

Abstract
Impairment of the ubiquitin-proteasome degradation system has recently been suggested to be related to the onset of neurodegenerative disorders such as Alzheimer′s disease and Parkinson's disease. In this study, we investigated whether intracellular α-synuclein affects proteasome activity in SH-SY5Y cells. To monitor intracellular proteasome activity, we used a reporter consisting of a short peptide degron fused to the carboxyl-terminus of green fluorescent protein (GFP-CL1), which is known to be degraded by proteasome. The level of intact GFP-CL1 was dramatically increased by coexpression of GFP-CL1 and α-synuclein, as judged by confocal microscopic and immunoblot analyses. Expression of two pathogenic mutants of α-synuclein, A30P and A53T, and phosphomimetic S129D mutant increased the intensities of GFP more effectively than did wild-type α-synuclein. GFP fluorescence in cells transfected with Δ73-83 mutant or β-synuclein, which does not assemble into filaments in vitro, was not changed as compared with that in cells expressing GFP-CL1 alone. Thus, the ability of α-synuclein to inhibit proteasome activity is related to its propensity to assemble into filaments. Furthermore, we observed that some compounds inhibiting α-synuclein filament formation in vitro prevented the α-synuclein-mediated proteasome dysfunction in cells transfected with both GFP-CL1 and α-synuclein. The cellular model expressing both GFP-CL1 and α-synuclein may be a useful tool to screen compounds protecting neurons from α-synuclein-mediated proteasome dysfunction.
Parkinson's disease (PD) is one of the most common neurodegenerative disorders. It is characterized by loss of dopaminergic neurons in the substantia nigra and by intracellular inclusion bodies known as Lewy bodies and Lewy neuritis. α-Synuclein is a natively unfolded protein and is deposited in a hyperphosphorylated form as the major component of these filamentous inclusions (1−3). Genetic studies have revealed that missense mutations (A30P, E46K, and A53T) in the α-synuclein gene cause familial forms of PD and dementia with Lewy bodies (DLB) (4−6). It has also been reported that multiplications (duplication and triplication) of α-synuclein gene cause an inherited form of PD and DLB (7−11). Thus, an increased level of intracellular α-synuclein is thought to contribute to the onset of familial PD.
Intracellular and extracellular protein deposits may cause neuronal cell death and may signal onset of many neurodegenerative diseases. We have reported that phosphorylated α-synuclein is partially ubiquitinated in α-synucleinopathy brains (12). We also found that ubiquitination of α-synuclein occurs in cultured cells at sites identical to those of filamentous α-synuclein ubiquitinated in vitro, suggesting that ubiquitination may be a late event in the formation of Lewy bodies (13). A recent paper confirmed our findings and showed that monoubiquitination and multiubiquitination at Lys12, 21, and 23 are detectable on phosphorylated α-synuclein in α-synucleinopathy brains (14). These findings indicate that the ubiquitin-proteasome system (UPS) is targeted to abnormal α-synuclein in the brains of patients but that ubiquitinated α-synuclein cannot be degraded by the UPS, suggesting that the UPS may not be effective to clear the abnormal form of α-synuclein.
The UPS functions in cellular quality control by degrading misfolded, unassembled, or damaged proteins that might create toxic intracellular aggregates (15,16). As the proteasome efficiently degrades multiubiquitinated proteins, the presence of elevated levels of ubiquitin conjugates in intracellular deposits of aggregated proteins in most neurodegenerative diseases suggests a linkage between UPS dysfunction and pathogenesis. Functional relationships between α-synuclein and the UPS have been described (17−19). Synder et al. reported that aggregated and monomeric α-synuclein inhibit proteasomal function in vitro(20). Lindersson et al. also observed proteasomal inhibition by α-synuclein filaments and oligomers in vitro(21). In cultured cells, it remains controversial whether expression of α-synuclein affects intracellular proteasome activity. Martin-Clemente et al. reported that α-synuclein expression level does not significantly affect proteasome function in PC12 cells (22), while Fujita et al. reported that proteasome activity was significantly decreased in cells overexpressing wild-type α-synuclein (23). Stefanis et al. described that expression of A53T mutant, but not wild-type α-synuclein, inhibits proteasome activity in PC12 cells (24). It was also reported that cell-produced α-synuclein oligomers are targeted to and impair the 26S proteasome (25).
Recently, Bence et al. reported impairment of the ubiquitin-proteasome system by the expression of an exon 1 fragment of huntingtin containing expanded polyglutamine homopolymer in cultured cells (26). They used a green fluorescent protein (GFP) reporter that is specifically degraded by the cellular proteasome. This fluorescent reporter system is also useful for facile detection of proteasome activity in Caenorhabditis elegans(27).
In this study, to investigate whether intracellular α-synuclein affects proteasome activity, we have established a more sensitive assay system to monitor proteasome activity in cells using the GFP reporter and demonstrated impairment of proteasome function by expression of α-synuclein in SH-SY5Y cells. We also found that two pathogenic mutations of α-synuclein (A30P and A53T) and S129D mutant, which mimics phosphorylation, inhibit proteasome activity more effectively than does wild-type α-synuclein, while Δ73-83 mutant and β-synuclein, which do not form fibrils in vitro, do not affect proteasome function. Furthermore, we used our cellular model to screen for compounds protecting cells from proteasome inhibition by α-synuclein and found that some polyphenols attenuate α-synuclein-induced proteasome impairment.
Experimental Procedures
Construction of Plasmids
The GFP-CL1 plasmid was constructed by digestion of pEGFP-C1 (Clontech) with BglII and SalI and ligation with hybridized oligonucleotides termed CL1s (5′-GATCCGCTTGTAAAAATTGGTTTTCTTCTTTATCTCATTTTGTTATTCATTTATAAG-3′) and CL1a (5′-TCGACTTATAAATGAATAACAAAATGAGATAAAGAAGAAAACCAATTTTTACAAGCG-3′). The resulting construct, the GFP gene containing an oligonucleotide encoding ACKNWFSSLSHFVIHL, was confirmed by DNA sequencing and designated pEGFP-CL1.
Human α-synuclein, β-synuclein, tau 3R1N, and 4R1N cDNAs in pRK172 vector were kind gifts from Dr. M. Goedert. The open reading frames of α-synuclein, β-synuclein, tau 3R1N, and 4R1N were subcloned into the mammalian expression vector pcDNA3. To construct plasmids encoding α-synuclein mutants A30P, A53T, S129A, and S129D, we performed site-directed mutagenesis using a site-directed mutagenesis kit (Strategene). We constructed a deletion mutant (residues 73−83: Δ73-83) of α-synuclein using PCR with the forward primer (5′-GGAGCAGGGAGCATTGCAGCA-3′) and the reverse primer (5′- CGTCACCACTGCTCCTCCAAC-3′) using pcDNA3-α-synuclein as a template. The amplified fragments were self-ligated, and the resulting plasmids designated pcDNA-α-synuclein Δ73-83. All constructs were verified by DNA sequencing.
Cell Culture and Plasmid Expression
SH-SY5Y cells were cultured in DMEM/F12 medium (Sigma) supplemented with 10% (v/v) fetal calf serum, Penicillin-Streptomycin-Glutamine (GIBCO), and MEM Non-Essential Amino Acids Solution (GIBCO). The cells were maintained at 37 °C in a humidified atmosphere of 5% (v/v) CO2. Cells were grown to 50% confluence in six-well culture dishes for transient expression and then transfected with various expression plasmids using FuGENE6 (Roche) according to the manufacturer′s instructions.
Confocal Immunofluorescence Microscopy
SH-SY5Y cells were grown on a coverslip (15 mm × 15 mm) and transfected with expression vector (total 1.3 μg, e.g., 1 μg of pcDNA3-α-synuclein plus 0.3 μg of pEGFP-CL1). After incubation for indicated times, transfected cells on the coverslips were fixed with 4% (w/v) paraformaldehyde in phosphate-buffered saline (PBS) for 30 min. The coverslips were then incubated in 50 mM NH4Cl in PBS for 10 min, and cell permeabilization was performed with 0.2% (v/v) Triton X-100 in PBS for 10 min. The cells were blocked for 30 min in 5% (w/v) BSA in PBS, then incubated with anti-α-synuclein monoclonal antibody, Syn102 (28) (1:500 dilution) for 1 h at 37 °C, followed by TRITC-labeled goat anti-mouse IgG (Sigma, 1:500 dilution) as the secondary antibody for 1 h at 37 °C. They were washed again and further incubated with TO-PRO-3 (Molecular Probes, 1:3000 dilution in PBS) for 45 min at 37 °C to stain nuclear DNA and analyzed using a LSM5 Pascal confocal laser microscope (Zeiss).
Sequential Extraction of Proteins and Immunoblotting
SH-SY5Y cells were grown in a six-well plate and transiently transfected with expression plasmids (total 1.3 μg). After incubation for 48−72 h, cells were harvested and lysed in TS buffer [50 mM Tris-HCl buffer, pH 7.5, 0.15 M NaCl, 5 mM ethylenediaminetetraacetic acid, 5 mM ethylene glycol bis(β-aminoethyl ether)-N,N,N,N-tetraacetic acid, and protease inhibitor cocktail (Roche)]. The lysate was centrifuged at 290,000g for 20 min at 4 °C, and the supernatant recovered as the TS-soluble fraction. The TS-insoluble pellet was lysed in TS buffer containing 1% (v/v) Triton X-100 (TX) and centrifuged at 290,000g for 20 min at 4 °C. The supernatant was collected as the TX-soluble fraction. The TX-insoluble pellet was further lyzed in SDS-sample buffer and heated for 5 min. Protein concentration was estimated by using the BCA Protein Assay Kit (Pierce). Samples (20 μg) were separated by 13.5% (v/v) SDS−PAGE using the Tris-tricine buffer system and proteins were transferred onto polyvinylidene difluoride membrane (Millipore). The blots were then blocked with 3% (v/v) gelatin and incubated overnight with anti-GFP monoclonal antibody (MBL), anti-Syn102, anti-human tau monoclonal antibody (HT7, Thermo Scientific) or anti-α-tublin monoclonal antibody (Sigma) in 10% (v/v) calf serum at a dilution of 1:1,000−5,000 at room temperature. After washing, blots were incubated with horseradish peroxidase-labeled secondary antibody (Bio-Rad) at a dilution of 1:10,000 in 10% (v/v) calf serum for 1 h at room temperature or a biotin-labeled secondary antibody (Vector) for 2 h at room temperature. Signals were detected using the chemiluminescence reagent Immunostar (Wako) or the ABC staining kit (Vector).
Measurement of Proteasome Activity Using a Peptide Substrate
SH-SY5Y cells transfected with expression plasmids were cultured for 72 h or treated with 20 μM MG132 for 6 h. Cells were harvested and cytosolic fraction was prepared as follows. Cells were resuspended in 100 μL of phosphate-buffered saline (PBS) and disrupted by sonication. Insoluble material was removed by ultracentrifugation at 290,000g for 20 min at 4 °C. The supernatant was assayed for proteasome activity by using a fluorescent peptide substrate, benzyloxycarbonyl-Leu-Leu-Glu-7-amido-4-methylcoumarin (z-LLE-MCA, Peptide Institute, Inc.). Aliquots (10 μL) were mixed with 0.1 M Tris-HCl buffer, pH 7.5 and water in a total volume of 1 mL. After preincubation at 37 °C for 10 min, 10 μL of 10 mM peptide substrate was added and the mixture was incubated for 30 min. A solution of 2% (v/v) acetic acid (1 mL) was added to the reaction mixture, and 7-amino-4-methylcoumarin (AMC) release was measured fluorometrically (excitation wavelength of 365 nm; emission wavelength of 460 nm). Enzyme activity was described as arbitrary unit/mg protein.
Screening for Compounds Preventing Proteasome Inhibition by α-Synuclein in Cultured Cells
SH-SY5Y cells were grown in six-well plates or on coverslips and transfected with both α-synuclein (1 μg) and GFP-CL1 plasmids (0.3 μg). Various compounds (all at 40 μM) were added to the culture medium 2 h after transfection. Cells were harvested after incubation for 72 h, and the immunoblot and/or confocal microscopic analyses were performed as above. A decrease in GFP level in transfected cells treated with a compound, compared with the GFP level in transfected cells without such treatment, indicated that the compound rescues cells from proteasome dysfunction caused by expression of α-synuclein.
Results
Cellular Expression of GFP-CL1
To establish a sensitive and convenient assay system for proteasome activity in cultured cells, we constructed GFP-CL1, which is GFP fused at the C-terminus with the 16-residue degron peptide CL1 (26,29). We transiently transfected SH-SY5Y cells with GFP or GFP-CL1, and analyzed the cells using confocal microscopy. As shown in Figure 1A, the fluorescence of GFP in cells transfected with GFP-CL1 was less than that in cells expressing GFP. The proteasome inhibitor MG132 increased fluorescence intensity in GFP-CL1-transfected cells (Figure 1A). Thus, we reconfirmed that the CL1 sequence specifically targeted normally stable GFP for efficient degradation by proteasome, as reported previously (26).
Figure 1.

GFP-CL1 is degraded by proteasome in SH-SY5Y cells. (A) SH-SY5Y cells grown on coverslips were transfected with GFP or GFP-CL1 using FuGENE6. After an overnight incubation with or without 1 μM MG132, cells were fixed (total 48 h after transfestion), stained with TO-PRO-3 (Blue), and analyzed by confocal microscopy. Scale bar = 100 μm. (B) SH-SY5Y cells were transfected with GFP or GFP-CL1 and treated overnight with or without 1 μM MG132. Cells were harvested, and cell lysate was fractionated as described in Experimental Procedures and analyzed by immunoblotting using anti-GFP antibody. TS, Tris-soluble fraction; TX, Triton X-100-soluble fraction; SDS, SDS-soluble fraction. (C) SH-SY5Y cells were transfected with GFP-CL1 and treated with MG132 (0−50 μM) for 6 h. The cells were harvested, and the TX-insoluble fraction was analyzed by immunoblotting with anti-GFP antibody.
To biochemically characterize the proteasomal degradation of GFP-CL1, we fractionated lysates of cells transfected with GFP-CL1 or GFP. The TS-, TX-, and SDS-soluble fractions were analyzed by immunoblotting using anti-GFP antibody. As shown in Figure 1B, almost all GFP was recovered in the TS fraction in cells transfected with GFP, although a little GFP was detected in the TX-soluble fraction. In contrast, in cells expressing GFP-CL1, only weak GFP immunoreactivity was detected in the TS- and TX-insoluble fractions because of GFP-CL1 degradation by proteasome in transfected cells. Interestingly, in cells expressing GFP-CL1 treated with MG132, a band of uncleaved GFP-CL1 was detected in the SDS-soluble fraction in addition to the weak bands seen in the TS- and TX-soluble fractions, suggesting that uncleaved GFP-CL1 unexpectedly aggregates in cells when proteasome activity is inhibited by MG132 treatment. We also reconfirmed that MG132 treatment effectively and dose-dependently blocked the degradation of GFP-CL1 (Figure 1C).
Expression of α-Synuclein Inhibits Cellular Proteasome Activity
To investigate whether expression of α-synuclein might affect proteasome function in cultured cells, we cotransfected both α-synuclein and GFP-CL1 into SH-SY5Y cells. To reconfirm that GFP-CL1 is effectively degraded by proteasome, cells transfected with GFP-CL1 were treated with several protease inhibitors. SDS-soluble fraction of cell lysate was subjected to immunoblot analysis using anti-GFP antibody. As shown in Figure 2A, increased levels of GFP-CL1 band were observed in cells coexpressing GFP-CL1 and α-synuclein (lane 6), indicating that proteasome activity is inhibited effectively by expression of α-synuclein. Similar results were obtained when we treated cells expressing GFP-CL1 with MG132 or lactacystin (lane 3 and 4). On the other hand, leucyl-leucinal (LL, a calpain inhibitor) or chloroquine (a lysosomal inhibitor) did not inhibit GFP-CL1 degradation (lanes 2 and 5). We then tested whether the proteasome inhibition by α-synuclein is dose-dependent, i.e., dependent on the α-synuclein expression level. SH-SY5Y cells were transfected with both pEGFP-CL1 (0.3 μg) and pcDNA3-α-synuclein (0, 0.2, 0.5, or 1 μg; total 1.3 μg plasmids), followed by immunoblot analysis. The result showed that the level of intact GFP-CL1 increased in parallel with increased expression of α-synuclein (Figure 2B). Furthermore, we tested whether expression of tau protein which is also an effective substrate for proteasome (30), inhibits intracellular proteasome activity in SH-SY5Y cells. We found that expression of tau protein (3R1N and 4R1N) did not affect the proteasome activity in SH-SY5Y cells, suggesting that the effect of α-synuclein on the proteasome activity observed in this study is not simply due to overexpression of a protein, but is specific for α-synuclein. We also confirmed the absence of significant cell death in our cellular model transiently expressing α-synuclein, suggesting that the decreased proteasome activity found in cells expressing α-synuclein is not caused by cellular damage due to the toxicity of expressed α-synuclein.
Figure 2.

Expression of α-synuclein inhibits proteasome activity in SH-SY5Y cells. (A) SH-SY5Y cells transfected with GFP-CL1 were treated with 1 μM LL, MG132, lactacystin, and chloroquine overnight. In the case of coexpression, cells were transfected with both GFP-CL1 and α-synuclein using FuGENE6. The cells were harvested, and the Triton X-100-insoluble and SDS-soluble fractions were prepared and analyzed by immunoblotting using an anti-GFP antibody. The results of quantitative analysis of the GFP-CL1 bands, expressed as means + SD (n = 3), are shown below the blot. (B) SH-SY5Y cells were transfected with GFP-CL1 (0.3 μg), α-synuclein (0, 0.2, 0.5, or 1 μg), and/or pcDNA3 empty vector (total plasmids = 1.3 μg) and incubated for 72 h. The cells were harvested, and then Tris-soluble (lower panels) and SDS-soluble fractions (upper panels) were prepared. Immunoblot analyses were performed using anti-GFP (upper panels) or anti-Syn102 (lower panels) antibodies. The results of quantitative analysis of the GFP-CL1 bands, expressed as means + SD (n = 3), are shown below the blot. (C) SH-SY5Y cells were transfected with both GFP-CL1 (0.3 μg) and α-synuclein (1 μg) or pcDNA3-tau 3R1N or 4R1N vector (1 μg) and incubated for 72 h. The cells were harvested, and the TX-insoluble fraction was analyzed by immunoblotting with anti-GFP antibody. The TS-soluble fraction was also analyzed by using anti-Syn102, anti-HT7, and anti-tubulin antibodies (as a loading control).
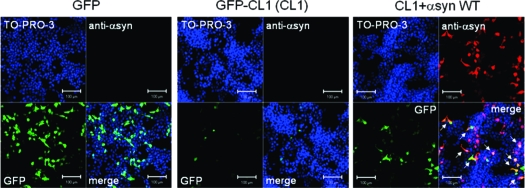
To visually monitor proteasome inhibition due to expression of α-synuclein, SH-SY5Y cells were transiently cotransfected with α-synuclein and GFP-CL1, and transfected cells were analyzed by confocal microscopy. As shown in Figure 3C, the numbers of GFP-positive cells were significantly increased in cells transfected with wild-type α-synuclein and GFP-CL1, compared with cells transfected with GFP-CL1 alone (Figure 3B). This is in good agreement with the above immunoblotting data (Figure 2), indicating that expression of α-synuclein inhibited proteasome activity.
Figure 3.

Expression of α-synuclein mutants affects proteasome activity in SH-SY5Y cells. SH-SY5Y cells grown on coverslips were transfected with GFP (A), GFP-CL1 (B), or both GFP-CL1 and wild-type α-synuclein (C), A30P (D), A53T (E), Δ73-83 (F), or β-synuclein (G). Cells were fixed 72 h after transfection and stained with anti-Syn102 (Red) and TO-PRO-3 (blue). Intracellular α-synuclein inhibits proteasome activity in cells expressing both GFP-CL1 and α-synuclein (arrows). Scale bar = 100 μm.
Taken together, these results showed that proteasome activity can be specifically and sensitively analyzed by monitoring the fluorescence intensity or immunoreactivity of GFP-CL1 in cultured cells and that the expression of α-synuclein impairs cellular proteasome function.
Effects of Expression of α-Synuclein Mutants and β-Synuclein on Proteasome Activity
Recombinant α-synuclein with a pathogenic mutation (A30P, A53T, or E46K) is more prone to assemble into filaments in vitro than the wild-type protein (31,32)). β-Synuclein and γ-synuclein (33) and the α-synuclein deletion mutant Δ73-83 (deletion of 73−83 residues) did not form fibrils in vitro (Nonaka et al., unpublished data). To study the effects of α-synuclein mutations on cellular proteasome activity, we transiently cotransfected these mutants and GFP-CL1 into SH-SY5Y cells and examined the cells using confocal microscopy. As shown in Figure 3, we found that increased GFP signals were observed in cells transfected with A30P (Figure 3D) or A53T (Figure 3E) compared with the signal seen in cells transfected with wild-type α-synuclein (Figure 3C), although the expression levels of the α-synuclein proteins were almost equal (Figure 3 and 4A). In contrast, little GFP fluorescence was detected in cells transfected with the α-synuclein deletion mutant Δ73-83 (Figure 3F) or β-synuclein (Figure 3G).
Figure 4.

Effects of expression of α-synuclein mutants on proteasome activity. (A, B) Immunoblot analyses of extracted proteins from cells transfected with both GFP-CL1 and wild-type α-synuclein or a mutant (A30P, A53T, S129A, S129D, Δ73-83 or β-synuclein). Cells were harvested 72 h after transfection, and Tris-soluble (lower panels) and SDS-soluble fractions (upper panels) were prepared. Immunoblot analyses were performed using anti-GFP (upper panels) or anti-Syn102 (lower panels) antibodies. The results of quantitative analysis of the GFP-CL1 bands, expressed as means + SD (n = 3), are shown below the blot.
We also quantitatively examined the effects of these α-synuclein mutations on proteasome activity using immunoblot analysis. SH-SY5Y cells were transfected with both GFP-CL1 and wild-type α-synuclein, or the A30P, A53T, or Δ73-83 mutant, or β-synuclein. Uncleaved GFP-CL1 was recovered in the TX-insoluble (SDS-soluble) fraction as described above. As shown in Figure 4A, the level of uncleaved GFP-CL1 in cells transfected with A53T was about 30% higher than that in cells transfected with wild-type α-synuclein (Figure 4A, lane 4). A slightly higher level of GFP-CL1 was detected in cells transfected with the A30P mutant (Figure 4A, lane 3) compared with that in cells transfected with wild-type α-synuclein (Figure 4A, lane 2). In contrast, the level of GFP-CL1 in cells transfected with Δ73-83 or β-synuclein was negligible, as was that in control cells (Figure 4A, lane 5 and 6). The expression levels of α-synuclein protein in Tris-soluble fractions were almost the same. These results showed that mutants of α-synuclein that are prone to aggregate in vitro have stronger inhibitory effects on cellular proteasome activity than does the wild-type protein and that Δ73-83 and β-synuclein, neither of which form fibrils in vitro, do not inhibit the proteasome.
Expression of Phosphomimetic α-Synuclein Mutant S129D Enhanced Its Inhibition of Proteasome Activity
Aggregated α-synuclein in brains of α-synucleinopathy patients is hyperphosphorylated at Ser129, and this post-translational modification facilitates self-aggregation in vitro(3). Altering Ser129 to the negatively charged residue Asp (S129D) to mimic phosphorylation significantly enhances α-synuclein toxicity in a Drosophila model (34). We asked whether phosphorylation of α-synuclein might affect proteasome activity in cultured cells. SH-SY5Y cells were transfected with GFP-CL1 and wild-type or mutant α-synuclein. Immunoreactivity of GFP-CL1 in cells transfected with S129A was similar to that of cells transfected with wild-type α-synuclein, while the levels of GFP-CL1 in cells expressing S129D mutant to mimic phosphorylation was significantly higher than those in cells transfected with the wild-type or S129A (Figure 4B). These results suggested that phosphorylation of Ser129 may facilitate inhibition of proteasome function by intracellular α-synuclein.
Comparison of Proteasome Activity Assay Using GFP-CL1 with a Conventional Cleaving Assay Using a Fluorescent Peptide Substrate
To compare the sensitivity of the assay of proteasome activity using GFP-CL1 with that of the conventional assay using a fluorescent peptide, we evaluated biochemically the proteasome activity of lysates from cells transfected with α-synuclein. SH-SY5Y cells were transfected with wild-type α-synuclein for 72 h, and proteasome activity in cell lysates was measured using a peptide substrate specific for proteasome, zLLE-MCA. As shown in Figure 5, zLLE-MCA-cleaving activity in cells transfected with wild-type α-synuclein was decreased slightly (∼10% inhibition) compared with that in cells transfected with empty vector, while MG132 had a potent inhibitory effect on proteasome activity (∼80% inhibition). On the other hand, as described above, the levels of GFP-CL1 in cells transfected with GFP-CL1 and α-synuclein were increased about 5 times compared with those in cells expressing GFP-CL1 (Figure 4A).
Figure 5.

Measurements of proteasome activity using a fluorescent peptide substrate. SH-SY5Y cells were transfected with pcDNA3 empty vector, wild-type α-synuclein (WT), A30P, A53T, S129D, Δ73-83 for 72 h or treated with 20 μM MG132 for 6 h. Cytosolic fractions were prepared and assayed using z-LLE-MCA as a substrate. The results were expressed as means + SD (n = 3).
As shown in Figure 5, the zLLE-MCA-cleaving activity in cells transfected with A30P was almost equal to that in cells expressing A53T, which in turn was decreased slightly compared with that in cells transfected with wild-type α-synuclein. Proteasome activity in cells transfected with Δ73-83 was found to be the same as that in cells transfected with empty vector. It was also revealed that S129D had a slightly stronger inhibitory effect on proteasome activity than did wild-type α-synuclein. Taken together, as compared with the results shown in Figure 3 and 4, these results clearly indicate that a method in this study by detecting the fluorescence of GFP-CL1 or immunoblot analysis using anti-GFP antibody is a simple and sensitive for monitoring proteasome activity in cells.
Small Molecular Compounds Attenuate Cellular Inhibition of Proteasome Activity by Expression of α-Synuclein
In this study, we have shown that proteasomal activity is impaired by expression of α-synuclein in SH-SY5Y cells. Impairment of proteasome function leads to induction of apoptosis, followed by cell death, so it is important to protect cells from such inhibition by intracellular α-synuclein. These results raised the possibility that our cellular model, in which both GFP-CL1 and α-synuclein are expressed, might be useful to screen for small molecules preventing proteasome inhibition mediated by expression of α-synuclein. Recently, we have reported that several compounds, including polyphenols, phenothiazines, and porphyrins, inhibit aggregation of recombinant α-synuclein in vitro(35). We therefore asked whether these compounds might affect proteasome inhibition by α-synuclein in cultured cells. Compounds of interest were added to the culture medium of cells transfected with both GFP-CL1 and α-synuclein 2 h after transfection. The cells were incubated for 72 h, harvested and analyzed by immunoblotting and confocal laser microscopy. Immunoblot analyses showed that the level of GFP-CL1 was decreased significantly when cells were treated with purpurogallin or myricetin (polyphenols), but not when they were treated with ferric-dehydroporphyrin IX (porphyrin) (Figure 6A). Cells treated with purpurogallin or myricetin displayed significant decreases in GFP-CL1 fluorescence by confocal microscopy (Figure 6B). Furthermore, we confirmed that exifone and gossypetin, plant polyphenols inhibiting fibril formation of α-synuclein in vitro(35), also attenuated α-synuclein-mediated proteasome inhibition (data not shown). We further observed that these small molecules did not affect α-synuclein expression (Figure 6A). These results indicate that these compounds may protect cells from proteasome dysfunction arising from expression of α-synuclein.
Figure 6.

Small molecules prevent proteasome dysfunction by intracellular α-synuclein. SH-SY5Y cells were transfected with GFP-CL1 alone or both GFP-CL1 and α-synuclein. Compounds (final 40 μM) were added 2 h after transfection. Cells were harvested after a 72-h incubation and analyzed by immunoblotting (A) and confocal microscopy (B). Immunoblot analyses of Tris-soluble (lower panels) and SDS-soluble fractions (upper panels) were performed using anti-GFP (upper panels) or anti-Syn102 (lower panels) antibodies. The decreased GFP-CL1 level in transfected cells treated with compound B or C, as compared with that of transfected cells treated with vehicle alone (DMSO), indicated that compound B or C, but not compound A, rescued cells from proteasome impairment by expression of α-synuclein.
Discussion
Recent studies have shown that aggregated and monomeric α-synuclein inhibit proteasomal function in vitro(20,21,36). In these reports, proteasome activity was measured in a cell-free system using fluorescent peptide substrates, and little is known about the effects of α-synuclein expression on proteasome activity in cultured cells. To examine this question, we tried to establish a sensitive and convenient method to measure proteasome activity using cultured cells transfected with a GFP-CL1 reporter plasmid. We further applied this system to show that transient coexpression of α-synuclein inhibits the proteasome activity of SH-SY5Y cells.
The GFP-CL1 reporter is a useful tool for the detection of proteasome activity in cells (26). Recently, Link et al. reported that the expression of GFP-CL1 in Caenorhabditis elegans muscle or neurons causes deposition of GFP-CL1 and leads to rapid paralysis (27). In this study, we also found that uncleaved GFP-CL1 was largely recovered in TX-insoluble fractions of SH-SY5Y cells transfected with GFP-CL1 and α-synuclein, while α-synuclein was recovered in TS- and TX-soluble fractions (Figure 1B and Figure S1 in Supporting Information), indicating that uncleaved GFP-CL1 was deposited, but α-synuclein was still soluble in transfected cells. Cell death was not detected in either cells transfected with GFP-CL1 alone or cells transfected with both GFP-CL1 and wild-type α-synuclein or mutants thereof, although Link et al. found that GFP-CL1 is toxic when expressed in C. elegans muscle cells (27). The discrepancy might be due to differences in the cell type used. Further studies are required in this regard. It is surprising that a relatively short addition of 16 residues to the C terminus of GFP can convert this normally soluble protein into a protein that strongly aggregates. We suggest that the C-terminal 16 residues may directly interfere with GFP folding, resulting in misfolded (but still fluorescent) forms that are prone to aggregate.
How does α-synuclein suppress intracellular proteasome activity? Several reports have described that α-synuclein may directly interfere with the proteasome. It was shown that α-synuclein binds to S6′ or S6a, a component of the 19S subunit in the 26S proteasome in cultured cells (23,37). It was also reported that aggregated α-synuclein, but not its monomer, effectively inhibited the 26S proteasome activity in vitro(20,21,25,36). Interestingly, we have shown that two pathogenic α-synuclein mutants, A30P and A53T, which are liable to form fibrils or oligomers in vitro, inhibited proteasome activity more strongly than did wild-type α-synuclein, while Δ73-83 mutant and β-synuclein, neither of which assemble into filaments in vitro, did not inhibit proteasome activity. Although we failed to detect any sarkosyl-insoluble aggregates of α-synuclein in cells transfected with wild-type or mutant α-synuclein (data not shown), these results raise the possibility that soluble prefibrillar intermediates, such as misfolded monomers, oligomers or protofibrils, may inhibit proteasome activity in transfected cells. Additional studies are needed to explore oligomers and protofibrils of α-synuclein in our cell model.
Phosphorylation seems to be a common mechanism controlling the neurotoxicity of aggregation-prone toxic proteins involved in neurodegenerative diseases such as tau (38,39), α-synuclein (3), and TDP-43 (40−42). We suggested that phosphorylation of α-synuclein may play a role in suppression of proteasome activity in cells. It was reported that expression of S129D mutant α-synuclein in Drosophila significantly enhances the toxicity (34). However, the mechanisms through which phosphomimetic S129D mutant causes cell death in the fly remain unknown. Phosphorylated α-synuclein at Ser129 is known to be more prone to aggregate than wild-type α-synuclein (3), and this may be related to its toxicity. Alternatively, inhibitory effects of the S129D mutant on intracellular proteasome activity, as shown in this study, may be involved in cell death.
Importantly, we have shown that intracellular α-synuclein may be toxic to neuronal cells because the protein causes proteasome dysfunction. It was previously reported that duplication and triplication of α-synuclein gene causes familial PD (7−11), indicating that overproduction of α-synuclein protein could lead to the onset of PD. It is reasonable to speculate that proteasome activity may also be affected in neurons of these patients in a manner similar to that shown in this study. It may be important to prevent α-synuclein-induced inhibition or decrease of proteasome activity. In this study, we screened for compounds protecting cells from the α-synuclein-mediated proteasome dysfunction and found that purpurogallin, myricetin, exifone, and gossypetin (polyphenols from plants) prevented proteasome impairment by α-synuclein. These compounds are thought to inhibit in vitro α-synuclein filament formation by stabilizing soluble prefibrillar intermediates (35). We speculate that these small molecules may interact with oligomeric forms of α-synuclein in cultured cells, although we can not exclude the possibility that these compounds directly regulate the activity of the proteasome. The precise mechanisms by which these polyphenols prevent α-synuclein-mediated proteasome dysfunction remain unknown. However, these compounds may be novel drug candidates for neurodegenerative diseases. Our cellular model system using coexpression of GFP-CL1 and α-synuclein should be a sensitive and useful tool for screening of compounds and drugs for the prevention and treatment of these disease.
Acknowledgments
We thank Drs. H. Mimuro and M. Masuda for helpful advice and discussions.
Supporting Information Available
Fractionation of cells expressing both of GFP-CL1 and α-synuclein. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by a Grant-in-Aid for Scientific Research on Priority Areas−Research on Pathomechanisms of Brain Disorders (to M.H., 20023038) and Grants-in-Aid for Scientific Research (B) (to M.H., 18300117) and (C) (to T.N., 19590297) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Supplementary Material
References
- Spillantini M. G.; Schmidt M. L.; Lee V. M.; Trojanowski J. Q.; Jakes R.; Goedert M. (1997) Alpha-synuclein in Lewy bodies. Nature 388, 839–840. [DOI] [PubMed] [Google Scholar]
- Baba M.; Nakajo S.; Tu P. H.; Tomita T.; Nakaya K.; Lee V. M.; Trojanowski J. Q.; Iwatsubo T. (1998) Aggregation of α-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am. J. Pathol. 152, 879–884. [PMC free article] [PubMed] [Google Scholar]
- Fujiwara H.; Hasegawa M.; Dohmae N.; Kawashima A.; Masliah E.; Goldberg M. S.; Shen J.; Takio K.; Iwatsubo T. (2002) α-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 4, 160–164. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos M. H.; Lavedan C.; Leroy E.; Ide S. E.; Dehejia A.; Dutra A.; Pike B.; Root H.; Rubenstein J.; Boyer R.; Stenroos E. S.; Chandrasekharappa S.; Athanassiadou A.; Papapetropoulos T.; Johnson W. G.; Lazzarini A. M.; Duvoisin R. C.; Di Iorio G.; Golbe L. I.; Nussbaum R. L. (1997) Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science 276, 2045–2047. [DOI] [PubMed] [Google Scholar]
- Kruger R.; Kuhn W.; Muller T.; Woitalla D.; Graeber M.; Kosel S.; Przuntek H.; Epplen J. T.; Schols L.; Riess O. (1998) Ala30Pro mutation in the gene encoding α-synuclein in Parkinson′s disease. Nat. Genet. 18, 106–108. [DOI] [PubMed] [Google Scholar]
- Zarranz J. J.; Alegre J.; Gomez-Esteban J. C.; Lezcano E.; Ros R.; Ampuero I.; Vidal L.; Hoenicka J.; Rodriguez O.; Atares B.; Llorens V.; Gomez Tortosa E.; del Ser T.; Munoz D. G.; de Yebenes J. G. (2004) The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 55, 164–173. [DOI] [PubMed] [Google Scholar]
- Singleton A. B.; Farrer M.; Johnson J.; Singleton A.; Hague S.; Kachergus J.; Hulihan M.; Peuralinna T.; Dutra A.; Nussbaum R.; Lincoln S.; Crawley A.; Hanson M.; Maraganore D.; Adler C.; Cookson M. R.; Muenter M.; Baptista M.; Miller D.; Blancato J.; Hardy J.; Gwinn-Hardy K. (2003) α-Synuclein locus triplication causes Parkinson's disease. Science 302, 841. [DOI] [PubMed] [Google Scholar]
- Chartier-Harlin M. C.; Kachergus J.; Roumier C.; Mouroux V.; Douay X.; Lincoln S.; Levecque C.; Larvor L.; Andrieux J.; Hulihan M.; Waucquier N.; Defebvre L.; Amouyel P.; Farrer M.; Destee A. (2004) Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet 364, 1167–1169. [DOI] [PubMed] [Google Scholar]
- Ibanez P.; Bonnet A. M.; Debarges B.; Lohmann E.; Tison F.; Pollak P.; Agid Y.; Durr A.; Brice A. (2004) Causal relation between α-synuclein gene duplication and familial Parkinson's disease. Lancet 364, 1169–1171. [DOI] [PubMed] [Google Scholar]
- Nishioka K.; Hayashi S.; Farrer M. J.; Singleton A. B.; Yoshino H.; Imai H.; Kitami T.; Sato K.; Kuroda R.; Tomiyama H.; Mizoguchi K.; Murata M.; Toda T.; Imoto I.; Inazawa J.; Mizuno Y.; Hattori N. (2006) Clinical heterogeneity of α-synuclein gene duplication in Parkinson's disease. Ann. Neurol. 59, 298–309. [DOI] [PubMed] [Google Scholar]
- Ikeuchi T.; Kakita A.; Shiga A.; Kasuga K.; Kaneko H.; Tan C. F.; Idezuka J.; Wakabayashi K.; Onodera O.; Iwatsubo T.; Nishizawa M.; Takahashi H.; Ishikawa A. (2008) Patients homozygous and heterozygous for SNCA duplication in a family with parkinsonism and dementia. Arch. Neurol. 65, 514–519. [DOI] [PubMed] [Google Scholar]
- Hasegawa M.; Fujiwara H.; Nonaka T.; Wakabayashi K.; Takahashi H.; Lee V. M.; Trojanowski J. Q.; Mann D.; Iwatsubo T. (2002) Phosphorylated α-synuclein is ubiquitinated in α-synucleinopathy lesions. J. Biol. Chem. 277, 49071–49076. [DOI] [PubMed] [Google Scholar]
- Nonaka T.; Iwatsubo T.; Hasegawa M. (2005) Ubiquitination of α-synuclein. Biochemistry 44, 361–368. [DOI] [PubMed] [Google Scholar]
- Anderson J. P.; Walker D. E.; Goldstein J. M.; de Laat R.; Banducci K.; Caccavello R. J.; Barbour R.; Huang J.; Kling K.; Lee M.; Diep L.; Keim P. S.; Shen X.; Chataway T.; Schlossmacher M. G.; Seubert P.; Schenk D.; Sinha S.; Gai W. P.; Chilcote T. J. (2006) Phosphorylation of Ser-129 is the dominant pathological modification of α-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 281, 29739–29752. [DOI] [PubMed] [Google Scholar]
- Ciechanover A.; Orian A.; Schwartz A. L. (2000) Ubiquitin-mediated proteolysis: biological regulation via destruction. Bioessays 22, 442–451. [DOI] [PubMed] [Google Scholar]
- Goldberg A. L. (2003) Protein degradation and protection against misfolded or damaged proteins. Nature 426, 895–899. [DOI] [PubMed] [Google Scholar]
- Tofaris G. K.; Razzaq A.; Ghetti B.; Lilley K. S.; Spillantini M. G. (2003) Ubiquitination of α-synuclein in Lewy bodies is a pathological event not associated with impairment of proteasome function. J. Biol. Chem. 278, 44405–44411. [DOI] [PubMed] [Google Scholar]
- Sampathu D. M.; Giasson B. I.; Pawlyk A. C.; Trojanowski J. Q.; Lee V. M. (2003) Ubiquitination of α-synuclein is not required for formation of pathological inclusions in α-synucleinopathies. Am. J. Pathol. 163, 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. W.; Corboy M. J.; DeMartino G. N.; Thomas P. J. (2003) Endoproteolytic activity of the proteasome. Science 299, 408–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder H.; Mensah K.; Theisler C.; Lee J.; Matouschek A.; Wolozin B. (2003) Aggregated and monomeric α-synuclein bind to the S6′ proteasomal protein and inhibit proteasomal function. J. Biol. Chem. 278, 11753–11759. [DOI] [PubMed] [Google Scholar]
- Lindersson E.; Beedholm R.; Hojrup P.; Moos T.; Gai W.; Hendil K. B.; Jensen P. H. (2004) Proteasomal inhibition by α-synuclein filaments and oligomers. J. Biol. Chem. 279, 12924–12934. [DOI] [PubMed] [Google Scholar]
- Martin-Clemente B.; Alvarez-Castelao B.; Mayo I.; Sierra A. B.; Diaz V.; Milan M.; Farinas I.; Gomez-Isla T.; Ferrer I.; Castano J. G. (2004) α-Synuclein expression levels do not significantly affect proteasome function and expression in mice and stably transfected PC12 cell lines. J. Biol. Chem. 279, 52984–52990. [DOI] [PubMed] [Google Scholar]
- Fujita M.; Sugama S.; Nakai M.; Takenouchi T.; Wei J.; Urano T.; Inoue S.; Hashimoto M. (2007) α-Synuclein stimulates differentiation of osteosarcoma cells: relevance to down-regulation of proteasome activity. J. Biol. Chem. 282, 5736–5748. [DOI] [PubMed] [Google Scholar]
- Stefanis L.; Larsen K. E.; Rideout H. J.; Sulzer D.; Greene L. A. (2001) Expression of A53T mutant but not wild-type α-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J. Neurosci. 21, 9549–9560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmanouilidou E., Stefanis L., Vekrellis K. (2008) Cell-produced α-synuclein oligomers are targeted to, and impair, the 26S proteasome, Neurobiol. Aging Epub ahead of print, DOI: 10.1016/j.neurobiolaging.2008.07.008. [DOI] [PubMed] [Google Scholar]
- Bence N. F.; Sampat R. M.; Kopito R. R. (2001) Impairment of the ubiquitin-proteasome system by protein aggregation. Science 292, 1552–1555. [DOI] [PubMed] [Google Scholar]
- Link C. D.; Fonte V.; Hiester B.; Yerg J.; Ferguson J.; Csontos S.; Silverman M. A.; Stein G. H. (2006) Conversion of green fluorescent protein into a toxic, aggregation-prone protein by C-terminal addition of a short peptide. J. Biol. Chem. 281, 1808–1816. [DOI] [PubMed] [Google Scholar]
- Tu P. H.; Galvin J. E.; Baba M.; Giasson B.; Tomita T.; Leight S.; Nakajo S.; Iwatsubo T.; Trojanowski J. Q.; Lee V. M. (1998) Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble α-synuclein. Ann. Neurol. 44, 415–422. [DOI] [PubMed] [Google Scholar]
- Gilon T.; Chomsky O.; Kulka R. G. (1998) Degradation signals for ubiquitin system proteolysis in Saccharomyces cerevisiae. EMBO J. 17, 2759–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David D. C.; Layfield R.; Serpell L.; Narain Y.; Goedert M.; Spillantini M. G. (2002) Proteasomal degradation of tau protein. J. Neurochem. 83, 176–185. [DOI] [PubMed] [Google Scholar]
- Conway K. A.; Harper J. D.; Lansbury P. T. (1998) Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease. Nat. Med. 4, 1318–1320. [DOI] [PubMed] [Google Scholar]
- Choi W.; Zibaee S.; Jakes R.; Serpell L. C.; Davletov B.; Crowther R. A.; Goedert M. (2004) Mutation E46K increases phospholipid binding and assembly into filaments of human α-synuclein. FEBS Lett. 576, 363–368. [DOI] [PubMed] [Google Scholar]
- Biere A. L.; Wood S. J.; Wypych J.; Steavenson S.; Jiang Y.; Anafi D.; Jacobsen F. W.; Jarosinski M. A.; Wu G. M.; Louis J. C.; Martin F.; Narhi L. O.; Citron M. (2000) Parkinson's disease-associated α-synuclein is more fibrillogenic than β- and γ-synuclein and cannot cross-seed its homologs. J. Biol. Chem. 275, 34574–34579. [DOI] [PubMed] [Google Scholar]
- Chen L.; Feany M. B. (2005) α-Synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat. Neurosci. 8, 657–663. [DOI] [PubMed] [Google Scholar]
- Masuda M.; Suzuki N.; Taniguchi S.; Oikawa T.; Nonaka T.; Iwatsubo T.; Hisanaga S.; Goedert M.; Hasegawa M. (2006) Small molecule inhibitors of α-synuclein filament assembly. Biochemistry 45, 6085–6094. [DOI] [PubMed] [Google Scholar]
- Snyder H.; Mensah K.; Hsu C.; Hashimoto M.; Surgucheva I. G.; Festoff B.; Surguchov A.; Masliah E.; Matouschek A.; Wolozin B. (2005) β-Synuclein reduces proteasomal inhibition by α-synuclein but not γ-synuclein. J. Biol. Chem. 280, 7562–7569. [DOI] [PubMed] [Google Scholar]
- Ghee M.; Fournier A.; Mallet J. (2000) Rat α-synuclein interacts with Tat binding protein 1, a component of the 26S proteasomal complex. J Neurochem 75, 2221–2224. [DOI] [PubMed] [Google Scholar]
- Morishima-Kawashima M.; Hasegawa M.; Takio K.; Suzuki M.; Yoshida H.; Titani K.; Ihara Y. (1995) Proline-directed and non-proline-directed phosphorylation of PHF-tau. J. Biol. Chem. 270, 823–829. [DOI] [PubMed] [Google Scholar]
- Hasegawa M.; Morishima-Kawashima M.; Takio K.; Suzuki M.; Titani K.; Ihara Y. (1992) Protein sequence and mass spectrometric analyses of tau in the Alzheimer′s disease brain. J. Biol. Chem. 267, 17047–17054. [PubMed] [Google Scholar]
- Nonaka T.; Arai T.; Buratti E.; Baralle F. E.; Akiyama H.; Hasegawa M. (2009) Phosphorylated and ubiquitinated TDP-43 pathological inclusions in ALS and FTLD-U are recapitulated in SH-SY5Y cells. FEBS Lett. 583, 394–400. [DOI] [PubMed] [Google Scholar]
- Inukai Y.; Nonaka T.; Arai T.; Yoshida M.; Hashizume Y.; Beach T. G.; Buratti E.; Baralle F. E.; Akiyama H.; Hisanaga S.; Hasegawa M. (2008) Abnormal phosphorylation of Ser409/410 of TDP-43 in FTLD-U and ALS. FEBS Lett. 582, 2899–2904. [DOI] [PubMed] [Google Scholar]
- Hasegawa M.; Arai T.; Nonaka T.; Kametani F.; Yoshida M.; Hashizume Y.; Beach T. G.; Buratti E.; Baralle F.; Morita M.; Nakano I.; Oda T.; Tsuchiya K.; Akiyama H. (2008) Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol. 64, 60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.