

Abstract
IL-10 is a potent immunomodulatory cytokine that affects innate and acquired immune responses. The immunological consequences of IL-10 production during pulmonary tuberculosis (TB) are currently unknown, although IL-10 has been implicated in reactivation TB in humans and with TB disease in mice. Using Mycobacterium tuberculosis-susceptible CBA/J mice, we show that blocking the action of IL-10 in vivo during chronic infection stabilized the pulmonary bacterial load and improved survival. Furthermore, this beneficial outcome was highly associated with the recruitment of T cells to the lungs and enhanced T cell IFN-γ production. Our results indicate that IL-10 promotes TB disease progression. These findings have important diagnostic and/or therapeutic implications for the prevention of reactivation TB in humans.
Interleukin-10 is a potent immunomodulatory cytokine that has been shown in vitro to directly or indirectly affect multiple cell types, including macrophages, monocytes, dendritic cells, CD4 T cells, and CD8 T cells (1). The dominant function of IL-10 is to deactivate macrophages, resulting in diminished Th1 cytokine production (2, 3), decreased production of reactive nitrogen or oxygen species (4), and limited Ag presentation (5), which may have far-reaching consequences on both innate and acquired immunity in vivo.
IL-10 has been identified as a correlate of susceptibility for tuberculosis (TB)3 in both mice and humans (6 –11), as well as susceptibility to Mycobacterium avium infection in murine models (12). In humans, IL-10 can be found in the serum (6, 7) and bronchoalveolar lavage fluid (8) of active TB patients and may be an important clinical biomarker of disease progression (9). In the murine model, IL-10-deficient C57BL/6 mice have a subtle yet increased early resistance to infection with Mycobacterium tuberculosis (M.tb) (10, 11). C57BL/6 mice are relatively resistant to infection with M.tb (13, 14) and a lack of IL-10 does not alter chronic infection in this strain (10, 11). Genetically manipulated C57BL/6 mice may not, therefore, accurately reflect a role for IL-10 in driving M.tb infection. We, and others, have previously defined the CBA/J mouse strain as relatively susceptible to M.tb infection, as it has increased bacterial load within the lung as infection progresses (13) and earlier mortality than does the C57BL/6 strain (14). Furthermore, disease progression in CBA/J mice has been associated with abundant IL-10 within the lung tissue (15). We have hypothesized that the increased production of IL-10 during late stages of a chronic M.tb infection in CBA/J mice exacerbates disease progression. This is supported by our previous findings that C57BL/6 mice engineered to overexpress IL-10 had accelerated disease progression following M.tb infection (15), confirming a clear relationship between IL-10 and poor disease outcome.
The increased amount of IL-10 produced by CBA/J mice during M.tb infection provides us with a natural model in which the role of IL-10 can be more accurately defined. In this study we used CBA/J mice to determine whether IL-10 can accelerate disease progression during an established chronic M.tb infection. Using anti-IL-10R1 Ab delivery starting at day 90 postinfection, we show that blockade of IL-10 activity led to improved control of M.tb bacterial load within the lung and enhanced Th1 immunity. These data demonstrate that increased IL-10 production exacerbates TB disease and provides supportive evidence that IL-10 can be a bio-marker of TB disease progression in humans.
Materials and Methods
Mice
Specific pathogen-free 8-wk-old female CBA/J and C57BL/6 mice (National Cancer Institute, Frederick, MD) were maintained in biosafety level 3 facilities and provided with sterile food and water ad libitum. All protocols were approved by The Ohio State University’s Institutional Laboratory Animal Care and Use Committee.
M.tb infection and quantification of bacterial load
M.tb Erdman (ATCC no. 35801) was obtained from American Type Culture Collection. Stocks were grown and mice infected with M.tb Erdman using an inhalation exposure system (Glas-Col) calibrated to deliver 50 –100 CFU to the lungs of each mouse, as previously described (16). At specific time points postinfection, the M.tb pulmonary load was determined as previously described (18).
Quantification of immune mediators
Lung homogenates were clarified for cytokine ELISAs using Ab pairs (BD Biosciences) as previously described (16). Nitrites/nitrates in lung tissue homogenates were quantified using the Greiss reaction. Briefly, Greiss reagent was added to equal volumes of clarified lung homogenate, absorbance was read at 550 nm, and values were compared with a sodium nitrite standard curve.
Ab administration
Anti-IL-10R1 mAb (1B1.3A) (Schering-Plough Biopharma) and whole rat IgG (Jackson ImmunoResearch Laboratories) or rat IgG1 isotype control mAb (GL113) (Schering-Plough Biopharma) was diluted to 2 mg/ml in PBS and stored at −80°C. Thawed stocks were maintained at 4°C for up to 1 mo. Ab administration was modified based on Silva et al. (17) as follows: at day 90 postinfection, 1 mg of anti-IL-10R1 (1B1.3A) or control Ab was injected into the peritoneal cavity of each mouse, followed by 0.2 mg at weekly intervals thereafter until the designated experimental time points. At the initiation of Ab treatment CBA/J mice consistently harbor 6.59 ± SE 0.12 log10 M.tb CFU (18). For animals in survival studies, Ab injection was continued weekly until signs of morbidity developed and mice were then euthanized.
Lung cell isolation and cell culture
Mice were euthanized by CO2 asphyxiation and single-cell suspensions obtained from the lung using collagenase/DNase as previously described (16). Viable cells were determined by trypan blue exclusion, counted, and resuspended at 2 × 106/ml. Lung cells (2.5 × 105) were cultured in duplicate with 10 μg/ml of OVA (Sigma-Aldrich), M.tb culture filtrate protein (National Institutes of Health, National Institute of Allergy and Infectious Diseases), or Con A (Sigma-Aldrich) for 72 h at 37°C. Supernatants were stored at −80°C until analysis. Cytokine production from cell culture supernatants was quantified by ELISA using Ab pairs and standards from BD Biosciences as described previously (16).
Flow cytometry
Isolated lung cells were suspended in deficient RPMI (Irvine Scientific) supplemented with 0.1% sodium azide (Sigma-Aldrich). Following 5–10 min of incubation at room temperature with Fc Block (clone 2.462; BD Biosciences), surface and intracellular targets were detected as previously described (16). Specific Abs and isotype controls were purchased from BD Biosciences: PerCP-Cy5.5 anti-CD3ε (145-2C11), allophycocyanin-Cy7 anti-CD4 (GK1.5), PeCy7 anti-CD8 (53-6.7), FITC anti-CD11a (2D7), allophycocyanin anti-CD11c (HL3), allophycocyanin anti-TCR-β-chain (H57597), PerCP anti-CD8 (53-6.7), anti-I-A/I-E (2G9), and PeCy7 anti-IFN-γ (XMG1.2). IFN-γ was determined according to the manufacturers instructions for intracellular cytokine staining (Cytofix/Cytoperm fixation/permeabilization solution kit with BD GolgiStop, BD Biosciences), following a 4-h incubation with anti-CD3 (145-2C11) and anti-CD28 (37.51). Samples were read using an LSRII flow cytometer and analyzed with FACSDiva software (BD Biosciences). For some experiments 100 μl of BrdU (FITC BrdU flow kit, BD Biosciences) was injected into the peritoneal cavity of each mouse 24 h before euthanasia. BrdU incorporation was detected by flow cytometry as per the manufacturer’s instructions (FITC BrdU flow kit).
Histology
The right cranial lung lobes from individual mice were inflated with 10% neutral buffered formalin before paraffin embedding, sectioning at 5 μm, and staining with H&E. Samples were evaluated in a double-blind fashion by a board-certified veterinary pathologist and graded as previously described (19).
Real-time PCR
Right medial lung lobes were homogenized in 1 ml of UltraSpec (Biotecx Laboratories) and frozen rapidly at −80°C. Total RNA was isolated by following the manufacturer’s instructions, and 1 μg was reverse transcribed using an Omniscript RT kit (Qiagen). Real-time PCR was performed with an iQ5 real-time PCR detection system (Bio-Rad) using Taq-Man gene expression assays for CIITA, IFN regulatory factor-1, or the 47-kDa GTPase LRG-47 (Applied Biosystems). The ΔΔCt method was used for relative quantification of mRNA expression; in this analysis 18S was used as an endogenous normalizer.
Statistics
Pairwise comparisons used the Student’s t-test; significance was defined as *, p < 0.05, **, p < 0.01, or ***, p < 0.001. Multigroup comparisons used one-way ANOVA with Tukey’s posttest; significance was defined as +, p < 0.05, ++, p < 0.01, or +++, p < 0.001. Log-rank analysis was used for survival studies.
Results
Anti-IL-10R1 treatment stabilized M.tb growth and improved survival in CBA/J mice
IL-10 production in the lungs of M.tb-infected CBA/J and C57BL/6 mice was determined. In comparison to the relatively resistant C57BL/6 mouse strain, IL-10 protein in CBA/J lungs was significantly higher at days 60, 120, and 150 days after M.tb infection (Fig. 1A). Furthermore, IL-10 continued to increase substantially over time during late infection (120 and 150 days) in CBA/J mice, while the levels stabilized in C57BL/6 mice (Fig. 1A). Similar increases were also detected in lung IL-10 mRNA (data not shown). Therefore, the immunocompetent inbred CBA/J mouse strain produces IL-10 during M.tb infection (15), allowing for the in vivo assessment of IL-10 function in a natural model of heightened M.tb susceptibility (14). To determine whether IL-10 directly influenced TB disease progression, we blocked the signaling chain of the IL-10 receptor by administering anti-IL-10R1 (1, 17) during chronic M.tb infection in CBA/J mice. M.tb-infected CBA/J mice were administered anti-IL-10R starting at day 90 postinfection. Day 90 was specifically chosen as the start point because this occurs before the substantial increase in lung IL-10 (Fig. 1A) and before loss of controlled M.tb growth (18) associated with increased morbidity in CBA/J mice (data not shown). Anti-IL-10R1 treatment was continued weekly throughout the experimental infection. The results showed a significant and predictable increase in bacterial burden in the lungs (15) of CBA/J mice that received control Ab (Fig. 1B), which approached 108 M.tb CFU, a level at which CBA/J mice rarely survive (data not shown). In contrast, treatment with anti-IL-10R1 stabilized the pulmonary bacterial load for up to 150 days (Fig. 1B). A separate group of mice was assigned to survival studies (Fig. 1C). Weekly Ab treatment was continued throughout survival experiments and mice were euthanized according to established morbidity criteria (13). Mice treated with anti-IL-10R1 survived ~70 days longer than did mice receiving control Ab (median survival of 196 days for anti-IL-10R1 treated vs 118 days for control antibody). Statistical significance was not achieved in the survival studies (p = 0.11); however, the small group size may have precluded this finding. Overall, these data indicate that blocking the activity of IL-10 in vivo in the CBA/J mouse strain during chronic infection with M.tb led to control of bacterial growth within the lung and extended survival. As expected, IL-10 blockade in the relatively resistant C57BL/6 mouse strain did not alter the M.tb lung load during the time frame studied (data not shown). This finding is consistent with results suggesting that IL-10 does not contribute to disease progression in C57BL/6 mice (10, 11).
FIGURE 1.
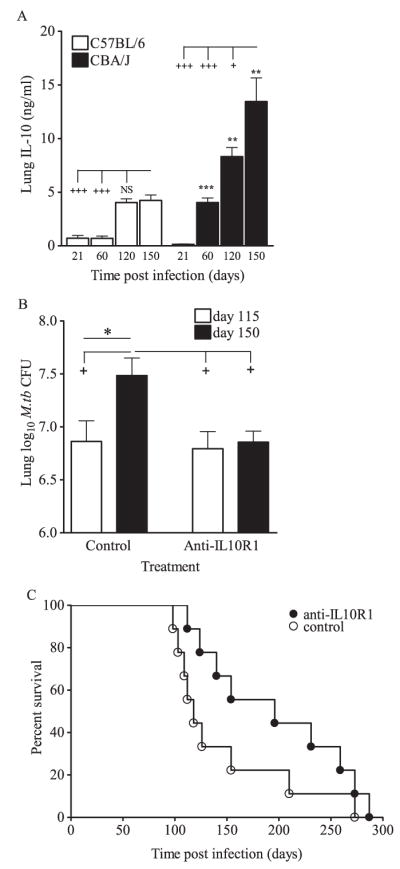
A role for IL-10 in driving disease progression in CBA/J mice. A, CBA/J and C57BL/6 mice were aerogenically infected with M.tb Erdman, euthanized at 21, 60, 120, and 150 days postinfection, lungs were homogenized, and IL-10 was detected by ELISA in clarified homogenates. Data are the means ± SD of one experiment with four mice per strain per time point analyzed by Student’s t test (**, p < 0.01 and ***, p < 0.001) at each time point and by one-way ANOVA with Tukey’s posttest for multigroup comparisons over time (+, p < 0.05 and +++, p < 0.001). B, CBA/J mice were aerogenically infected with M.tb Erdman and injected weekly with anti-IL-10R1 or control Ab starting at day 90 postinfection. Mice were euthanized at days 115 and 150, lungs homogenized and plated onto 7H11 agar, and M.tb CFU were determined after 21 days incubation at 37°C. Data are the means ± SEM of five independent experiments each with three to six mice per treatment group per time point, analyzed pairwise by Student’s t test (*, p < 0.05) and by one-way ANOVA with Tukey’s posttest for multigroup comparisons (+, p < 0.05). Total numbers of mice: control group 115 days, n = 26; control group 150 days, n = 25; anti-IL-10R group 115 days, n = 22; anti-IL-10R group day 150, n = 22. For survival, separate groups of anti-IL-10R1-treated mice and controls were euthanized when signs of morbidity developed (C) and statistical significance was determined by log-rank analysis. Survival data are combined from two independent experiments.
Anti-IL-10R1-treated CBA/J mice have increased cell density in the lung
We next determined whether improved control of infection was associated with increased T cell numbers within the lung. After 115 days no significant differences in total numbers of viable cells, CD4 T cells, or CD8 T cells were observed (Fig. 2). In contrast, at 150 days postinfection the total numbers of lung cells, CD4 T cells, and CD8 T cells were increased in mice that received anti-IL-10R1, although statistical significance was not reached. To determine whether increased cell numbers in anti-IL-10R1-treated CBA/J mice resulted from T cell proliferation, mice were injected with BrdU 24 h before euthanasia, and BrdU incorporation was determined by intracellular flow cytometry. Data from three independent experiments failed to identify a consistent increase in the number of proliferating CD4 or CD8 T cells in the lung (data not shown). To determine whether cell recruitment contributed to the increased number of T cells in anti-IL-10R1-treated mice, T cells from the lung were analyzed for CD11abright expression. At day 150 postinfection, the numbers of CD4 and CD8 T cells expressing CD11abright (Fig. 3) were significantly increased in mice treated with anti-IL-10R1. In contrast, mice treated with control Ab showed moderate increases in CD11abright T cells. Importantly, within both CD4 and CD8 T cell populations, multivariate analysis showed a significant increase in CD11abright expression across all groups and time points as a result of anti-IL-10R1 treatment. These data strongly suggest that IL-10 blockade promotes T cell recruitment into the lungs.
FIGURE 2.

Anti-IL-10R1 treatment increased lung cellularity during chronic M.tb infection in CBA/J mice. Mice were aerogenically infected with M.tb Erdman, injected weekly with anti-IL-10R1 or control Ab starting at day 90 postinfection, and euthanized at days 115 and 150. Isolated lung cells were counted, fixed, and labeled with anti-CD3, anti-CD4, and anti-CD8 for flow cytometric analysis. Total number of cells isolated (A), number of CD4 T cells (B), and number of CD8 T cells (C) are the means ± SEM of five independent experiments, each with three to six mice per group per time point, analyzed by Student’s t test for pairwise comparisons and one-way ANOVA with Tukey’s posttest for multigroup comparisons. Total numbers of mice: control group 115 days, n = 27; control group 150 days, n = 24; anti-IL-10R group 115 days, n = 24; anti-IL-10R group day 150, n = 22.
FIGURE 3.
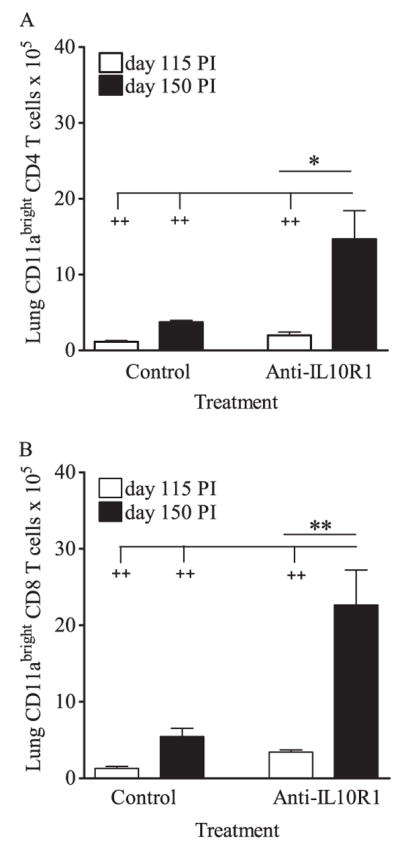
Anti-IL-10R1 treatment of CBA/J mice increased the numbers of CD11abright T cells in the lung during chronic M.tb infection. Mice were aerogenically infected with M.tb Erdman, injected weekly with anti-IL-10R1 or control Ab starting at day 90 postinfection, and euthanized at days 115 and 150. Isolated lung cells were counted, fixed, and labeled with anti-CD3, anti-CD4, anti-CD8, and anti-CD11a for flow cytometric analysis. Numbers of CD11abright CD4 (A) and CD8 (B) T cells are the means ± SEM and are representative of two independent experiments, each with four to five mice per group per time point, with pairwise analyses using Student’s t test (*, p < 0.05 and **, p < 0.001) or multigroup analysis using one-way ANOVA with Tukey’s posttest (+, p < 0.05; ++, p < 0.01; +++, p < 0.001).
IFN-γ production from lung cells of anti-IL-10R1-treated CBA/J mice
Two methods were used to determine the functional effects of IL-10 blockade on lung cells during chronic M.tb infection. First, lung cell suspensions were cultured for 4 h with anti-CD3 and anti-CD28 to stimulate TCR-mediated crosslinking and signaling in the presence of GolgiStop to allow for intracellular accumulation of IFN-γ. The proportion (%) of IFN-γ+ producing CD4 and CD8 T cells within the lungs of mice treated with anti-IL-10R1 was not consistently statistically increased (data not shown). Additionally, when the absolute numbers of IFN-γ+ T cells were calculated at day 115, there were no significant differences between control and anti-IL-10R1-treated mice (Fig. 4, A and B). In contrast, after 150 days of infection there was a substantial increase in the number of IFN-γ+ T cells in the lungs of anti-IL-10R1-treated CBA/J mice. Statistical significance between anti-IL-10R1-treated and control Ab treated mice was observed in the CD8 T cell population (Fig. 4B), and a trend for more IFN-γ+ cells was observed within the CD4 T cells population (Fig. 4A, p = 0.08). These data indicate that anti-IL-10R1 treatment during chronic M.tb lung infection increases the number of CD4 and CD8 T cells within the lung that have the potential to produce IFN-γ.
FIGURE 4.
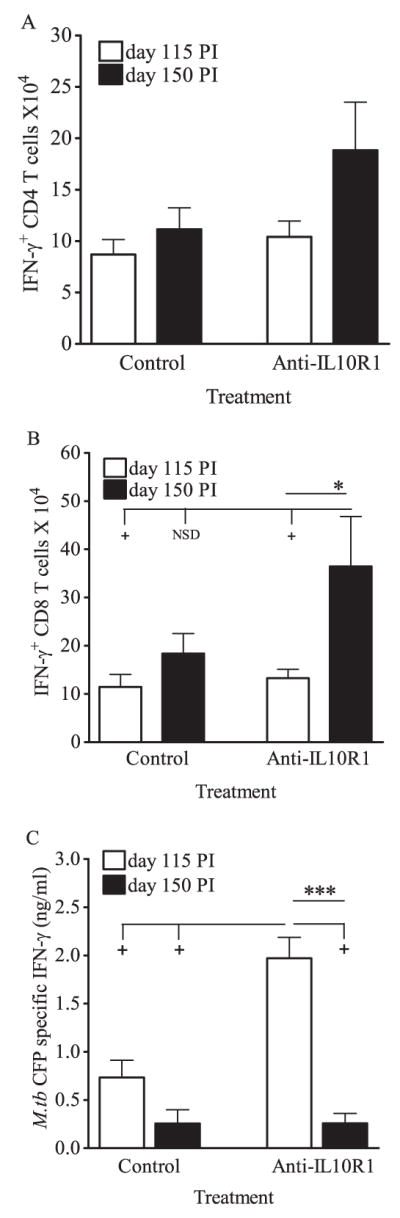
Anti-IL-10R1 treatment improved IFN-γ production during chronic M.tb infection of CBA/J mice. Mice were aerogenically infected with M.tb Erdman, injected weekly with anti-IL-10R1 or control Ab starting at day 90 postinfection, and euthanized at days 115 and 150. Isolated lung cells were stimulated with anti-CD3 and anti-CD28 for 4 h, fixed, labeled with anti-TCRβ, anti-CD4, and anti-CD8, permeabilized, and labeled with anti-IFN-γ for intracellular flow cytometry. Numbers of IFN-γ+ CD4 T cells (A) and IFN-γ+ CD8 T cells (B) are the means ± SEM of four independent experiments, each with three to six mice per group per time point, with pairwise analyses using Student’s t test (*, p < 0.05) and multigroup comparisons using one-way ANOVA (+, p < 0.05; NSD, no significant difference). Total numbers of mice: control group 115 days, n = 19; control group 150 days, n = 16; anti-IL-10R group 115 days, n = 20; anti-IL-10R group day 150, n = 18. C, Isolated lung cells were stimulated ex vivo with M.tb culture filtrate protein, and secreted Ag-specific IFN-γ in supernatants was quantified by ELISA. Data are the means ± SEM of two independent experiments, each with four to five mice per treatment group per time point, with pairwise analyses by Student’s t test (***, p < 0.001) and multigroup comparisons using one-way ANOVA (+, p < 0.05). Total numbers of mice: control group 115 days, n = 9; control group 150 days, n = 8; anti-IL-10R group 115 days, n = 9; anti-IL-10R group day 150, n = 8.
To evaluate Ag-specific responses ex vivo, lung cells were cultured with M.tb culture filtrate protein and IFN-γ production was determined by ELISA. As anticipated (15), Ag-specific IFN-γ production was low in lung cultures from CBA/J mice that had received control Ab at each time point tested (Fig. 4C). At day 115 postinfection, lung cell cultures from anti-IL-10R1-treated mice had significantly increased IFN-γ production relative to the control group (Fig. 4C), suggesting that IL-10R1 treatment enhanced numbers/function of lung Ag-specific cells during the first 25 days of anti-IL-10R1 therapy. Interestingly, IFN-γ production in anti-IL-10R1-treated mice declined to levels similar to the controls by day 150.
Altered cytokine production in the lung following anti-IL-10R1 treatment
To quantify cytokine production within the lung during infection, cytokine levels were directly measured in lung homogenates. Mice receiving control Ab showed stable but elevated levels of IFN-γ (Fig. 5A) within the lung at both time points analyzed. Mice receiving anti-IL-10R1 treatment had significantly less IFN-γ protein within the lung after 115 days infection relative to isotype-treated mice; however, these levels increased significantly by 150 days (Fig. 5A). Therefore, anti-IL-10R1 treatment led to more T cells capable of Ag-specific IFN-γ production (Fig. 4), but these cells were not continually producing IFN-γ in vivo (Fig. 5A).
FIGURE 5.
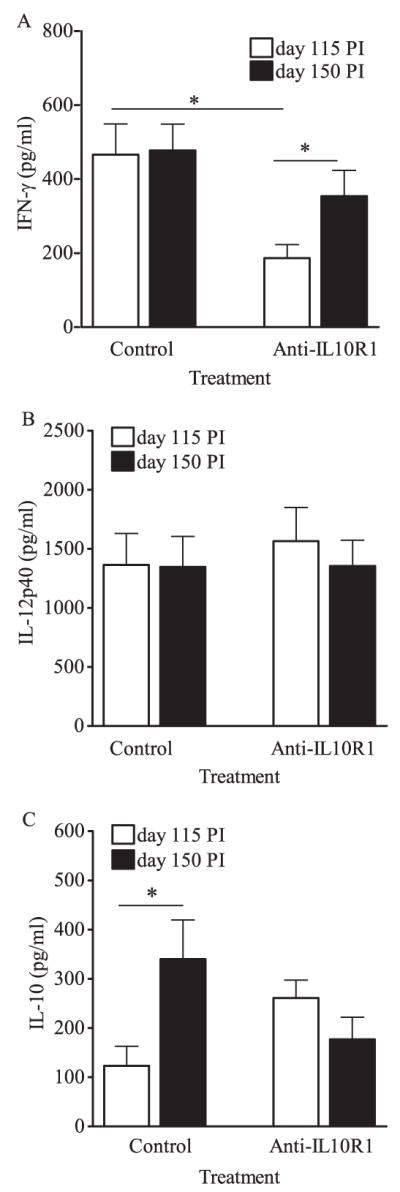
Cytokines in lung tissue following anti-IL-10R1 treatment during chronic M.tb infection. CBA/J mice were aerogenically infected with M.tb Erdman and injected weekly with anti-IL-10R1 or control Ab starting at day 90 postinfection. Mice were euthanized at days 115 and 150, lungs homogenized, and IFN-γ (A), IL-12p40 (B), and IL-10 (C) quantified by ELISA in clarified supernatants. Data shown are the means ± SEM of three (A and C) or four (B) independent experiments, each with three to six mice per treatment group per time point, and analyzed in pairs by Student’s t test (*, p < 0.05). Total numbers of mice (A and C): control group 115 days, n = 17; control group 150 days, n = 17; anti-IL-10R group 115 days, n = 14; anti-IL-10R group day 150, n = 14. Total numbers of mice (B): control group 115 days, n = 22; control group 150 days, n = 18; anti-IL-10R group 115 days, n = 22; anti-IL-10R group day 150, n = 17.
No consistent differences were found for IL-12p40 protein (Fig. 5B), TNF (data not shown), or reactive nitrogen species (data not shown) attributable to anti-IL-10R1 treatment. Additionally, the numbers of lung CD11c+ cells and their activation status were assessed. There were no consistent statistically significant differences in proportions or absolute numbers of lung CD11c+ (data not shown). To assess whether in vivo IL-10 blockade increased MHCII expression on CD11c+ cells, the mean fluorescent intensity of surface MHCII was determined; however, IL-10 blockade did not affect MHCII expression at the level of global CD11c+ lung cells. Overall, these results suggest that macrophage function remains intact during IL-10 blockade in M.tb-infected CBA/J mice or alternatively reflects the limitations of detection of our assays. As anticipated (15), IL-10 protein levels within the lungs of mice receiving control Ab significantly increased between 115 and 150 days (Fig. 5C). In contrast, anti-IL-10R1-treated mice did not exhibit an increase in IL-10 protein in the lung, perhaps reflecting a feedback loop in IL-10 regulation (Fig. 5C).
To determine potential mechanisms by which increased T cell-derived IFN-γ could affect M.tb burden during IL-10 blockade, downstream effectors of IFN-γ signaling were quantified by RT-PCR. No significant differences in mRNA levels for CIITA, IFN regulatory factor 1, or the 47-kDa GTPase LRG-47 were detected (data not shown). These results suggest that, at least at the tissue level, IL-10 blockade does not affect transcription of these molecules.
Discussion
Our data provide evidence that IL-10 production during chronic M.tb infection promotes bacterial growth and exacerbation of disease in a susceptible CBA/J mouse model of infection. This was shown by blocking the biological action of IL-10 in vivo using anti-IL-10R1 Abs that were delivered during chronic M.tb infection. Anti-IL-10R1 treatment led to a stabilization of the lung bacillary load, increased median survival time, and an increased number of cells within the lung by day 150 of infection. Enhanced cellularity within the lung was associated with augmentation of CD8 and CD4 T cells that expressed CD11abright and/or were capable of secreting IFN-γ. Overall, these data indicate that IL-10 production during infection with M.tb leads to the poor T cell responses and impaired control of infection in CBA/J mice. Furthermore, these deficiencies can be overcome by blocking the action of IL-10 in vivo.
Treatment with anti-IL-10R1 led to an increased number of CD4 and CD8 T cells within the lung that were capable of producing IFN-γ, which for CD8 T cells was significantly higher than for cells from control-treated mice. Analysis of IFN-γ within the whole-lung homogenate supported our findings, with a significant increase in IFN-γ between days 115 and 150 of infection in anti-IL-10R1-treated mice. Why control treated mice had increased IFN-γ in whole-lung homogenates at day 115 is uncertain. It is possible that IFN-γ was increased as a result of subtle differences in lung bacterial growth or turnover that were not detected by the CFU assay. Importantly, Ag-specific IFN-γ secretion ex vivo showed that lung cells from anti-IL-10R1 mice secreted significantly more IFN-γ after 115 days of infection than did lung cells from control mice. We can hypothesize that this early availability (day 115) of Ag-specific T cells contributed to the stable bacillary load within the lungs of CBA/J mice treated with anti-IL-10R1 Ab. Why we saw a significant decrease in Ag-specific IFN-γ secretion during the later (day 150) time point is unclear. This finding, however, was seen in multiple experiments and could reflect a restoration of phenotype to the control Ab-treated group. Alternatively, our data suggest that a reduced bacterial load in anti-IL-10R1-treated mice led to fewer actively secreting Ag-specific cells in the lung or that CD8 T cells were the dominant source of IFN-γ within the lung as infection progressed.
IL-10 is an antiinflammatory cytokine that can dampen an overly exuberant immune response and potentially prevent tissue damage (1). Indeed, the increased cellularity within the lungs of anti-IL-10R1-treated CBA/J mice alludes to a role for IL-10 in preventing detrimental pulmonary inflammation. Anti-IL-10R1 treatment did not, however, lead to any overt histopathological changes in the lung (data not shown), and therefore we consider these increases to be reflective of enhanced protective immunity rather than a failure to dampen inflammatory responses. Furthermore, the lung cell numbers recovered from anti-IL-10R1-treated CBA/J mice were comparable to C57BL/6 mice (data not shown), an inbred mouse strain with relative resistance to M.tb. In fact, we have previously shown that C57BL/6 mice have more CD11abright CD4 T cells in the lungs during M.tb infection than do CBA/J mice (13) and, again, the increased number of CD11abright T cells in anti-IL-10R1-treated CBA/J mice suggests acquisition of a protective immune response. Furthermore, Ely et al. have implied that CD11abright T cells localize to lung tissue during an Ag-specific immune response (20). In conjunction with our findings that IL-10 blockade did not consistently alter lung T cell proliferation, these results suggest that IL-10 may be a significant factor in the inhibition of T cell recruitment to the lung in CBA/J mice.
The failure to reach statistical significance in some of our data may be a consequence of the variable time frame that CBA/J mice progress to active disease (14), and the choice of only two selected time points for analysis, which cannot account for the wax and wane of immunity that has been described by others (21). Additionally, the extensive Ab treatments may have stimulated anti-rat Ab production and a loss of functional activity of the anti-IL-10R Ab, reducing the long-term biological impact of the Ab treatment (preliminary studies showed some mice generated anti-rat Abs after multiple injections (data not shown)). We can only speculate as to the outcome of infection/survival if anti-IL-10R1 treatment remained effective for the long term. Alternative strategies to study the role of IL-10 during chronic M.tb infection, such as IL-10-deficient CBA/J mice, could provide valuable tools to assess the relative role of IL-10 in disease progression.
It is important to acknowledge that we are manipulating a complex in vivo system, and our readout of a protective response is based on what is currently known about immunity during M.tb infection. Molecules such as IL-12, TNF, and NO are essential for control of M.tb infection (22–25), and consequently these were the parameters that we measured. We found little difference in the expression of these known protective molecules when we manipulated the action of IL-10. What is significant in this study, however, is that despite failing to find significant differences in some immunological parameters, we could significantly modify the course of infection (lung bacterial load) by blocking the action of IL-10. The possibility therefore exists that enhanced protection is associated with alternate cells/molecules that have yet to be clearly defined.
In summary, we have shown that modulation of IL-10 in vivo using a mouse strain that naturally produces abundant IL-10 during infection with M.tb leads to stabilization of the lung bacterial load, extended survival, and enhanced T cell responses within the lung. It is important to recognize that the detection of an improved phenotype following the in vivo manipulation of one single immunological mediator is a highly significant finding. To date, in vivo removal of cytokines during M.tb infection have rarely shown a beneficial effect (26, 27), and the depletion or absence of a single cytokine typically impairs resistance (22, 23, 25, 28, 29). Our finding that IL-10 blockade altered the CBA/J susceptibility phenotype of M.tb infection has particular relevance to studies in humans where a spectrum of susceptibility and immune responses can often be observed.
Acknowledgments
M.tb Ags were provided by Colorado State University through National Institutes of Health National Institute of Allergy and Infectious Diseases contract HHSN266200400091C, titled “Tuberculosis Vaccine Testing and Research Materials.”
Footnotes
Support was provided by the American Lung Association (CI-8609-N) and National Institutes of Health Grants R01 (AI-064522) and T32 (RR007073). Contents of this paper are the authors’ responsibility and do not necessarily represent views of the National Center for Research Resources or the National Institutes of Health. Schering-Plough Biopharma is supported by the Schering Plough Corporation.
Abbreviations used in this paper: TB, tuberculosis; M.tb, Mycobacterium tuberculosis.
Disclosures
Rene de Waal Malefyt is an employee of Schering-Plough Biopharma.
References
- 1.Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 2.de Waal Malefyt R, Abrams J, Bennett B, Figdor C, de Vries J. Interleukin 10 (IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J Exp Med. 1991;174:1209 –1220. doi: 10.1084/jem.174.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fiorentino D, Zlotnik A, Mosmann T, Howard M, O’Garra A. IL-10 inhibits cytokine production by activated macrophages. J Immunol. 1991;147:3815–3222. [PubMed] [Google Scholar]
- 4.Bogdan C, Vodovotz Y, Nathan C. Macrophage deactivation by interleukin 10. J Exp Med. 1991;174:1549 –1555. doi: 10.1084/jem.174.6.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koppelman B, Neefjes JJ, de Vries JE, de Waal Malefyt R. Interleukin-10 down-regulates MHC class II alphabeta peptide complexes at the plasma membrane of monocytes by affecting arrival and recycling. Immunity. 1997;7:861– 871. doi: 10.1016/s1074-7613(00)80404-5. [DOI] [PubMed] [Google Scholar]
- 6.Olobo JO, Geletu M, Demissie A, Eguale T, Hiwot K, Aderaye G, Britton S. Circulating TNF-α, TGF-β, and IL-10 in tuberculosis patients and healthy contacts. Scand J Immunol. 2001;53:85–91. doi: 10.1046/j.1365-3083.2001.00844.x. [DOI] [PubMed] [Google Scholar]
- 7.Verbon A, Juffermans NP, van Deventer SJH, Speelman P, van Deutekom H, van der Poll T. Serum concentrations of cytokines in patients with active tuberculosis (TB) and after treatment. Clin Exp Immunol. 1999;115:110 –113. doi: 10.1046/j.1365-2249.1999.00783.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonecini-Almeida MG, Ho JL, Boechat N, Huard RC, Chitale S, Doo H, Geng J, Rego L, Lazzarini LCO, Kritski AL, et al. Down-modulation of lung immune responses by interleukin-10 and transforming growth factor-β (TGF-β) and analysis of TGF-β receptors I and II in active tuberculosis. Infect Immun. 2004;72:2628 –2634. doi: 10.1128/IAI.72.5.2628-2634.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jamil B, Shaid F, Hasan Z, Nasir N, Razzaki T, Dawood G, Hussain R. Interferon-γ/IL-10 ratio defines disease severity in pulmonary and extra-pulmonary tuberculosis. Tuberculosis. 2007;87:279 –287. doi: 10.1016/j.tube.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 10.North RJ. Mice incapable of making IL-4 or IL-10 display normal resistance to infection with Mycobacterium tuberculosis. Clin Exp Immunol. 1998;113:55–58. doi: 10.1046/j.1365-2249.1998.00636.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jung YJ, Ryan L, LaCourse R, North RJ. Increased interleukin-10 expression is not responsible for failure of T helper 1 immunity to resolve airborne Mycobacterium tuberculosis infection in mice. Immunology. 2003;109:295–299. doi: 10.1046/j.1365-2567.2003.01645.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roque S, Nobrega C, Appelberg R, Corrcia-Neves M. IL-10 underlies distinct susceptibility of BALB/c and C57BL/6 mice to Mycobacterium avium infection and influences efficacy of antibiotic therapy. J Immunol. 2007;178:8028–8035. doi: 10.4049/jimmunol.178.12.8028. [DOI] [PubMed] [Google Scholar]
- 13.Turner J, Gonzalez-Juarrero M, Saunders BM, Brooks JV, Marietta P, Ellis DL, Frank AA, Orme IM. Immunological basis for reactivation of tuberculosis in mice. Infect Immun. 2001;69:3264 –3270. doi: 10.1128/IAI.69.5.3264-3270.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Medina E, North RJ. Mycobacterium tuberculosis and relationship to major histocompatibility complex haplotype and Nramp1 genotype. Immunology. 1998;93:270 –274. doi: 10.1046/j.1365-2567.1998.00419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Turner J, Gonzalez-Juarrero M, Ellis DL, Basaraba RJ, Kipnis A, Orme IM, Cooper AM. In vivo IL-10 production reactivates chronic pulmonary tuberculosis in C57BL/6J mice. J Immunol. 2002;169:6343– 6351. doi: 10.4049/jimmunol.169.11.6343. [DOI] [PubMed] [Google Scholar]
- 16.Vesosky B, Flaherty DK, Turner J. Th1 cytokines facilitate CD8-T-cell-mediated early resistance to infection with Mycobacterium tuberculosis in old mice. Infect Immun. 2006;74:3314 –3324. doi: 10.1128/IAI.01475-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Silva RA, Pais TF, Appelberg R. Blocking the receptor for IL-10 improves antimycobacterial chemotherapy and vaccination. J Immunol. 2001;167:1535–1541. doi: 10.4049/jimmunol.167.3.1535. [DOI] [PubMed] [Google Scholar]
- 18.Beamer GL, Flaherty DK, Vesosky B, Turner J. Peripheral blood interferon-γ release assays predict lung responses and Mycobacterium tuberculosis disease outcome in mice. Clin Vaccine Immunol. 2008;15:474 – 483. doi: 10.1128/CVI.00408-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flaherty DK, Vesosky B, Beamer GL, Stromberg P, Turner J. Exposure to Mycobacterium avium can modulate established immunity against Mycobacterium tuberculosis infection generated by Mycobacterium bovis BCG vaccination. J Leukocyte Biol. 2006;80:1262–1271. doi: 10.1189/jlb.0606407. [DOI] [PubMed] [Google Scholar]
- 20.Ely KH, Cookenham T, Roberts AD, Woodland DL. Memory T cell populations in the lung airways are maintained by continual recruitment. J Immunol. 2006;176:537–543. doi: 10.4049/jimmunol.176.1.537. [DOI] [PubMed] [Google Scholar]
- 21.Lazarevic V, Nolt D, Flynn JL. Long-term control of Mycobacterium tuberculosis infection is mediated by dynamic immune responses. J Immunol. 2005;175:1107–1117. doi: 10.4049/jimmunol.175.2.1107. [DOI] [PubMed] [Google Scholar]
- 22.Flynn J, Chan J, Triebold K, Dalton D, Stewart T, Bloom B. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J Exp Med. 1993;178:2249 –2254. doi: 10.1084/jem.178.6.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cooper AM, Magram J, Ferrante J, Orme IM. Interleukin-12 (IL-12) is crucial to the development of protective immunity in mice intravenously infected with Mycobacterium tuberculosis. J Exp Med. 1997;186:39 – 45. doi: 10.1084/jem.186.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacMicking JD, North RJ, LaCourse R, Mudgett JS, Shah SK, Nathan CF. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc Natl Acad Sci USA. 1997;94:5243–5248. doi: 10.1073/pnas.94.10.5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Flynn J, Goldstein M, Chan J, Triebold K, Pfeffer K, Lowenstein C. Tumor necrosis factor α is required in the protective immune response against M. tuberculosis in mice. Immunity. 1995;2:561–572. doi: 10.1016/1074-7613(95)90001-2. [DOI] [PubMed] [Google Scholar]
- 26.Roy E, Brennan J, Jolles S, Lowrie DB. Beneficial effect of anti-interleukin-4 antibody when administered in a murine model of tuberculosis. Tuberculosis. 2007;88:197–202. doi: 10.1016/j.tube.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 27.Pearl JE, Khader SA, Solache A, Gilmartin L, Ghilardi N, deSauvage F, Cooper AM. IL-27 signaling compromises control of bacterial growth in mycobacteria-infected mice. J Immunol. 2004;173:7490 –7496. doi: 10.4049/jimmunol.173.12.7490. [DOI] [PubMed] [Google Scholar]
- 28.Saunders BM, Frank AA, Orme IM, Cooper AM. Interleukin-6 induces early gamma interferon production in the infected lung but is not required for generation of specific immunity to Mycobacterium tuberculosis infection. Infect Immun. 2000;68:3322–3326. doi: 10.1128/iai.68.6.3322-3326.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chakravarty SD, Zhu G, Tsai MC, Mohan VP, Marina S, Kirschner DE, Huang L, Flynn J, Chan J. Tumor necrosis factor blockade in chronic murine tuberculosis enhances granulomatous inflammation and disorganizes granulomas in the lungs. Infect Immun. 2008;76:916 –926. doi: 10.1128/IAI.01011-07. [DOI] [PMC free article] [PubMed] [Google Scholar]