuclei remained elevated thereafter. Labeled cells were sparsely distributed in the OE, with clusters apparent (Fig. 2G). Contralateral counts did not differ from labeling in saline-treated rats out to 3 weeks, and labeling did not differ between the left and right OE in these controls. However at 8 weeks, counts of TUNEL+ nuclei in the OE on the untreated side were elevated compared to control counts (data not shown). As reported previously, OMP-IR appeared to degrade rapidly in dying OSNs, but we identified apoptotic mature neurons at both 2 and 8 weeks after lesion using combined immunodetection (Figs. 2H-I, Holcomb et al., 1995). Based on OMP- and GAP-43-immunostaining, overall numbers of mature neurons were reduced by 3 weeks, while immature neurons were increased, although the extent of change was not uniform throughout the OE (Fig. 2K-L). By 8 weeks, expression of OMP-IR had partially recovered, however ventral portions of the septum remained sparsely populated by mature cells. GAP43-IR was elevated throughout the epithelium from 2-8 weeks.
Quantitative changes in OMP and GAP-43 expression
To quantify changes in OMP and GAP-43 expression, we carried out in situ tissue labeling to measure the density of radiolabeled-cRNA hybridization within the OE (Ardiles et al., 2007), and again, compared the progression of changes observed with those produced by bulbectomy (Verhaagen et al., 1990). Rapid loss of OMP mRNA occurred with bulbectomy, reaching a low point at 6 days, when levels measured ∼21% of control values (Figs. 3A-B), recovering to 45% when followed out to 3 weeks. Changes with NMDA lesion developed more gradually, and were of limited magnitude. Hybridization of OMP cRNA ipsilateral to the treated bulb began to decrease at 6 days, at which time GAP-43 mRNA levels were elevated. Overall, OMP cRNA hybridization density did not decline beyond 85% of contralateral levels at any time after NMDA when averaged throughout the entire OE, but more notable changes in expression occurred within restricted areas (Fig. 3B). In these regions, labeling “thinned” out. Variations may reflect the sparing that occurred in the most caudal and rostral bulb. OMP and GAP-43 mRNA levels began returning to control levels by 8 weeks, following re-innervation of the bulb by OSNs born post-lesion. This was paralleled by changes in immunoreactive protein (Fig. 3D). Shifts in the mature and immature OSN populations were accompanied by changes in the size of the OE ipsilateral to NMDA. Reductions in OE thickness with NMDA were detected at 6 days, when the OE measured 93% (+/-7%, SEM) of contralateral measures, and reached a minimum thickness of 86% (+/-8%) at three weeks. By 8 weeks, depth had recovered to 93% (+/-11%). With bulbectomy, rapid atrophy occurred; at 2 days depth was reduced to 62% of normal (+/-13%, SEM), recovering to 72% (+/-7%) of contralateral measures by 3 weeks.
Figure 3.

OMP and GAP-43 expression in the target-deprived OE. A. OMP mRNA expression declines rapidly with bulbectomy, and gradually with NMDA. B. Changes in GAP-43 mRNA expression are complementary to OMP, increasing over time with both lesion treatments. C. Localization of OMP mRNA at 6 days after bulbectomy (top) and 3 weeks after NMDA (bottom). The deprived olfactory epithelium (OE) is on the left. With NMDA, labeling thins out in areas of reduced OMP expression (arrows) compared to labeling on the untreated side. D. OMP-immunoreactivity (IR) in the contralateral OE (top) and deprived OE (bottom) at 8 weeks after NMDA. Images were taken from the OE lining the septum. *p<0.05; **p<0.01, Tukey-Kramer test. Bar = 1.2 mm in C, 30 μm in D.
Sensory axons in the neuron-depleted bulb
Both GAP-43+ and OMP+ axons remained present in the ONL of all NMDA-treated bulbs, with GAP-43+ fibers predominating by 3 weeks (Fig. 4). During the first 2 weeks, OMP+ glomeruli in the lesioned bulb atrophied, with sensory fibers either eliminated or retracted into the ONL. GAP-43+ axons, normally restricted to the nerve layer, arborized within shrunken glomeruli, along with OMP+ fibers (Fig. 4C). At 3 weeks, portions of the bulb lacked identifiable glomeruli altogether, with sensory axons restricted to the nerve layer. In other areas, glomerular structures of irregular size, shape and distribution were present, with varied composition in terms of OMP+ and GAP-43+ axons (Fig. 4F). Many, though not all, contained fractin+ axons, indicative of caspase-3-mediated neurodegeneration (Fig. 4G, Suurmeijer et al., 1999). The majority of foci were comprised of both immature and mature axons, but a small number contained only OMP+ or GAP-43+ fibers (Figs. 4H-I). Within mixed glomeruli, GAP+ axons were often distributed around a core of axons expressing OMP (Fig. 4H), although other foci displayed the opposite distribution. These patterns in mature/immature axon distribution (GAP43+ foci, mixed glomeruli) are reminiscent of the innervation patterns that emerge over the course of early bulb development (Kim and Greer, 2000).
Figure 4.

Sensory projections in the lesioned bulb. A. Horizontal section showing the beginnings of cavitation in the lesioned bulb. B-E. At 2 weeks, glomeruli appear shrunken and irregular, with most containing a mixture of OMP+ and GAP-43+ fibers (arrows in C and E), in contrast to the OMP+/GAP-43-glomeruli (gl) seen normally (B and D). F. At later time points, glomerular structures of various sizes and axon composition are seen. The asterisk in F indicates the lesion cavity below the medial side of the bulb fragment, and the arrow indicates axons traveling deep within the tissue. G. Fractin immunostaining showing degenerating axons within a glomerulus. H. GAP-43+ axons (red, arrow) are distributed around OMP+ axons (green) within a small glomerulus (arrow). I. Other foci contain only GAP-43+ axons (arrow). J. BrdU+ cells (red, arrow) deep within the lesioned bulb 3 weeks after BrdU treatment. K. Some of these cells also express NeuN (green). L. Large numbers of mature (green) and immature axons (red) penetrate the lesioned bulb at 8 weeks, where they terminate in irregular glomeruli. M. Small numbers of NeuN+ nuclei (red, solid arrow) can be seen near OMP+ glomeruli (gl, green). Staining along the margin of the lesion cavity below is due to non-specific labeling of degenerating cells (open arrow). N. A thin strip of tissue along the lateral side of the remnant contains TH-IR neurons (arrows) and neuropil within glomerular structures. The asterisk indicates the lesion cavity. O-P. Examples of BrdU+/TH+ cells in the lesioned bulb 5 weeks after BrdU. onl, olfactory nerve layer. Bar = 500 μm in A, 190 μm in B-E, 140 μm in F, 25 μm in G, 70 μm in H-I, 60 μm in J, 125 μm in L, 75 μm in M, 175 μm in N, 18 μm in K, O-P. Magenta-green version of this figure available as supplementary figure 2.
Tissue shrinkage shortened the distance between the nerve layer and lesion core, and from 3 weeks onward, sensory afferents penetrated far enough to approach the lesion cavity. Here they coalesced into foci along the outer extent of its glial margins, or diverted to travel parallel to the network of astrocytes distributed around and extending outward from it (Figs. 4F and L). NeuN+ cells were scattered in the deep bulb remnant, particularly caudal to the cavity, and were also observed associated with some, but not all, glomerular structures, including those near the lesion margins (Fig. 4M). At 8 weeks, a partial, irregular GCL was present in the caudal remnant and in some areas this extended outward toward the ONL, with glomerular foci contained in it. Following a single dose of BrdU at 3 weeks, we identified BrdU+/NeuN+ neurons within the reconstituting granule cell layer that were generated in the SVZ/RMS, migrated to the bulb, and survived for at least 2 weeks (Figs. 4J-K). At 5 weeks, we observed rare examples of BrdU+/TH+ neurons (as well as more frequent BrdU-/TH+ cells) in the lesioned bulb (Figs. 4O-P), both in regions containing glomerular foci, and in deeper locations within the irregular granule cell layer. Most of these exhibited dendrites.
Ultrastructural features of olfactory axons with limited targets
The continued presence of sensory axons within the neuron-depleted bulb, as seen with immunostaining at the light microscopic level, led us to examine these more closely using electron microscopy. We chose to evaluate the lesioned bulb at 3 weeks, at a time when the axons of sensory neurons born after lesion would have reached the bulb remnant. Moreover, increased OE TUNEL labeling, reduced OMP expression, and accumulation of fractin-IR indicated that a subpopulation of OSNs were undergoing programmed cell death at this time. Examination of NMDA-injected bulbs revealed ensheathing glia-enclosed axon bundles in the ONL, containing individual axon terminals in the process of dissolution that appeared electron lucent and swollen (Fig. 5). Many contained swollen mitochondria and some processes appeared to be undergoing filamentous degeneration (Fig. 5C). Different ONL locations displayed variations in the density of degenerating profiles, with some areas appearing heavily populated (Fig. 5A). Similar pale, swollen axons were observed in control bulbs as well, though much less frequently. In spite of the paucity of bulb neurons, numerous sensory axons with normal morphology were also present in the nerve layer of lesioned bulbs. Deep to this, the characteristic glomerular neuropil seen in control bulbs was lacking and probable microglial cells were encountered, many of which contained electron-dense and electron-lucent inclusion bodies indicative of phagocytosis (Fig. 5). Fibrous astrocytes were identified within the tissue parenchyma (Fig. 5D), and lining lesion cavity (not shown). As shown in figure 5F, dendritic processes were occasionally encountered in portions of the remnant, and when present, these received numerous synaptic contacts from sensory axons.
Figure 5.

Ultrastructural changes in the lesioned bulb. A. An ensheathing glial cell (oeg) with processes extending around a bundle of sensory axons along the outer margin of the olfactory nerve layer. Interspersed between normal, darker axon profiles are numerous pale, enlarged axons that appear to be degenerating (asterisks). B. Higher magnification of the boxed region in A showing electron-lucent axons, some with swollen mitochondria (arrow). C. Hypertrophied axon showing signs of neurofilamentous degeneration. D. An astrocyte extending a process filled with fibrils (asterisk) within the lesioned bulb. E. Putative microglial cell (m) containing inclusion bodies (asterisk) indicative of phagocytosis. I. A region in the damaged bulb in which sensory axons make synapses (arrows) on surviving dendritic processes. Bars = 1 μm in A, C, D, F; 0.5 μm in B; 2 μm in E.
Proliferation in the target-deprived OE
Coordinate regulation of neuronal numbers within the normal OE links increased sensory neuron apoptosis with up-regulation of neurogenesis (Carr and Farbman, 1993; Calof et al., 1998; Cowan and Roskams, 2002). By localizing expression of Ki-67, we observed that delayed apoptosis was indeed followed by a 3-fold increase in the numbers of proliferating cells 2 weeks after NMDA lesion, compared to saline treatment (Fig. 6). Our cell counts were limited to the OE lining the septum to ensure sampling of matched left-right regions, but increased proliferation was readily apparent throughout the entire deprived OE, and in both lesioned and control rats, labeled cells tended to cluster in groups. A more modest increase (1.7 fold) also occurred in the contralateral epithelium, an effect also seen with unilateral bulbectomy (Schwob et al., 1992; Carr and Farbman, 1993).
Figure 6.

A-D. Increased cell proliferation in the target-deprived OE is seen with Ki-67-immunostaining at 2 weeks post-NMDA (A-B) and with BrdU labeling (C-D; administered at 3 weeks). E-F. Quantitative changes in the numbers of proliferating cells in the OE are similar when measured with both techniques. *p<0.01, Student’s t-test. Bar = 35 μm in A-B, 29 μm in C-D.
Life-span of sensory neurons in the target-deprived OE
Schwob and colleagues (1992) have thoroughly detailed the chronic changes in sensory neuron life-span that occur in the rat OE following bulbectomy, and to facilitate comparisons to this work, we carried out BrdU labeling in NMDA-treated rats, using similar survival intervals (4 days, 2, and 5 weeks). BrdU was given at 3 weeks post-lesion, after the peak period of bulb neuron death and the initial wave of reactive gliosis. By this time the remnant had achieved its shrunken, cavitated condition, yet was still contacted by sensory axons, as seen with both immunostaining and electron microscopy.
Counts of BrdU-labeled cells at 4 days confirmed the results of our Ki-67 analysis; increased proliferation produced a 2.1-fold increase in the number of cells that survived short-term (Table 2 and Fig. 6). Many of these new cells survived out to 2 weeks in the target-deprived OE, when the numbers of labeled cells still exceeded the number present on the untreated side (Table 2 and Fig. 7). Between 4 days and 2 weeks, ∼48% of BrdU-labeled cells were lost from the deprived OE (32.1 cells/mm vs. 16.7 cells/mm), while cell loss in the contralateral and normal OE averaged 30-32%. The loss of about half of the new cells contrasts with the chronic situation seen after bulbectomy, when by this time, ∼90% of new cells have died, and more survivors remain on the un-operated side (Schwob et al., 1992). Whereas BrdU-labeled cells at 4 days were confined to the deep OE, at 2 weeks, most stained nuclei were positioned superficial to this, consistent with their development as sensory neurons (Fig. 7).
Table 2.
Cell survival in the olfactory epithelium:
| Mean BrdU+ cells/mm +/- SEM: | |||
|---|---|---|---|
| Treatment and side |
4 days | 2 wks | 5 wks |
| All Saline | 15.0 +/- 1.9 | 10.2 +/- 0.6 | 4.8 +/- 0.4 |
| NMDA left | 17.9 +/- 2.1 | 12.6 +/- 0.5* | 3.9 +/- 0.8 |
| NMDA right | 32.1 +/- 1.1* | 16.7 +/- 0.9* | 5.4 +/- 0.7* |
N=2 for saline-treated rats. N=4 for NMDA-treated rats. Total counts from eight sections per animal, as described in Materials and Methods.
p<0.05, right NMDA compared to left NMDA cell counts, left NMDA compared to saline cell counts, Students t-tests.
Figure 7.

Long-term neuronal survival in the deprived OE. A-D. BrdU-labeling demonstrates that substantial numbers of surviving cells are present in the deprived OE at both 2 and 5 weeks after target depletion. E. Photomicrograph showing co-localization of BrdU with OMP-IR in the deprived OE at 5 weeks (arrows). F-G. Confocal microscope images of 5-week-old OMP+ sensory neurons generated after bulb NMDA lesion (arrows, 2.8 μm optical section thickness). Bar = 78 μm in A-D, 25 μm in E, 18 μm in F-G. Magenta-green version of this figure available as supplementary figure 3.
Unexpectedly, some BrdU+ cells survived in the target-deprived OE out to 5 weeks (Table 2). Between 2 and 5 weeks, NMDA-treated rats lost ∼68% of BrdU+ cells in the deprived OE (16.7 cells/mm vs. 5.4 cells/mm), while the loss in saline-treated rats was ∼53%. However, due to increased proliferation, the mean density of surviving cells in the deprived OE was essentially equivalent to controls at this time (5.4 cells/mm vs. 4.8 cells/mm). The percentage of BrdU+ cells lost between 4 days and 5 weeks was 83% ipsilateral to lesion, compared to 68% in saline controls. The location of surviving cells within the deprived OE suggested they were sensory neurons. We therefore utilized combined labeling for OMP- and BrdU-IR to verify that some five-week-old survivors were mature OSNs generated after lesion. Total numbers of BrdU+/OMP+ cells (mean +/- SEM) counted in eight serial sections in saline animals averaged 213 +/-23 (SEM) per side. Counts from the untreated side of NMDA rats averaged 158 +/- 25, while the mean count ipsilateral to NMDA was 380 +/-78. Taken together with the gradual recovery of OMP expression, our results suggest that the remnant bulb tissue and/or nerve pathway is able to support the long-term survival of OSNs even when the availability of target neurons is limited, and that this support is lacking when scar tissue produced by bulbectomy prevents axons from reaching the CNS.
Trophic factor expression levels in the lesioned bulb
The extent of recovery we observed in the OSN population prompted us to investigate the availability of neurotrophic factors in the lesioned bulb using semi-quantitative RT-PCR. As a first assessment, we focused on an array of factors known to be synthesized in the normal bulb (MacKay-Sim and Chuah, 2001). These assays were carried out using bulb tissue collected three weeks after NMDA infusion, again because acute lesion effects had subsided by this time, and because the axons of sensory neurons born 1-2 weeks after NMDA would encounter this particular target environment. Analysis of standard curves indicated that the relative abundance of the mRNAs in the normal rat bulb was as follows: IGF-1>FGF1>FGF2>CNTF>BDNF/NGF>NT3. NT3 expression was very low in all samples and was excluded from further analysis. As shown in figure 8, levels of NGF and BDNF, both synthesized by bulb neurons, were reduced by NMDA treatment. BDNF was of particular interest due to sensory neuron expression of the Trk B receptor (Roskams et al., 1996; Feron et al., 2008). In contrast to the neurotrophins, expression of other factors increased with lesion. FGF2 and IGF-1 demonstrated significant elevations in expression, while CNTF and FGF1 levels remained relatively unchanged (Fig. 8). Though not statistically significant, we also detected contralateral changes in trophic factor expression, which may contribute to some of the contalateral effects we report here. The current analysis was limited to a select group of trophic factors, and does not address the role of other factors, or other types of stabilizing signals, such as adhesion molecules, in OE recovery after bulb lesion.
Figure 8.
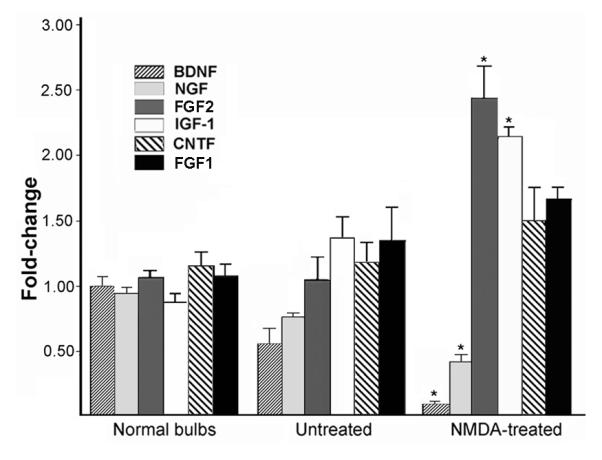
Changes in relative expression levels of neurotrophic factor mRNAs as measured by semi-quantitative PCR. Values were normalized to beta-actin and bars represent group means +/- SEM (n=4). All assays were carried out using individual bulb samples, not pooled tissue. Data for normal bulbs represent the combined results from left and right bulbs, which were not significantly different (p>0.05, paired t-tests). Changes in lesioned bulbs (NMDA-treated) as well as contralateral bulbs (untreated) are apparent. *p<0.01, t-tests, NMDA vs. untreated and vs. normal.
DISCUSSION
The lifespan of olfactory sensory neurons in vivo does not appear to be predetermined. Epigenetic factors impact their survival, among them, peripheral exposure to substances that access the nasal cavity where the cell bodies are located, and interactions with the CNS where their axons project (Schwob, 2002). The latter is evidenced by their reduced life-span following bulbectomy (Schwob et al., 1992; Carr and Farbman, 1993), but even in the intact system OSNs do not typically survive beyond 8 weeks (Farbman, 1990), so the extent to which target neurons, or other individual cell types, specifically contribute to survival mechanisms in OSNs is not clear. According to classic model of neurotrophic support as applied to the olfactory pathway, shortly after their axons contact the bulb, sensory neurons obtain necessary retrograde signals from the bulb neurons they innervate to promote their continued survival. In a continuously regenerating system such as this, where survival is limited even under normal circumstances, we anticipated that any reductions in target support would dramatically reduce OSN lifespan. However, as we demonstrate here, the sensory population was surprisingly resilient when postsynaptic neurons were depleted.
There are two phases of OSN death that follow bulbectomy (Schwob, 2002; Hayward et al., 2004). The acute phase consists of neurons that undergo apoptosis within the first 5 days in response to axotomy. Cell death is detectable by 12 hrs, and peaks at 2-3 days post-lesion (Holcomb et al., 1995; Calof et al., 1998). Studies by Roskam and colleagues have shown that this injury triggers rapid caspase activation, first evident in the axonal compartment, then moving retrogradely to the cell body (Cowan and Roskams, 2001; Carson et al., 2005). Neuronal death then signals an increase in progenitor proliferation that produces a large cohort of immature, GAP-43+ neurons. The second phase occurs as developing axons from these new neurons fail to locate target tissue, a situation that resembles developmental neuron death. This occurs when the new cells are 6-12 days of age, as they are making the transition to OMP-expressing mature neurons (Schwob et al, 1992: Carr and Farbman, 1993; Hayward et al., 2004). With chronic disconnection, this cycle of enhanced neurogenesis, differentiation, and premature death continues. Expression of Bcl-2, under control of the OMP promoter, can rescue sensory neurons during this phase, and restore most of the mature population after bulbectomy, supporting the hypothesis that bulb-mediated trophic mechanisms normally contribute to maintenance of these cells (Schwob et al., 1992; Allsopp et al., 1993; Hayward et al., 2004).
The pattern of OE cell death after NMDA differs distinctly from that which follows bulbectomy. As there is no axotomy, there is no acute OSN death, and bulb neuron loss is followed by delayed, gradual increases in epithelial apoptosis. Sensory neuron age may determine the temporal pattern of death, with those already nearing the end of their lifespan affected first, or those potentially comprising a vulnerable subpopulation, such as those expressing the retinoic acid-degrading enzyme Cyp26B1 (Hagglund et al., 2006). Alternatively, resistant neurons may be less sensitive due to levels of receptor expression, or have secured a prior supply of trophic factor that is still in the process of intracellular transport and survival signaling. Beginning ∼1 week after NMDA, apoptosis increases and continues to increase gradually thereafter. A correlated reduction in OMP expression is evident by 2 weeks, and as with bulbectomy, cell death stimulates increased progenitor proliferation and an expansion of the immature OSN population. That sensory neurons are part of the apoptotic population is confirmed by our co-localization of TUNEL and OMP-IR, and by fractin immunostaining of sensory axon terminals in the lesioned bulb. Fractin is generated by the cleavage of actin by activated caspase-3 (Suurmeijer et al., 1999), and its expression in the lesioned bulb at 2-8 weeks suggests that, as at earlier time points after NMDA, apoptotic signaling is initiated in sensory terminals (Cowan et al., 2001). To some extent, the condition of the epithelium at 3 weeks after NMDA shares similarities with the chronic bulbectomy condition, and like bulbectomy, the vulnerable neurons at this time may be those unable to locate appropriate targets (Verhaagen et al., 1990; Schwob et al., 1992; Carr and Farbman, 1993). However apoptosis levels remain lower, and mature, OMP-expressing neurons persist in significant numbers. Moreover, rather than stabilizing at this stage or atrophying further, the mature population partially recovers over the next 5 weeks.
In the chronic bulbectomy model, only a small fraction of neurons (∼10%) born ipsilateral to surgery survive for 2 weeks (Schwob et al., 1992; Hayward et al., 2004). In the NMDA-treated rat, where new axons have access to the lesioned bulb, elevated neurogenesis produced a cohort of survivors at this time that outnumbered those in control epithelia. Most of these two-week-old neurons are likely to be mature, or in transition from their immature state, and their survival suggests their stabilization by factors and/or cell interactions in the remaining tissue. Target removal by bulbectomy does not delay the time course of OSN maturation (Schwob et al., 1992), however we cannot rule out a change in maturation rate with NMDA, so it remains possible that some of these cells are immature and not yet sensitive to target deprivation. If this were the case, we would expect the vast majority of these to be eliminated during the ensuing 3 weeks. Though proportionally more cells were eliminated from the deprived epithelium (68%) compared to normal (53%), numbers of surviving cells were similar (5.4 vs. 4.8 cells/mm). This demonstrates that the neuron-depleted pathway supports a substantial population of five-week-old sensory neurons, as we confirmed with BrdU/OMP labeling. Contralateral increases in cell proliferation and death, similar to those reported with bulbectomy, also occurred (Schwob et al., 1992; Carr and Farbman, 1993; Hayward et al., 2004). The exact mechanisms underlying this response are not known, however there were subtle changes in trophic factor expression in the untreated bulb. Patterns of activity regulate the expression of some CNS factors and bulb NMDA damage may alter contralateral bulb activity through commissural connections (Shieh and Gosh, 1999).
Indirect evidence that neurotrophic factors from the bulb support OSNs in vivo has been provided by studies of bulbectomized transgenic mice in which signaling pathways that mediate apoptosis with trophic factor deprivation are disrupted. These include the beneficial effects of caspase-3, p75 and BAX gene knockout, and the protective effects of Bcl-2 over-expression (Cowan et al., 2001; Robinson et al., 2003; Hayward et al., 2004; Watt et al., 2004; Carson et al., 2005). Paradoxically, analyses of neurotrophic factor knockout mice have not identified an essential bulb factor or cell source. Interpretation of effects seen in these animals has been complicated by the fact that some factors are expressed in both the OE and bulb, cause early death when eliminated, exert developmental effects on one or both structures, and that within trophic factor gene families, members exhibit coincident or overlapping expression and can activate multiple receptors that also show coincident expression (Guthrie and Gall, 1991; Deckner et al., 1993; Roskams et al., 1996; Kornblum et al., 1998; MacKay-Sim and Chuah, 2000; Nef et al., 2001; Carter and Roskams, 2002; Feron et al., 2008).
By eliminating bulb neurons, we had hoped to narrow down the source potential survival cues, and provide evidence that would assign this function to target neurons. Unexpectedly, significant numbers of OSNs born after lesion matured and survived for what constitutes a significant portion of the normal OSN lifespan; far more than survive after bulbectomy. This finding is reminiscent of the survival of adult basal forebrain neurons following removal of hippocampal target neurons (Sofroniew et al., 1990). Target neuron elimination clearly is not the equivalent of complete bulb removal, and leaves other, potentially supportive cells available. These include olfactory glia, interneurons generated after lesion, small numbers of surviving neurons, and cells outside the bulb proper. Trophic factors from these sources may act individually or in combination to support OSNs. The major neuronal populations contacted by sensory axons are the mitral, tufted and periglomerular cells. In situ localization of mRNA in these populations has shown that they synthesize a number of different neurotrophic factors, including NGF, BDNF, IGF-1, FGF1, and TGF-alpha (Mackay-Sim and Chuah, 2000), and their expression presumably would be reduced by neuron ablation. We show here by Q-PCR that BDNF and NGF expression are in fact significantly decreased. CNTF mRNA levels are not altered, consistent with OEG serving as the chief source of this factor. IGF-1 and FGF2 expression, and to a lesser extent FGF1, were increased with lesion, and these changes may reflect the presence of reactive glia in the bulb remnant. Reactive astrocytes up-regulate expression of FGF2, and reactive microglia express IGF-1 in brain regions undergoing neurodegeneration (Guthrie et al., 1995). FGF2 is also constitutively expressed by OEG, and the supportive role of these unique glial cells in the olfactory system has been long recognized (Mackay-Sim and Chuah, 2000; Schwob, 2002). Glial secretion of these factors in proximity to sensory axons within the lesioned bulb may support those subpopulations that express FGF and IGF-1 receptors (MacKay-Sim and Chuah, 200; Ferrari et al., 2003).
Although there was catastrophic loss of bulb neurons with NMDA (Fig. 1J), scattered NeuN+ cells remained and these survivors (or newly-added interneurons) may be of a subclass that provides a specific, essential factor. Alternatively, they may up-regulate expression of a given factor in response to increased input, and we identified numerous sensory synapses on dendrites in the lesioned bulb at the ultrastructural level. Sparing of the posterior portion of medial bulb would support sensory afferents that normally project here, as well as inappropriate axons that invade over time as innervation topography breaks down (Ardiles et al., 2007). Other neuronal sources include the new neurons that managed to infiltrate the damaged bulb over time, in spite of the reactive gliosis and cavitation that followed lesion. Their presence indicates that neurons generated in the SVZ continue to migrate to the bulb, and over the weeks that follow lesion, some survive and succeed in reaching areas containing sensory axons. These may provide acceptable targets for regenerating sensory axons, and it is easy to envision that with more limited types of bulb damage, this neuronal replacement could work to partially restore functional circuitry and support innervation.
Target-deprived OSNs may also succeed in deriving sufficient trophic support from sources outside the bulb proper, for example from centrifugal afferents, or from neurons located in more posterior regions. Following neonatal bulbectomy, the absence of scar tissue allows olfactory axons to penetrate into a number of forebrain areas, including the AON and RMS (Graziadei et al., 1978; Guthrie and Leon, 1989). In a prior study, we observed regenerating axons in the AON of NMDA-lesioned mice at 8 weeks (Ardiles et al., 2007), however we did not observe this pattern in the rats that survived to 8 weeks in the present study. Another source may be cerebrospinal fluid (CSF); a recent report of trophic factor expression in the SVZ noted that the choroid plexus expresses NT4, making it likely that CSF within the lesion cavity contains this TrkB-binding neurotrophin (Galvao et al., 2008). Finally, the sensory neurons themselves may utilize paracrine mechanisms of support, and come to rely more heavily on these when targets are limited. Bulbectomy synchronously kills mature OSNs and few survive afterward, while following NMDA lesion, a large subpopulation remains present. Mature OSNs express trophic factors, among them the neurotrophins, and therefore may exchange survival signals within the OE or nerve (Vigers et al., 2003; Simpson et al., 2003; Feron et al., 2008).
The critical need to maintain sensory function has provided the olfactory system with neuronal replacement mechanisms that allow for adaptation to the sensory world, but these can also be mobilized in response to damage. Here we demonstrate that sensory projections to the bulb persist, and OSNs survive long-term, when bulb target neurons are depleted in vivo. This suggests that sensory neurons can obtain sufficient trophic support from remaining or alternative sources, and highlights the remarkable ability of the system to adapt to injury.
Supplementary Material
Other acknowledgements
Special thanks to Katty Sahakian, Walter Hoover and Dr. Zhiyin Shan for technical assistance, Drs. S. Watson and C. Isgor for the Ki-67 antibody, and Dr. F. Margolis for the OMP plasmid cDNA.
This work was supported by NIH/NIGMS grant S06GM073621 to KMG.
LITERATURE CITED
- Abercrombie M. Estimation of nuclear population from microtome sections. Anat. Rec. 1946;94:239–47. doi: 10.1002/ar.1090940210. [DOI] [PubMed] [Google Scholar]
- Allsopp TE, Wyatt S, Paterson HF, Davies AM. The proto-oncogene bcl-2 can selectively rescue neurotrophic factor-dependent neurons from apoptosis. Cell. 1993;73:295–307. doi: 10.1016/0092-8674(93)90230-n. [DOI] [PubMed] [Google Scholar]
- Alvarez-Buylla A, Herrera DG, Wichterle H. The subventricular zone: source of neuronal precursors for brain repair. Prog Brain Res. 2000;127:1–11. doi: 10.1016/s0079-6123(00)27002-7. 2000. [DOI] [PubMed] [Google Scholar]
- Ardiles Y, de la Puente R, Toledo R, Isgor C, Guthrie K. Response of olfactory axons to loss of synaptic targets in the adult mouse. Exp. Neurol. 2007;207:275–288. doi: 10.1016/j.expneurol.2007.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer S, Rasika S, Han J, Mauduit C, Raccurt M, Morel G, Jourdan F, Benahmed M, Moyse E, Patterson PH. Leukemia inhibitory factor is a key signal for injury-induced neurogenesis in the adult mouse olfactory epithelium. J Neurosci. 2003;23:1792–803. doi: 10.1523/JNEUROSCI.23-05-01792.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beites CL, Kawauchi S, Crocker CE, Calof AL. Identification and molecular regulation of neural stem cells in the olfactory epithelium. Exp Cell Res. 2005;306:309–16. doi: 10.1016/j.yexcr.2005.03.027. [DOI] [PubMed] [Google Scholar]
- Biffo S, Verhaagen J, Schrama LH, Schotman P, Danho W, Margolis FL. B-50/GAP43 expression correlates with process outgrowth in the embryonic mouse nervous system. Eur J Neurosci. 1990;2:487–499. doi: 10.1111/j.1460-9568.1990.tb00440.x. [DOI] [PubMed] [Google Scholar]
- Burek MJ, Oppenheim RW. Programmed cell death in the developing nervous system. Brain Pathol. 1996;6:427–46. doi: 10.1111/j.1750-3639.1996.tb00874.x. [DOI] [PubMed] [Google Scholar]
- Calof AL, Rim PC, Askins KJ, Mumm JS, Gordon MK, Iannuzzelli P, Shou J. Factors regulating neurogenesis and programmed cell death in mouse olfactory epithelium. Ann N Y Acad Sci. 1998;30(855):226–9. doi: 10.1111/j.1749-6632.1998.tb10571.x. [DOI] [PubMed] [Google Scholar]
- Capurso SA, Calhoun ME, Sukhov RR, Mouton PR, Price DL, Koliatsos VE. Deafferentation causes apoptosis in cortical sensory neurons in the adult rat. J Neurosci. 1997;17:7372–84. doi: 10.1523/JNEUROSCI.17-19-07372.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr VM, Farbman AI. The dynamics of cell death in olfactory epithelium. Exp Neurol. 1993;124:308–314. doi: 10.1006/exnr.1993.1201. [DOI] [PubMed] [Google Scholar]
- Carson C, Saleh M, Fung FW, Nicholson DW, Roskams AJ. Axonal dynactin p150Glued transports caspase-8 to drive retrograde olfactory receptor neuron apoptosis. J Neurosci. 2005;25:6092–104. doi: 10.1523/JNEUROSCI.0707-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter LA, Roskams AJ. Neurotrophins and their receptors in the primary olfactory neuraxis. Microsc Res Tech. 2002;58:189–96. doi: 10.1002/jemt.10148. [DOI] [PubMed] [Google Scholar]
- Christensen MD, Holbrook EH, Costanzo RM, Schwob JE. Rhinotopy is disrupted during the re-innervation of the olfactory bulb that follows transection of the olfactory nerve. Chem Senses. 2001;26:359–69. doi: 10.1093/chemse/26.4.359. [DOI] [PubMed] [Google Scholar]
- Cho JY, Min N, Franzen L, Baker H. Rapid down-regulation of tyrosine hydroxylase expression in the olfactory bulb of naris-occluded adult rats. J Comp Neurol. 1996;369:264–76. doi: 10.1002/(SICI)1096-9861(19960527)369:2<264::AID-CNE7>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Cowan CM, Thai J, Krajewski S, Reed JC, Nicholson DW, Kaufmann SH, Roskams AJ. Caspases 3 and 9 send a pro-apoptotic signal from synapse to cell body in olfactory receptor neurons. J Neurosci. 2001;21:7099–109. doi: 10.1523/JNEUROSCI.21-18-07099.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan CM, Roskams AJ. Apoptosis in the mature and developing olfactory neuroepithelium. Microscopy Res Tech. 2002;58:204–215. doi: 10.1002/jemt.10150. [DOI] [PubMed] [Google Scholar]
- Costanzo RM. Comparison of neurogenesis and cell replacement in the hamster olfactory system with and without a target (olfactory bulb) Brain Res. 1984;307:295–301. doi: 10.1016/0006-8993(84)90483-9. [DOI] [PubMed] [Google Scholar]
- Deckner ML, Frisén J, Verge VM, Hökfelt T, Risling M. Localization of neurotrophin receptors in olfactory epithelium and bulb. Neuroreport. 1993;5:301–4. doi: 10.1097/00001756-199312000-00030. [DOI] [PubMed] [Google Scholar]
- Farbman AI. Olfactory neurogenesis: genetic or environmental controls? Trends Neurosci. 1990;13:362–365. doi: 10.1016/0166-2236(90)90017-5. [DOI] [PubMed] [Google Scholar]
- Ferrari CC, Johnson BA, Leon M, Pixley SK. Spatiotemporal distribution of the insulin-like growth factor receptor in the rat olfactory bulb. Neurochem Res. 2003;28:29–43. doi: 10.1023/a:1021639926941. [DOI] [PubMed] [Google Scholar]
- Feron F, Bianco J, Ferguson I, Mackay-Sim A. Neurotrophin expression in the adult olfactory epithelium. Brain Res. 2008;1196:13–21. doi: 10.1016/j.brainres.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Galvão RP, Garcia-Verdugo JM, Alvarez-Buylla A. Brain-derived neurotrophic factor signaling does not stimulate subventricular zone neurogenesis in adult mice and rats. J Neurosci. 2008;28:13368–83. doi: 10.1523/JNEUROSCI.2918-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graziadei PPC, Levine RR, Graziadei GA. Regeneration of olfactory axons and synapse formation in the forebrain after bulbectomy in neonatal mice. Proc Natl Acad Sci USA. 1978;75:5230–5234. doi: 10.1073/pnas.75.10.5230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grill RJ, Pixley SK. In vitro generation of adult rat olfactory sensory neurons and regulation of maturation by coculture with CNS tissues. J Neurosci. 1997;17:3120–7. doi: 10.1523/JNEUROSCI.17-09-03120.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie KM, Leon M. Induction of tyrosine hydroxylase expression in rat forebrain neurons. Brain Res. 1989;497:117–31. doi: 10.1016/0006-8993(89)90977-3. [DOI] [PubMed] [Google Scholar]
- Guthrie KM, Gall CM. Differential expression of mRNAs for the NGF family of neurotrophic factors in the adult rat central olfactory system. J Comp Neurol. 1991;313:93–102. doi: 10.1002/cne.903130107. [DOI] [PubMed] [Google Scholar]
- Guthrie KM, Nguyen T, Gall CM. Insulin-like growth factor-1 mRNA is increased in deafferented hippocampus: spatiotemporal correspondence of a trophic event with axon sprouting. J Comp Neurol. 1995;352:147–60. doi: 10.1002/cne.903520111. [DOI] [PubMed] [Google Scholar]
- Hägglund M, Berghard A, Strotmann J, Bohm S. Retinoic acid receptor-dependent survival of olfactory sensory neurons in postnatal and adult mice. J Neurosci. 2006;26:3281–91. doi: 10.1523/JNEUROSCI.4955-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward MD, Bocchiaro CM, Morgan JI. Expression of Bcl-2 extends the survival of olfactory receptor neurons in the absence of an olfactory bulb. Mol Brain Res. 2004;132:221–234. doi: 10.1016/j.molbrainres.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Higuchi H, Yang HY, Sabol SL. Rat neuropeptide Y precursor gene expression. mRNA structure, tissue distribution, and regulation by glucocorticoids, cyclic AMP, and phorbol ester. J Biol Chem. 1988;263:6288–95. [PubMed] [Google Scholar]
- Holcomb JD, Mumm JS, Calof AL. Apoptosis in the neuronal lineage of the mouse olfactory epithelium: regulation in vivo and in vitro. Dev Biol. 1995;172:307–323. doi: 10.1006/dbio.1995.0025. [DOI] [PubMed] [Google Scholar]
- Isgor C, Watson SJ. Estrogen receptor alpha and beta mRNA expressions by proliferating and differentiating cells in the adult rat dentate gyrus and subventricular zone. Neuroscience. 2005;134:847–56. doi: 10.1016/j.neuroscience.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Iwai N, Zhou Z, Roop DR, Behringer RR. Horizontal basal cells are multipotent progenitors in normal and injured adult olfactory epithelium. Stem Cells. 2008;26:1298–1306. doi: 10.1634/stemcells.2007-0891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawauchi S, Beites CL, Crocker CE, Wu HH, Bonnin A, Murray R, Calof AL. Molecular signals regulating proliferation of stem and progenitor cells in mouse olfactory epithelium. Dev Neurosci. 2004;26:166–80. doi: 10.1159/000082135. [DOI] [PubMed] [Google Scholar]
- Keller A, Margolis FL. Immunological studies of the rat olfactory marker protein. J Neurochem. 1975;24:1101–6. doi: 10.1111/j.1471-4159.1975.tb03883.x. [DOI] [PubMed] [Google Scholar]
- Keller A, Margolis FL. Isolation and characterization of rat olfactory marker protein. J Neurochem. 1976;251:6232–7. [PubMed] [Google Scholar]
- Kim H, Greer CA. The emergence of compartmental organization in olfactory bulb glomeruli during postnatal development. J Comp Neurol. 2000;422:297–311. [PubMed] [Google Scholar]
- Kornblum HI, Hussain R, Wiesen J, Miettinen P, Zurcher SD, Chow K, Derynck R, Werb Z. Abnormal astrocyte development and neuronal death in mice lacking the epidermal growth factor receptor. J Neurosci Res. 1998;53:697–717. doi: 10.1002/(SICI)1097-4547(19980915)53:6<697::AID-JNR8>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Leung CT, Coulombe PA, Reed RR. Contribution of olfactory neural stem cells to tissue maintenance and regeneration. Nature Neurosci. 2007;10:720–716. doi: 10.1038/nn1882. [DOI] [PubMed] [Google Scholar]
- Mackay-Sim A, Chuah MI. Neurotrophic factors in the primary olfactory pathway. Prog Neurobiol. 2000;62:527–59. doi: 10.1016/s0301-0082(00)00009-5. [DOI] [PubMed] [Google Scholar]
- Mahalik TJ. Apparent apoptotic cell death in the olfactory epithelium of adult rodents: death occurs at different developmental stages. J Comp Neurol. 1996;372:457–64. doi: 10.1002/(SICI)1096-9861(19960826)372:3<457::AID-CNE8>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Nef S, Lush ME, Shipman TE, Parada LF. Neurotrophins are not required for normal embryonic development of olfactory neurons. Dev Biol. 2001;234:80–92. doi: 10.1006/dbio.2001.0240. [DOI] [PubMed] [Google Scholar]
- Newman MP, Féron F, Mackay-Sim A. Growth factor regulation of neurogenesis in adult olfactory epithelium. Neuroscience. 2000;99:343–50. doi: 10.1016/s0306-4522(00)00194-9. [DOI] [PubMed] [Google Scholar]
- Robinson AM, Conley DB, Kern RC. Olfactory neurons in bax knockout mice are protected from bulbectomy-induced apoptosis. Neuroreport. 2003;14:1891–4. doi: 10.1097/00001756-200310270-00002. [DOI] [PubMed] [Google Scholar]
- Roskams AJ, Bethel MA, Hurt KJ, Ronnet GV. Sequential expression of Trks A, B and C in the regenerating olfactory epithelium. J Neurosci. 1996;16:1294–1307. doi: 10.1523/JNEUROSCI.16-04-01294.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmued LC, Albertson C, Slikker W., Jr Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 1997;751:37–46. doi: 10.1016/s0006-8993(96)01387-x. [DOI] [PubMed] [Google Scholar]
- Schwob JE, Szumowski KE Mieleszko, Stasky AA. Olfactory sensory neurons are trophically dependent on the olfactory bulb for their prolonged survival. J. Neurosci. 1992;12:3896–3919. doi: 10.1523/JNEUROSCI.12-10-03896.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwob JE. Neural regeneration and the peripheral olfactory system. Anat Rec (New Anat) 2002;269:33–49. doi: 10.1002/ar.10047. [DOI] [PubMed] [Google Scholar]
- Shetty RS, Bose SC, Nickell MD, McIntyre JC, Hardin DH, Harris AM, McClintock TS. Transcriptional changes during neuronal death and replacement in the olfactory epithelium. Mol Cell Neurosci. 2005;30:583–600. [PubMed] [Google Scholar]
- Shieh PB, Ghosh A. Molecular mechanisms underlying activity-dependent regulation of BDNF expression. J Neurobiol. 1999;41:127–34. [PubMed] [Google Scholar]
- Simpson PJ, Wang E, Moon C, Matarazzo V, Cohen DR, Liebl DJ, Ronnett GV. Neurotrophin-3 signaling maintains maturational homeostasis between neuronal populations in the olfactory epithelium. Mol Cell Neurosci. 2003;24:858–74. doi: 10.1016/j.mcn.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV, Galletley N, Isacson O, Svendsen CN. Survival of adult basal forebrain cholinergic neurons after loss of target neurons. Science. 1990;247:338–342. doi: 10.1126/science.1688664. [DOI] [PubMed] [Google Scholar]
- Suurmeijer AJ, van der Wijk J, van Veldhuisen DJ, Yang F, Cole GM. Fractin immunostaining for the detection of apoptotic cells and apoptotic bodies in formalin-fixed and paraffin-embedded tissue. Lab Invest. 1999;79:619–20. [PubMed] [Google Scholar]
- Verhaagen J, Oestreicher AB, Grillo M, Khew-Goodall Y-S, Gispen WH, Margolis F. Neuroplasticity in the olfactory system: differential effects of central and peripheral lesions of the primary olfactory pathway on the expression of B50/GAP43 and the olfactory marker protein. J Neurosci Res. 1990;26:31–44. doi: 10.1002/jnr.490260105. [DOI] [PubMed] [Google Scholar]
- Vigers AJ, Böttger B, Baquet ZC, Finger TE, Jones KR. Neurotrophin-3 is expressed in a discrete subset of olfactory receptor neurons in the mouse. J Comp Neurol. 2003;463:221–35. doi: 10.1002/cne.10752. [DOI] [PubMed] [Google Scholar]
- Watt WC, Sakano H, Lee ZY, Reusch JE, Trinh K, Storm DR. Odorant stimulation enhances survival of olfactory sensory neurons via MAPK and CREB. Neuron. 2004;41:955–67. doi: 10.1016/s0896-6273(04)00075-3. [DOI] [PubMed] [Google Scholar]
- Weiler E, Farbman AI. Mitral cell loss following lateral olfactory tract transection increases proliferation density in rat olfactory epithelium. Eur J Neurosci. 1999;11:3265–3275. doi: 10.1046/j.1460-9568.1999.00748.x. [DOI] [PubMed] [Google Scholar]
- Wu HH, Ivkovic S, Murray RC, Jaramillo S, Lyons KM, Johnson JE, Calof AL. Autoregulation of neurogenesis by GDF11. Neuron. 2003;37:197–207. doi: 10.1016/s0896-6273(02)01172-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.