

Summary
The development of left ventricular cardiomyocyte hypertrophy in response to increased hemodynamic load and neurohormonal stress is initially a compensatory response. However, persistent stress eventually leads to dilated heart failure, which is a common cause of heart failure in human hypertensive and valvular heart disease. We have recently reported that Rho-associated coiled-coil containing protein kinase 1 (ROCK1) homozygous knockout mice exhibited reduced cardiac fibrosis and cardiomyocyte apoptosis, while displaying a preserved compensatory hypertrophic response to pressure overload. In this study, we have tested the effects of ROCK1 deficiency on cardiac hypertrophy, dilation, and dysfunction. We have shown that ROCK1 deletion attenuated left ventricular dilation and contractile dysfunction, but not hypertrophy, in a transgenic model of Gαq overexpression-induced hypertrophy which represents a well-characterized and highly relevant genetic mouse model of pathological hypertrophy. Although the development of cardiomyocyte hypertrophy was not affected, ROCK1 deletion in Gαq mice resulted in a concentric hypertrophic phenotype associated with reduced induction of hypertrophic markers indicating that ROCK1 deletion could favorably modify hypertrophy without inhibiting it. Furthermore, ROCK1 deletion also improved contractile response to β-adrenergic stimulation in Gαq transgenic mice. Consistent with this observation, ROCK1 deletion prevented down-regulation of type V/VI adenylyl cyclase expression, which is associated with the impaired β-adrenergic signaling in Gαq mice. The present study establishes for the first time a role for ROCK1 in cardiac dilation and contractile dysfunction.
Keywords: Rho kinase, ROCK1 knockout mice, cardiac dilation, pathological cardiac hypertrophy, contractile dysfunction
Introduction
The development of postnatal cardiomyocyte hypertrophy, also named pathological hypertrophy, is the common response when heart endures increased hemodynamic load (as with chronic hypertension or valvular stenosis), myocardial injury, and neurohormonal stress. Pathological hypertrophy is characterized by increases in myocyte size and protein content, and a change in gene expression profiles from adult to a “fetal-like” program [1, 2]. Although this adaptation is initially a compensatory response, persistent stress eventually leads to a decompensate congestive heart failure, which is characterized by chamber dilation and myocardial dysfunction. Due to the high mortality of heart failure, there is a growing interest in identifying the signaling mechanisms underlying the development of cardiac hypertrophy and the transition to heart failure.
The Gq class of heterotrimeric G proteins is one important signal transducer that is responsible for the development of cardiac hypertrophy and subsequent cardiac decompensation. This class of proteins couple membrane receptors for neurohumoral factors (i.e., α1-adrenergic agonists, angiotensin II, endothelin, and prostaglandin F2α) in response to the cardiac hypertrophic response. Previous studies demonstrated that Gq signaling is necessary for pressure-overload induced cardiac hypertrophy [3, 4]. Transgenic overexpression of the α subunit of Gq in the myocardium is a well-characterized in vivo pathological hypertrophy model which recapitulates many cellular, molecular, and functional characteristics of pressure overload-induced hypertrophy including left ventricular dilation, depressed ventricular contractility at baseline and in response to β-adrenergic receptor stimulation, diminished basal and agonist stimulated adenylyl cyclase activity, and activation of a fetal cardiac gene program [5, 6]. This transgenic mouse model offers a means of studying the signaling mechanisms in myocardium responsible for cardiac dilation and contractile dysfunction in hypertrophic but not in failing phase. A number of studies have delineated several potential mechanisms by which Gαq overexpression leads to cardiomyocyte hypertrophy, cardiac dilation and dysfunction. These mechanisms include increased expression and activity of protein kinase C (PKC)α [7], decreased adenylyl cyclase expression and catalytic activity [8, 9], and increased activation of the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated protein kinase (ERK) kinase kinase 1 (MEKK1)/c-Jun N-terminal kinase (JNK) signaling pathway [10].
Rho-associated coiled-coil containing protein kinase (ROCK) is a downstream mediator of RhoA GTPase, and is believed to play a critical role in mediating the effects of RhoA on stress fiber formation, smooth muscle contraction, cell adhesion, membrane ruffling, and cell motility. Recent in vivo studies using ROCK inhibitors, Y27632 and fasudil, suggest a role of ROCK in mediating cardiac hypertrophy and remodeling [11-14]. These pharmacological inhibitors are not able to distinguish between the two isoforms of the family, ROCK1 and ROCK2 [15-17]. We have recently shown that ROCK1 homozygous deficient mice developed cardiac myocyte hypertrophy in response to pressure overload induced by transverse aortic constriction, indicating that ROCK1 is not critical for the development of cardiac hypertrophy [18]. Interestingly, these mice exhibited significantly reduced interstitial fibrosis [18] and cardiomyocyte apoptosis [19]. This observation is consistent with another recent study using ROCK1 haploinsufficient mice which did not show decreased hypertrophy but decreased perivascular fibrosis induced by angiotensin II [20].
A number of the previous in vitro studies performed in cultured rat neonatal cardiomyocytes have shown that RhoA/ROCK pathway cooperated with Gαq pathway for the induction of cardiomyocye hypertrophy by G protein-coupled receptor agonists [21-24]. However, the in vivo interaction between these pathways has not been demonstrated. The combination of the well-characterized transgenic Gαq cardiac hypertrophy model with the ROCK1 deficient mice provides an opportunity to examine the potential in vivo interaction between ROCK1 and Gαq pathways and to test effects of ROCK1 deletion on the development of dilated cardiomyopathy induced by intrinsic cardiac effects in Gαq overexpression transgenic mice.
Materials and Methods
All experiments were conducted in accordance with the National Institutes Health “Guide for the Care and Use of Laboratory Animals” and were approved by the Institutional Animal Care and Use Committee at Indiana University School of Medicine.
Mouse models
Generation and characterization ROCK1-/- mice have been described previously [18]. Transgenic FVB mice overexpressing Gαq in cardiomyocytes (Gαq, 25-copy line) have been characterized previously [5, 6]. These transgenic mice were mated with ROCK1-/- (FVB background) to generate Gαq/ROCK1+/- mice, which were then bred with ROCK-/- mice to generate Gαq/ROCK1-/- mice. Southern blot and PCR analyses were carried out for ROCK1-/- genotyping as previously described [18] and PCR analysis was carried out for the Gαq transgene [10].
Echocardiography
Cardiac dimension and contractility were evaluated by noninvasive transthoracic echocardiography with a Vevo 770 high resolution Imaging System (VisualSonics Inc, Toronto). Mice were anesthetized by inhaling isofluorane (1–2% via nosecore) and maintained on platform at 37°C during ultrasound scanning with a 35 MHz mechanical transducer. In some cases, mice were studied before and 5 min after intraperitoneal injection of isoproterenol (100 ng/g body weight). Left ventricular chamber dimension and wall thickness at systolic and diastolic phases were measured from M-mode recordings at the level of the papillary muscles of the left ventricle in systole and diastole. Heart rate was recorded simultaneously. Fractional shortening was calculated from left ventricular end-systolic and end-diastolic dimensions (LVESD and LVEDD, respectively) using the formula: (LVEDD-LVESD)/LVEDD × 100.
Histology
Assessment of cardiac hypertrophy was performed as previously described [25]. Total heart weight, lung weight and liver weight were indexed to tibial length. For histology, hearts were perfused with PBS followed by 10% buffered formalin and embedded in paraffin. Tissue sections were then stained with hematoxylin/eosin for initial evaluation, picrosirius red to identify collagen fibers, immunostaining for laminin to measure mycocyte size, and TUNEL staining to monitor myocyte apoptosis. The picrosirius red-stained slides were scanned by using a microscope equipped with a digital camera [26], and quantitative evaluation was performed with Image-Pro software (MediaCybernetics, Inc. Maryland). The collagen-stained area was calculated as a percentage of the total myocardial area as previously described [18]. Immunostaining for laminin was performed by using rabbit primary antibody (Sigma), followed by biotinylated goat anti-rabbit IgG. The secondary antibody was visualized with Vectastain Elite ABC kit (Vector Laboratories). Myocyte cross sectional area was calculated by using Image-Pro (about 200 myocytes and 4 hearts per genotype). For TUNEL analysis, paraffin sections were processed using the ApopTag Fluorescence In Situ Apoptosis Detection kit (Chemicon International) as previously described [27].
Protein analysis
Protein samples were prepared as previously described [27]. Separation of cytosolic and membrane fractions was performed as reported [27]. Protein expression of ROCK1 was examined using a rabbit anti-ROCK1 polyclonal antibody (H-85) (Santa Cruz), and also by a mouse anti-ROCK1 monoclonal antibody (BD Transduction laboratories). Protein expression of ROCK2 was examined using a mouse anti-ROCK2 monoclonal antibody (BD Transduction Laboratories). Other primary antibodies included rabbit polyclonal antibodies to Gαq (E-17) (sc-393), PKCα (C-20) (sc-208), β1-adrenergic receptor (β1-AR) (sc-568), β2-adrenergic receptor (β2-AR) (sc-570), adenylyl cyclase V/VI (sc-590) (Santa Cruz), mouse monoclonal antibodies to PKCε (BD Transduction Laboratories), rabbit polyclonal antibodies to phospho-MYPT1 (myosin phosphatase targeting subunit 1) (Thr696) (Upstate Cell Signaling Solution), and rabbit polyclonal antibodies to p44/42 MAPK, SAPK/JNK, p38 MAPK, phospho-p44/42 MAPK (Thr202/Tyr204), phospho-SAPK/JNK (Thr183/Tyr185), phospho-p38 MAPK (Thr180/Tyr182), ERM (ezrin, radixin, and moesin), and phospho-ERM (Cell Signaling). Equal loading of proteins was confirmed by Ponceau red staining of the membrane after immunoblotting and by actin immunoreactivity.
Gene expression analysis
Total RNA was extracted from ventricular tissues by using TRIZOL (Gibco-BRL, Gaithersburg, Maryland) and RNAeasy (Qiagen). Gene expression analyses by Affymetrix mouse genomic 430 2.0 arrays and by real-time RT-PCR were performed as previously described [18]. TaqMan primers and probes for mouse GAPDH, ROCK1, ROCK2, atrial natriuretic factor (ANF), skeletal α-actin, β-myosin heavy chain (MHC), αMHC, adenylyl cyclase V and adenylyl cyclase VI were purchased from Applied Biosystems (Foster City, CA).
Statistical analysis
Data are reported as means ± SE. Comparisons between groups were analyzed by Student's t-test or ANOVA as appropriate, with P < 0.05 considered as significant.
Results
Disruption of ROCK1 did not prevent development of cardiomyocyte hypertrophy induced by cardiac-specific overexpression of Gαq
The first step was to determine whether ROCK1 plays a role in mediating Gαq-induced hypertrophic response in vivo. We generated Gαq/ROCK1-/- compound mice as described in Materials and Methods. Western blot analysis of ventricular tissue homogenate from sex (male) and age-matched (12 weeks) wild type (WT), Gαq, ROCK1-/-, and Gαq/ROCK1-/- mice confirmed that Gαq expression level was increased in Gαq and Gαq/ROCK1-/- mice and ROCK1 expression was absent in ROCK1-/- and Gαq/ROCK1-/- mice (Fig. 1). ROCK1 deficiency did not change the expression level of Gαq transgene in ventricular tissue (Fig. 1) and similar results were obtained from at least six hearts in each group. In addition, ROCK2 expression remained similar in all four groups. Phosphorylation of ERM proteins (known substrates of ROCK) was assessed to examine whether Gαq overexpression alters ROCK activity and no significant difference was detected among all four groups (Fig. 1). In addition, phosphorylation of MYPT1, another known substrate of ROCK, was also similar among all four groups (data not shown). These results suggest that the total level of ROCK activity is not significantly altered by ROCK1 deletion or Gαq overexpression.
Figure 1. Western blot analysis.
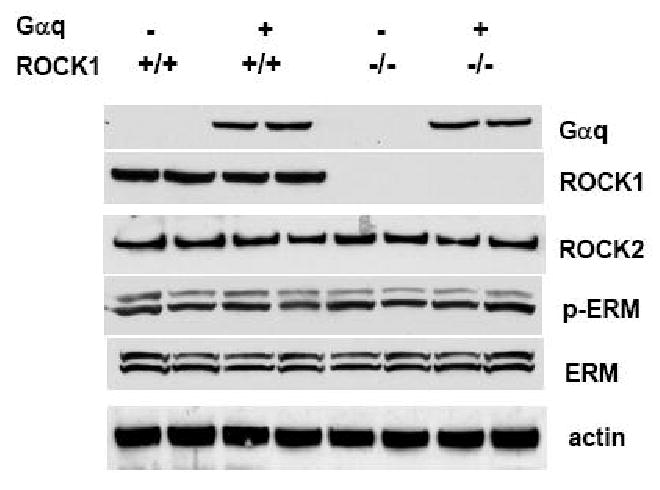
Protein levels of Gαq, ROCK1, and ROCK2 were determined by Western blot analysis of ventricular homogenates (50 μg per lane) from four groups of 12-week-old male mice with indicated genotype. Deletion of ROCK1 was confirmed in ROCK1-/- or Gαq/ROCK1-/- mice. Gαq expression was invariant among Gαq and Gαq/ROCK1-/- hearts. No significant difference was detected for ROCK activity assessed by Western blot analysis for phosphorylated ERM and total ERM among these groups. An anti-actin antibody was used to confirm equal loading. Similar results were obtained from at least six hearts in each group.
Gαq-induced cardiac hypertrophic response was then assessed by cardiac mass, cardiomyocyte size, and echocardiography in sex and age-matched mice. As described below, ROCK1-/- mice were indistinguishable from WT mice for all the physiological, morphological, and biochemical analyses performed in the present study. As previously reported [6], an enlarged heart was seen in Gαq transgenic mice with an increased ratio of heart weight/tibial length compared with non-transgenic littermates at 12 weeks of age (Table 1). The Gαq/ROCK1-/- compound mice also exhibited a significant increase in heart weight/tibial length ratio compared with ROCK1-/- or WT mice. However, the extent of increase in heart weight/tibial length ratio of Gαq/ROCK1-/- compound mice was mildly reduced compared with that of Gαq transgenic mice, but this difference was not statistically significant (P = 0.272). Consistent with morphometric data, the calculated left ventricular mass obtained by echocardiography was also significantly increased in Gαq/ROCK1-/- compound mice compared with ROCK1-/- or WT mice (Table 2). A trend toward less left ventricular mass than Gαq mice (P = 0.127) was again noticed in Gαq/ROCK1-/- compound mice. Left ventricular cardiomyocyte cross sectional area, which is another index of hypertrophy, showed the same pattern, with significant increase in Gαq/ROCK1 compound mice compared with ROCK1-/- and WT mice, and this increase in Gαq/ROCK1 compound mice was only slightly reduced when compared with Gαq mice (P = 0.063) (Fig. 2). Taken together, these results indicate that ROCK1 deletion did not prevent Gαq-induced cardiomyocyte hypertrophy. This observation is consistent with our previous report that ROCK1 deletion did not prevent the development of myocardial and cardiomyocyte hypertrophy induced by pressure overload [18].
Table 1. Morphometric analysis of Gαq, Gαq/ROCK1-/-, ROCK1-/- and WT mice.
| WT (n=16) |
Gαq (n=16) |
ROCK1-/- (n=14) |
Gαq/ROCK1-/- (n=17) |
|
|---|---|---|---|---|
| Body weight (g) | 24.4±0.6 | 24.7±0.5 | 24.9±0.4 | 25.1±0.6 |
| Whole heart weight (mg) | 128±4 | 158±11* | 136±5 | 157±6* |
| Lung weight (mg) | 182±7 | 204±7* | 179±6 | 177±8# |
| Liver weight (mg) | 1,126±41 | 1,134±40 | 1,164±34 | 1,129±36 |
| Tibial length (mm) | 19.8±0.4 | 19.1±0.3 | 19.7±0.8 | 19.9±0.9 |
| Heart weight/tibial length (mg/mm) | 6.50±0.23 | 8.23±0.52* | 6.89±0.21 | 7.87±0.28* |
| Lung weight/tibial length (mg/mm) | 9.27±0.49 | 10.73±0.41* | 9.08±0.28 | 8.89±0.35# |
| Liver weight/tibial length (mg/mm) | 56.9±2.6 | 59.5±2.5 | 58.9±1.7 | 56.7±1.8 |
Data are presented as mean ± SE.
P < 0.01 for Gαq vs. WT or Gαq/ROCK1-/- vs. ROCK1-/-.
P < 0.01 for Gαq/ROCK1-/- vs. Gαq.
Table 2. Echocardiographic parameters of Gαq, Gαq/ROCK1-/-, ROCK1-/- and WT mice.
| WT (n=12) |
Gαq (n=12) |
ROCK1-/- (n=12) |
Gαq/ROCK1-/- (n=12) |
|
|---|---|---|---|---|
| LV mass (mg) | 87.4±5.9 | 108.9±4.4** | 91.1±4.5 | 104.1±5.5* |
| LVEDD (mm) | 2.97±0.07 | 4.08±0.07** | 3.18±0.08 | 3.33±0.14## |
| LVESD (mm) | 1.46±0.06 | 2.97±0.05** | 1.61±0.05 | 1.77±0.05## |
| LVPWd (mm) | 0.83±0.06 | 0.71±0.04* | 0.84±0.05 | 0.93±0.04* ## |
| LVPWs (mm) | 1.19±0.05 | 1.04±0.04* | 1.21±0.06 | 1.36±0.06* ## |
| IVSd (mm) | 0.88±0.04 | 0.73±0.06* | 0.86±0.03 | 0.92±0.03# |
| IVSs (mm) | 1.23±0.05 | 1.11±0.03* | 1.25±0.06 | 1.33±0.05# |
| LV fractional shortening (%) | 50.6±0.4 | 27.1±0.3** | 48.3±0.5 | 46.7±0.4## |
| LV ejection fraction (%) | 79.5±1.8 | 55.3±1.1** | 78.5±1.5 | 77.8±0.5## |
| Heart rate (bpm) | 467±3 | 238±6** | 463±7 | 368±4** ## |
Data are presented as mean ± SE. LVESD and LVEDD: left ventricular end-systolic and end-diastolic diameters; LVPWs and LVPWd: end-systolic and end-diastolic LV posterior wall thickness; IVSs and IVSd: end-systolic and end-diastolic intraventricular septal thickness.
P < 0.05,
P < 0.01 for Gαq vs. WT or Gαq/ROCK1-/- vs. ROCK1-/-.
P < 0.05,
P < 0.01 for Gαq/ROCK1-/- vs. Gαq.
Figure 2. ROCK1 deletion did not prevent myocyte hypertrophy induced by Gαq.
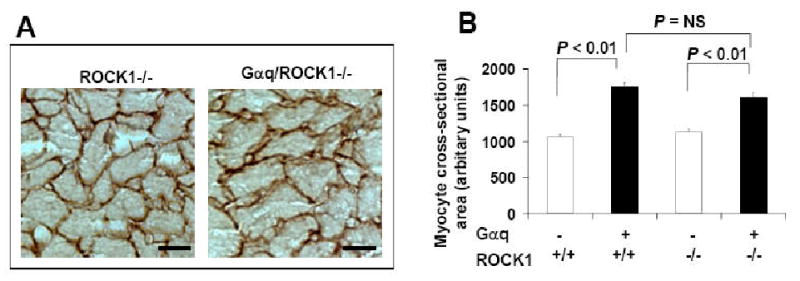
A. Representative images of laminin staining of heart sections from 12-week-old male mice. B. Quantitative analysis of cardiac myocyte area measured from laminin stained sections. Each column represents results obtained from approximately 200 myocytes from four hearts per group. Bar, 10 μm.
Although the extent of cardiac hypertrophy in Gαq/ROCK1-/- mice was similar to that in Gαq mice, an intermediate phenotype of hypertrophic markers induction was observed in Gαq/ROCK1-/- compound mice (Table 3). The levels of ANF, skeletal α-actin, and βMHC assessed by real-time RT-PCR were significantly increased in Gαq/ROCK1-/- compound mice compared with WT or ROCK1-/- mice. However, induction of these hypertrophic markers in Gαq/ROCK1-/- mice was significantly reduced compared with Gαq mice. This intermediate induction of some of these hypertrophic markers in Gαq/ROCK1-/- mice was also observed in microarray analysis (Table 3). Interestingly, ROCK1 deletion was found to be associated with decreased induction of hypertrophic markers induced by pressure overload after 3 weeks of transverse aortic banding [18]. Together, our observations point out that ROCK1 partially mediates induction of embryonic marker genes by Gαq activation or pressure overload.
Table 3. Gene expression analysis.
| Microarray (Fold over WT) |
Real time RT-PCR (Fold over WT) |
|||||
|---|---|---|---|---|---|---|
| Gαq n = 3 |
Gαq/ROCK1-/- n = 3 |
ROCK1-/- n = 3 |
Gαq n = 6 |
Gαq/ROCK1-/- n = 6 |
ROCK1-/- n = 6 |
|
| ANF | ND | ND | ND | 9.22* | 5.22*# | -1.04 |
| βMHC | 10.61* | 5.12*# | -1.13 | 6.64* | 3.77*# | -1.09 |
| αSK | 2.77* | 2.14* | -2.63* | 3.48* | 2.71*# | -2.01* |
| PKCα | 2.02* | 2.01* | 1.05 | ND | ND | ND |
| AC 5 | 1.13 | 1.08 | 1.03 | -1.07 | -1.02 | -1.04 |
| AC 6 | 1.1 | 1.15 | 1.07 | 1.02 | 1.05 | 1.02 |
| Gαq | 26.9* | 28.9* | -1.06 | ND | ND | ND |
| ROCK1 | 1.13 | -4.05*# | -4.67* | 0.94 | -3.97*# | -4.72* |
| ROCK2 | 1.03 | 1.02 | 1.05 | 1.04 | 1.05 | 1.02 |
αSK, skeletal α-actin; AC, adenylyl cyclase.
P < 0.05 Gαq vs. WT or Gαq/ROCK1-/- vs. WT or ROCK1-/- vs. WT.
P < 0.05 for Gαq/ROCK1-/- vs. Gαq. ND: not determined.
ROCK1 deletion prevented left ventricular dilation and dysfunction induced by cardiac-specific overexpression of Gαq
Ventricular dimension and contractile function were assessed by echocardiography. Consistent with the results of previously reported studies [6], Gαq mice developed left ventricular chamber dilation and contractile dysfunction (Fig. 3A, Table 2). In contrast, left ventricular dimension and left ventricular contractile function were preserved in Gαq/ROCK1-/- mice indicated by normalization of end-systolic and end-diastolic dimensions and left ventricular fractional shortening in these mice compared to WT and ROCK1-/- values (Fig. 3A, Table 2). In addition, left ventricular posterior wall thickness was significantly increased in the Gαq/ROCK1-/- mice compared with WT and ROCK1-/- mice (Table 2). These echocardiographic parameters indicate that Gαq/ROCK1-/- mice underwent concentric cardiac hypertrophy with preserved contractile function whereas Gαq mice developed cardiac dilation with impaired contractile function. Moreover, morphological analysis also supported a concentric hypertrophic phenotype in Gαq/ROCK1-/- mice (Fig. 3B). Finally, morphometric analysis (Table 1) showed a slight but significant increase in lung weight in Gαq mice indicating mild lung congestion as previously reported [28]. This phenotype was completely rescued in Gαq/ROCK1-/- mice (Table 1), supporting improved cardiac function in Gαq/ROCK1-/- mice.
Figure 3. ROCK1 deletion prevented left ventricular dilation in Gαq mice.
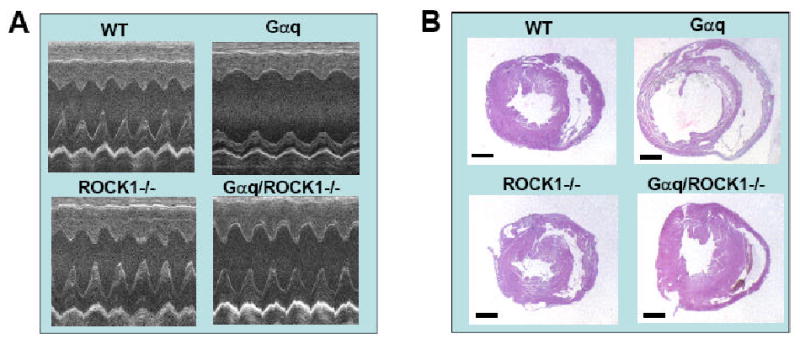
A. Representative M-mode echocardiographic images of 12-week-old male mice with indicated genotype. Note the normalization of left ventricular systolic and diastolic dimensions in compound Gαq/ROCK1-/- mice compared with Gαq mice. B. Representative images of hematoxylin/eosin staining of heart sections, showing concentric remodeling in Gαq/ROCK1-/- and dilation in Gαq mice. Bar, 1 mm.
Gαq mice had lower mean heart rates compared with WT mice (238±6 beats/min vs. 467±3 beats/min) as previously reported [6]. The mean heart rate of Gαq/ROCK1-/- (368±4 beats/min) mice was significantly higher than that of Gαq mice, but still significantly lower than those of WT (467±3 beats/min) and ROCK1-/- (463±7 beats/min) mice. The increased heart rate in Gαq/ROCK1-/- mice in comparison with Gαq mice unlikely contributed to the improved cardiac contractile function in Gαq/ROCK1-/- mice, because transgenic expression of regulator of G protein signaling 4 [29] or deletion of MEKK1 [10] completely prevented the Gαq-induced left ventricular dilation and contractile dysfunction without rescuing the heart rate.
Besides the normalized ventricular contractile function at baseline, Gαq/ROCK1-/- mice also exhibited improved contractility in response to β-adrenergic receptor stimulation when compared with Gαq mice (Fig. 4). Gαq mice exhibited an impaired response to isoproterenol as previously reported [30], and the concurrent deletion of ROCK1 improved left ventricular fractional shortening toward normal. However, a mild but significant reduction in left ventricular fractional shortening was observed in Gαq/ROCK1-/- group compared with ROCK1-/- or WT group in the presence of isoproterenol.
Figure 4. ROCK1 deletion increased basal and isoproterenol-stimulated ventricular function in Gαq mice.
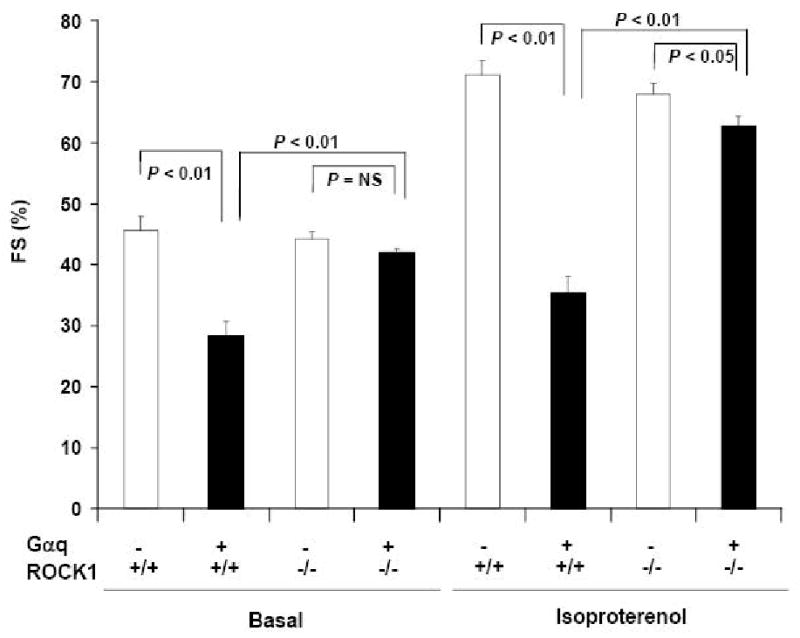
Fractional shortening (FS) was determined by echocardiography in four groups of 12-week-old male mice with indicated genotype before and after administration of isoproterenol (n = 6-8 in each group). Basal and isoproterenol-stimulated left ventricular fractional shortening was reduced in Gαq group compared with WT group. Concurrent ROCK1 deletion increased both basal and isoproterenol-stimulated left ventricular fractional shortening toward normal, and FS was significantly different between Gαq/ROCK1-/- and Gαq groups (P < 0.01).
Additional histological analysis of the heart indicated that fibrosis was not consistently observed in the Gαq and Gαq/ROCK1-/- groups. This observation is consistent with previous reports for the Gαq transgenic mice under these conditions [6, 10, 30]. In addition, there was no significant increase in cardiomyocyte apoptosis in Gαq and Gαq/ROCK1-/- groups compared with WT or ROCK1-/- groups as the TUNEL positivity of cardiomyocytes was not significantly different among the four groups (WT 0.084±0.007%, Gαq 0.101±0.011%, ROCK1-/- 0.082±0.008%, Gαq/ROCK1-/- 0.089±0.012%, n = 4 for each group). This observation is consistent with previous reports showing increased cardiomyocyte apoptosis in Gαq hearts compared with WT hearts in the peripartum period, but not under baseline conditions [31-33].
ROCK1 deletion did not inhibit MAPK activation and PKCα activation induced by cardiac-specific overexpression of Gαq
To investigate potential mechanisms underlying the beneficial effects of concurrent deletion of ROCK1 on cardiac structure and contractile function in Gαq mice, we have examined the effects of ROCK1 deletion on several signaling molecules previously reported to be involved in Gαq-induced cardiomyopathy. Gαq, in its active form, activates phosphatidylinositol-specific phospholipase C (PLC), resulting in inositol triphosphate-mediated calcium release and diacylglycerol-mediated activation of PKC. Furthermore, Gαq overexpression has also been associated with increased activation of JNK [10], up-regulation of PKCα expression and activity [34], increased activation of PKCε [6], uncoupling of β-adrenergic receptor from Gs, an increase in Giα, and down-regulation of adenylyl cyclase expression [34].
Activation of MEKK1 has previously been reported to mediate Gαq-induced hypertrophic response, chamber dilation, and contractile dysfunction [10]. A significant increase in JNK activation was detected in Gαq mice at 3 weeks of age [10], but not observed at 12 weeks [5, 6, 10]. To test the effects of ROCK1 deletion on JNK activation, Western blots with anti-phospho antibodies reactive to activated ERK, JNK, and p38 MAPK were performed with ventricular homogenates of 4 groups of mice at 3 weeks of age. JNK activation was significantly increased in Gαq mice as well as in Gαq/ROCK1-/- mice when compared with WT and ROCK1-/- mice (Fig. 5A, C). A significant increase in ERK activation was also observed in Gαq mice as well as in Gαq/ROCK1-/- mice (Fig. 5A, B), indicating that ROCK1 deletion could not prevent activation of MAPK by Gαq.
Figure 5. ROCK1 deletion did not inhibit MAPK activation and PKCα activation induced by Gαq.
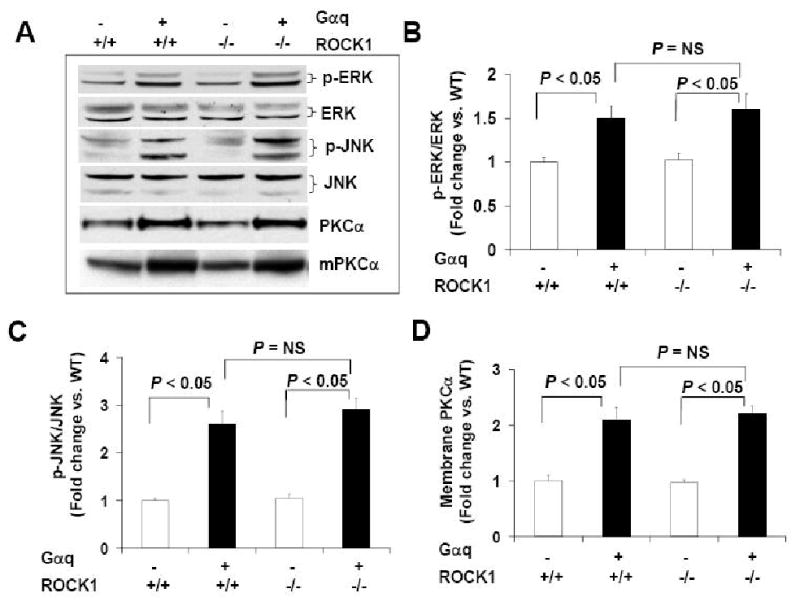
A. Representative images of Western blot analysis of ERK (∼42, 44 kDa) and phosphorylated ERK (p-ERK) (∼42, 44 kDa), JNK (∼46, 54 kDa) and phosphorylated JNK (p-JNK) (∼46, 54 kDa) in ventricular homogenates from 3-week-old male mice and Western blot analysis of PKCα (∼80 kDa) in ventricular homogenates or in the membrane fraction (mPKCα) of ventricular homogenates of 12-week-old male mice with indicated genotype. B-D. Densitometric analysis of immunoreactive bands of p-ERK/ERK, p-JNK/JNK, PKCα (n = 4-6 in each group), expressed as fold change relative to WT group. An equal amount of protein samples (50 μg) was applied to each lane.
Increased PKCα expression and activity in Gαq mouse hearts were considered as a contributory factor to cardiac hypertrophy and impaired cardiac contractile function [7, 34]. The inactive and active PKCα are preferentially localized in the cytosol and at the cell membrane respectively [7, 34]. Western blot analyses showed that PKCα protein level was significantly increased in total ventricular homogenates as well as in the membrane fraction of Gαq and Gαq/ROCK1-/- mouse hearts compared to WT and ROCK1-/- mouse hearts (Fig. 5A, D). Microarray analysis showed a 2-fold increase in PKCα mRNA in Gαq and Gαq/ROCK1-/- mouse hearts compared with WT mouse hearts (Table 3). These results indicate that ROCK1 deletion did not prevent up-regulation of the expression and activation of PKCα by cardiac-specific Gαq overexpression.
ROCK1 deletion rescued down-regulation of adenylyl cyclase type V/VI induced by cardiac-specific overexpression of Gαq
A significant decrease in the cardiac expression of type V/VI adenylyl cyclases was reported to be a major mechanism underlying impaired ventricular contractile function and β-adrenergic signaling in Gαq overexpressed hearts [34]. Moreover, transgenic expression of type V adenylyl cyclase [8] or type VI adenylyl cyclase [9] restored ventricular contractile function and normalized β-adrenergic signaling. Western blot analysis was performed with an antibody against type V/VI adenylyl cyclase because type V or VI specific antibodies are not commercially available. In adult cardiac tissues, both type V and VI adenylyl cyclases are the predominant adenylyl cyclase isoforms. Western blot showed reduced immunoreactive signal in Gαq mouse hearts compared to WT mouse hearts (Fig. 6A, B) in agreement with previous studies [34]. Interestingly, expression of type V/VI adenylyl cyclases was not significantly different from Gαq/ROCK1-/- hearts compared with WT hearts (Fig. 6A, B). We observed that mRNA levels of type V or VI adenylyl cyclase were not affected by Gαq overexpression or ROCK1 deletion (Table 3), indicating that down-regulation of type V/VI adenylyl cyclases by Gαq is a post-transcriptional event. In addition, we examined the expression levels of other critical signaling molecules in β-adrenergic receptor signaling such as β1 and β2-adrenergic receptors. We found their expression levels were not affected by Gαq overexpression as previously reported [34], or by ROCK1 deletion (Fig. 6A). These results indicate that ROCK1 deletion rescued this critical molecular defect induced by cardiac-specific overexpression of Gαq, consistent with improved contractile function by ROCK1 deletion.
Figure 6. ROCK1 deletion prevented down-regulation of type V/VI adenylyl cyclases induced by Gαq.
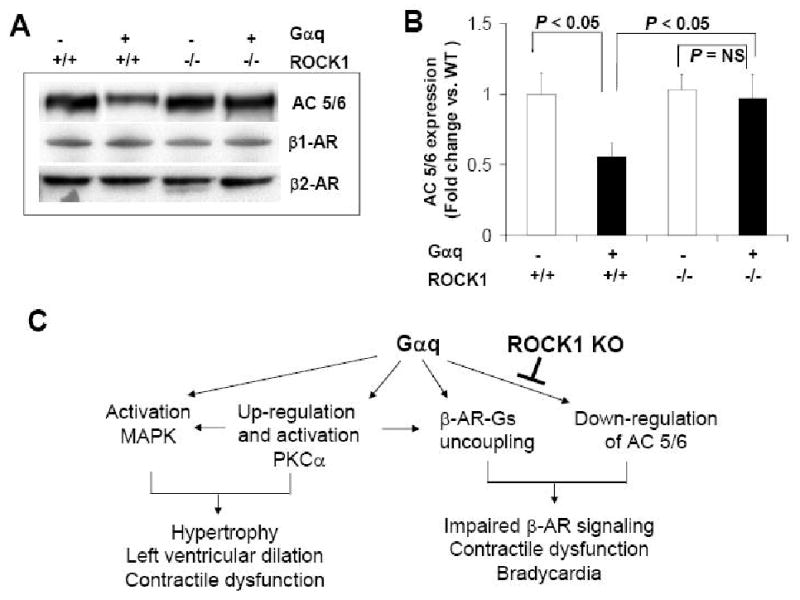
A. Representative images of Western blot analysis of β1-adrenergic receptor (β1-AR) (∼65 kDa), β2-adrenergic receptor (β2-AR) (∼60 kDa), and type V/VI adenylyl cyclases (AC 5/6) (∼130 kDa) in ventricular homogenates of 12-week-old male mice with indicated genotype. B. Densitometric analysis of immunoreactive bands of AC 5/6 (n = 4-6 in each group), expressed as fold change relative to WT group. An equal amount of protein samples (50 μg) was applied to each lane. C. Schematic summary of the relationship between Gαq-activated and ROCK1-mediated signaling pathways in vivo. ROCK1 deletion has no effect on Gαq-PKC-MAPK signaling pathway for Gαq-induced hypertrophic response. The preventive effect of ROCK1 deletion on contractile dysfunction in Gαq mice is mediated at least in part through preventing down-regulation of type V/VI adenylyl cyclases.
Discussion
The importance of Gαq-mediated signaling for the development of cardiomyocyte hypertrophy is well recognized and cardiomyocyte-specific overepxression of Gαq transgenic mice serve as a well-characterized in vivo model to dissect signaling pathways responsible for the development of cardiac hypertrophy and the transition to heart failure [35]. In this study, we have shown that ROCK1 deletion prevented or attenuated a variety of pathological characteristics of Gαq mice, such as left ventricular dilation, contractile dysfunction, bradycardia, impaired contractile response to β-adrenergic stimulation, and induction of hypertrophic markers. The present study is the first to demonstrate an in vivo role of ROCK1 in an animal model of dilated cardiomyopathy and contractile dysfunction.
It is worth noting that most of the analyses in the present study were performed in male mice at 12 weeks, the age when the pathological phenotypes induced by Gαq overexpression have been well-characterized in other studies. ROCK1 deletion did not prevent the development of cardiomyocyte hypertrophy induced by Gαq overexpression in the present study, or by pressure overload in our previous study [18], indicating that ROCK1 is not required for the initial compensatory response to hypertrophic stimuli. Previous genetic manipulations targeted on proximal players of the Gαq pathways, of particular importance, Gαq-PLC-PKC-MAPK pathway, have shown that cardiac hypertrophy was attenuated by inhibition of Gαq activity [29], inhibition of PKCα activation [7], or by inhibition of MAPK activation [10]. Consistent with the preserved hypertrophic phenotype, ROCK1 deletion neither affected the activation of MAPK, nor the up-regulation and activation of PKCα (Fig. 5). On the other hand, Gαq overexpression had no detectable effect on ROCK1 expression and activation (Fig. 1). These observations do not provide support for direct biochemical interactions between Gαq and ROCK1 for Gαq-induced hypertrophic response in vivo (Fig. 6C). Our in vivo observation is contrasted to the previous studies performed in cultured rat neonatal cardiomyocytes showing that Rho/ROCK pathway cooperated with Gαq-PLC-PKC-MAPK pathway for the induction of cardiomyocye hypertrophy by G protein-coupled receptor agonists [21-24]. This discrepancy may relate to the in vivo complexity of the hypertrophic responses or to the presence of ROCK2 isoform. Future in vivo loss-of-function study of the ROCK2 isoform is necessary to address this discrepancy.
Although the cardiac hypertrophy induced by Gαq was not reduced by ROCK1 deletion, the structure and function of this hypertrophic heart were substantially improved, suggesting that ROCK1 inhibition could favorably modify hypertrophy without inhibiting it. ROCK1 deletion prevented cardiac dilation and preserved contractile function at baseline condition, and also improved contractile response to β-adrenergic stimulation. The mechanism underlying the Gαq-induced cardiac dilation is not clear and is likely reflecting the deleterious consequences of myocardial hypertrophy, as reduced cardiac dilation has been found to be associated with inhibition of cardiac hypertrophy in several studies [10, 29, 30, 36]. However, it is not the case in the present study as ROCK1 deletion prevented cardiac dilation without attenuating cardiac hypertrophy at baseline condition. These results suggest that although ROCK1 could not interfere with proximal Gαq signaling events implicated in cardiac hypertrophy, it may modify distal remodeling events in this pathological cardiac hypertrophy model.
It has been recently shown that ROCK1 deletion reduced interstitial fibrosis [18] and cardiomyocyte apoptosis [19] induced by pressure overload, and that ROCK1 haploinsufficiency also decreased perivascular fibrosis induced by angiotensin II [20]. However, the present study suggests that the beneficial effect of ROCK1 deletion on cardiac structure in Gαq transgenic hearts at baseline condition may be mediated by a mechanism other than inhibition of cardiomyocyte apoptosis and cardiac fibrosis as no significant increase in cardiomyocyte apoptosis and fibrosis was detected. These observations are consistent with the previous reports showing that cardiomyocyte apoptosis and fibrosis were increased in the peripartum Gαq mice [5, 32, 33], but not at baseline condition [6, 7, 10].
The present study suggests that the preventive effect of ROCK1 deletion on contractile dysfunction in Gαq mice is mediated at least in part through preventing down-regulation of type V/VI adenylyl cyclases (Fig. 6C). Impaired β-adrenergic receptor coupling in Gαq mice contributes to the impaired contractile function in this model [6]. Among the potential mechanisms of β-adrenergic receptor dysfunction, a significant decrease in type V/VI adenylyl cyclase expression (about 45%) and adenylyl cyclase activity (about 45%) is likely to be a predominant mechanism, as transgenic replacement of type V or type VI adenylyl cyclase in Gαq mice normalized basal and β-adrenergic agonist-stimulated adenylyl cyclase activities, improved contractile function, and increased heart rate, but did not reduce cardiac hypertrophy [8, 34]. Additionally, transgenic expression of type VI adenylyl cyclase in Gαq mice also normalized basal and β-adrenergic agonist-stimulated adenylyl cyclase activities, improved cardiac contractility, and increased heart rate, but did not reduce cardiac dilation [9]. Elevated PKCα expression and activity in Gαq mouse hearts also possibly contribute to β-adrenergic receptor phosphorylation and uncoupling [7, 34]. However, deleting ROCK1 did not prevent up-regulation of PKCα expression in Gαq mouse hearts, and therefore, the rescue effect of ROCK1 deletion on cardiac contractile function in Gαq mice appears to be independent of PKCα expression and activity. It is worth noting that it is generally believed that desensitization or inhibition of β-adrenergic pathway is protective in heart failure and recent studies with adenylyl cyclase V knockout mice have demonstrated a pro-apoptotic role for adenylyl cyclase V under several pathological conditions [37, 38]. In contrast, transgenic expression of adenylyl cyclase VI or V resulted in cardiac protection in Gαq model [8, 9] as mentioned above and transgenic expression of adenylyl cyclase VI also resulted in cardiac protection in myocardial infarction model [39]. These observations point out the necessity to further dissect downstream signaling pathways mediating β-adrenergic signaling in cardiomyopathy in which ROCK1 may play a role.
The mechanism underlying the down-regulation of type V/VI adenylyl cyclases in Gαq mice is not clear, and appears to be a post-transcriptional event as the mRNA levels were not affected. A number of post-translational modifications of type V and VI adenylyl cyclases have been reported including glycosylation, nitrosylation, and phosphorylation by PKA and PKC, which may affect their activity and turnover [40]. A slight increase in molecular weight for adenylyl cyclases was detected only in Gαq ventricular homogenates, but not in WT or Gαq/ROCK1-/- groups (Fig. 6A), suggesting increased post-translational modifications of the type V/VI adenylyl cyclases by Gαq overexpression, which could be attenuated by ROCK1 deletion.
Given the remarkable beneficial effects of ROCK1 deletion on cardiac structure and function observed in Gαq transgenic model, it will be of interest to determine whether ROCK1 deficiency can preserve cardiac structure and function in other pathophysiological settings. In the previous studies, the effect of ROCK1 deficiency [18] and haploinsufficiency [20] on myocardial dilation and dysfunction could not be determined as myocardial structure and function were preserved in control and ROCK1 knockout mice under the study conditions. Given the multiple cellular effects of ROCK1 deletion on pathological remodeling including inhibition of cardiomyocyte apoptosis and fibrosis observed in the previous studies [18-20] as well as preservation of β-adrenergic coupling observed in the current study, ROCK1 deletion will most likely exhibit protective effects in a number of other pathophysiological settings.
In conclusion, ROCK1 deficiency preserved cardiac structure and function in a well-characterized model of pathological cardiac hypertrophy. These observations strongly support a role for ROCK1 in the transition from pathological hypertrophy to heart failure, therefore suggesting that ROCK1 may be an important therapeutic target for heart failure.
Acknowledgments
This work was supported by American Heart Association Scientist Development Grant (to L.W.), National Institutes of Health grants (to L.W.), and by the Riley Children's Foundation and the Lilly Endowment. We thank Dr. Loren J. Field for many insightful comments on this study. We are grateful to Dr. Michael Rubart for comments on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sadoshima J, Izumo S. The cellular and molecular response of cardiac myocytes to mechanical stress. Annu Rev Physiol. 1997;59:551–71. doi: 10.1146/annurev.physiol.59.1.551. [DOI] [PubMed] [Google Scholar]
- 2.Dorn GW, 2nd, Robbins J, Sugden PH. Phenotyping hypertrophy: eschew obfuscation. Circ Res. 2003;92:1171–5. doi: 10.1161/01.RES.0000077012.11088.BC. [DOI] [PubMed] [Google Scholar]
- 3.Akhter SA, Luttrell LM, Rockman HA, Iaccarino G, Lefkowitz RJ, Koch WJ. Targeting the receptor-Gq interface to inhibit in vivo pressure overload myocardial hypertrophy. Science. 1998;280:574–7. doi: 10.1126/science.280.5363.574. [DOI] [PubMed] [Google Scholar]
- 4.Wettschureck N, Rutten H, Zywietz A, Gehring D, Wilkie TM, Chen J, et al. Absence of pressure overload induced myocardial hypertrophy after conditional inactivation of Galphaq/Galpha11 in cardiomyocytes. Nat Med. 2001;7:1236–40. doi: 10.1038/nm1101-1236. [DOI] [PubMed] [Google Scholar]
- 5.Adams JW, Sakata Y, Davis MG, Sah VP, Wang Y, Liggett SB, et al. Enhanced Galphaq signaling: a common pathway mediates cardiac hypertrophy and apoptotic heart failure. Proc Natl Acad Sci U S A. 1998;95:10140–5. doi: 10.1073/pnas.95.17.10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.D'Angelo DD, Sakata Y, Lorenz JN, Boivin GP, Walsh RA, Liggett SB, et al. Transgenic Galphaq overexpression induces cardiac contractile failure in mice. Proc Natl Acad Sci U S A. 1997;94:8121–6. doi: 10.1073/pnas.94.15.8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hahn HS, Marreez Y, Odley A, Sterbling A, Yussman MG, Hilty KC, et al. Protein kinase Calpha negatively regulates systolic and diastolic function in pathological hypertrophy. Circ Res. 2003;93:1111–9. doi: 10.1161/01.RES.0000105087.79373.17. [DOI] [PubMed] [Google Scholar]
- 8.Tepe NM, Liggett SB. Transgenic replacement of type V adenylyl cyclase identifies a critical mechanism of beta-adrenergic receptor dysfunction in the G alpha q overexpressing mouse. FEBS Lett. 1999;458:236–40. doi: 10.1016/s0014-5793(99)01147-3. [DOI] [PubMed] [Google Scholar]
- 9.Roth DM, Gao MH, Lai NC, Drumm J, Dalton N, Zhou JY, et al. Cardiac-directed adenylyl cyclase expression improves heart function in murine cardiomyopathy. Circulation. 1999;99:3099–102. doi: 10.1161/01.cir.99.24.3099. [DOI] [PubMed] [Google Scholar]
- 10.Minamino T, Yujiri T, Terada N, Taffet GE, Michael LH, Johnson GL, et al. MEKK1 is essential for cardiac hypertrophy and dysfunction induced by Gq. Proc Natl Acad Sci U S A. 2002;99:3866–71. doi: 10.1073/pnas.062453699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kobayashi N, Horinaka S, Mita S, Nakano S, Honda T, Yoshida K, et al. Critical role of Rho-kinase pathway for cardiac performance and remodeling in failing rat hearts. Cardiovasc Res. 2002;55:757–67. doi: 10.1016/s0008-6363(02)00457-1. [DOI] [PubMed] [Google Scholar]
- 12.Satoh S, Ueda Y, Koyanagi M, Kadokami T, Sugano M, Yoshikawa Y, et al. Chronic inhibition of Rho kinase blunts the process of left ventricular hypertrophy leading to cardiac contractile dysfunction in hypertension-induced heart failure. J Mol Cell Cardiol. 2003;35:59–70. doi: 10.1016/s0022-2828(02)00278-x. [DOI] [PubMed] [Google Scholar]
- 13.Higashi M, Shimokawa H, Hattori T, Hiroki J, Mukai Y, Morikawa K, et al. Long-term inhibition of Rho-kinase suppresses angiotensin II-induced cardiovascular hypertrophy in rats in vivo: effect on endothelial NAD(P)H oxidase system. Circ Res. 2003;93:767–75. doi: 10.1161/01.RES.0000096650.91688.28. [DOI] [PubMed] [Google Scholar]
- 14.Hattori T, Shimokawa H, Higashi M, Hiroki J, Mukai Y, Tsutsui H, et al. Long-term inhibition of Rho-kinase suppresses left ventricular remodeling after myocardial infarction in mice. Circulation. 2004;109:2234–9. doi: 10.1161/01.CIR.0000127939.16111.58. [DOI] [PubMed] [Google Scholar]
- 15.Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 1997;389:990–4. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]
- 16.Ishizaki T, Uehata M, Tamechika I, Keel J, Nonomura K, Maekawa M, et al. Pharmacological properties of Y-27632, a specific inhibitor of rho-associated kinases. Molecular pharmacology. 2000;57:976–83. [PubMed] [Google Scholar]
- 17.Breitenlechner C, Gassel M, Hidaka H, Kinzel V, Huber R, Engh RA, et al. Protein kinase A in complex with Rho-kinase inhibitors Y-27632, Fasudil, and H-1152P: structural basis of selectivity. Structure (Camb) 2003;11:1595–607. doi: 10.1016/j.str.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 18.Zhang YM, Bo J, Taffet GE, Chang J, Shi J, Reddy AK, et al. Targeted deletion of ROCK1 protects the heart against pressure overload by inhibiting reactive fibrosis. Faseb J. 2006;20:916–25. doi: 10.1096/fj.05-5129com. [DOI] [PubMed] [Google Scholar]
- 19.Chang J, Xie M, Shah VR, Schneider MD, Entman ML, Wei L, et al. Activation of Rho-associated coiled-coil protein kinase 1 (ROCK-1) by caspase-3 cleavage plays an essential role in cardiac myocyte apoptosis. Proc Natl Acad Sci U S A. 2006;103:14495–500. doi: 10.1073/pnas.0601911103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rikitake Y, Oyama N, Wang CY, Noma K, Satoh M, Kim HH, et al. Decreased perivascular fibrosis but not cardiac hypertrophy in ROCK1+/- haploinsufficient mice. Circulation. 2005;112:2959–65. doi: 10.1161/CIRCULATIONAHA.105.584623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoshijima M, Sah VP, Wang Y, Chien KR, Brown JH. The low molecular weight GTPase Rho regulates myofibril formation and organization in neonatal rat ventricular myocytes. Involvement of Rho kinase. J Biol Chem. 1998;273:7725–30. doi: 10.1074/jbc.273.13.7725. [DOI] [PubMed] [Google Scholar]
- 22.Kuwahara K, Saito Y, Nakagawa O, Kishimoto I, Harada M, Ogawa E, et al. The effects of the selective ROCK inhibitor, Y27632, on ET-1-induced hypertrophic response in neonatal rat cardiac myocytes--possible involvement of Rho/ROCK pathway in cardiac muscle cell hypertrophy. FEBS Lett. 1999;452:314–8. doi: 10.1016/s0014-5793(99)00680-8. [DOI] [PubMed] [Google Scholar]
- 23.Wei L. Lysophospholipid signaling in cardiac myocyte hypertrophy. J Mol Cell Cardiol. 2004;36:465–8. doi: 10.1016/j.yjmcc.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 24.Yanazume T, Hasegawa K, Wada H, Morimoto T, Abe M, Kawamura T, et al. Rho/ROCK pathway contributes to the activation of extracellular signal-regulated kinase/GATA-4 during myocardial cell hypertrophy. J Biol Chem. 2002;277:8618–25. doi: 10.1074/jbc.M107924200. [DOI] [PubMed] [Google Scholar]
- 25.Wei L, Taffet GE, Khoury DS, Bo J, Li Y, Yatani A, et al. Disruption of Rho signaling results in progressive atrioventricular conduction defects while ventricular function remains preserved. Faseb J. 2004;18:857–9. doi: 10.1096/fj.03-0664fje. [DOI] [PubMed] [Google Scholar]
- 26.Soonpaa MH, Field LJ. Assessment of cardiomyocyte DNA synthesis in normal and injured adult mouse hearts. Am J Physiol. 1997;272:H220–6. doi: 10.1152/ajpheart.1997.272.1.H220. [DOI] [PubMed] [Google Scholar]
- 27.Wei L, Imanaka-Yoshida K, Wang L, Zhan S, Schneider MD, DeMayo FJ, et al. Inhibition of Rho family GTPases by Rho GDP dissociation inhibitor disrupts cardiac morphogenesis and inhibits cardiomyocyte proliferation. Development. 2002;129:1705–14. doi: 10.1242/dev.129.7.1705. [DOI] [PubMed] [Google Scholar]
- 28.Sakata Y, Hoit BD, Liggett SB, Walsh RA, Dorn GW., 2nd Decompensation of pressure-overload hypertrophy in G alpha q-overexpressing mice. Circulation. 1998;97:1488–95. doi: 10.1161/01.cir.97.15.1488. [DOI] [PubMed] [Google Scholar]
- 29.Rogers JH, Tsirka A, Kovacs A, Blumer KJ, Dorn GW, 2nd, Muslin AJ. RGS4 reduces contractile dysfunction and hypertrophic gene induction in Galpha q overexpressing mice. J Mol Cell Cardiol. 2001;33:209–18. doi: 10.1006/jmcc.2000.1307. [DOI] [PubMed] [Google Scholar]
- 30.Dorn GW, 2nd, Tepe NM, Lorenz JN, Koch WJ, Liggett SB. Low- and high-level transgenic expression of beta2-adrenergic receptors differentially affect cardiac hypertrophy and function in Galphaq-overexpressing mice. Proc Natl Acad Sci U S A. 1999;96:6400–5. doi: 10.1073/pnas.96.11.6400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adams JW, Pagel AL, Means CK, Oksenberg D, Armstrong RC, Brown JH. Cardiomyocyte apoptosis induced by Galphaq signaling is mediated by permeability transition pore formation and activation of the mitochondrial death pathway. Circ Res. 2000;87:1180–7. doi: 10.1161/01.res.87.12.1180. [DOI] [PubMed] [Google Scholar]
- 32.Yussman MG, Toyokawa T, Odley A, Lynch RA, Wu G, Colbert MC, et al. Mitochondrial death protein Nix is induced in cardiac hypertrophy and triggers apoptotic cardiomyopathy. Nat Med. 2002;8:725–30. doi: 10.1038/nm719. [DOI] [PubMed] [Google Scholar]
- 33.Hayakawa Y, Chandra M, Miao W, Shirani J, Brown JH, Dorn GW, 2nd, et al. Inhibition of cardiac myocyte apoptosis improves cardiac function and abolishes mortality in the peripartum cardiomyopathy of Galpha(q) transgenic mice. Circulation. 2003;108:3036–41. doi: 10.1161/01.CIR.0000101920.72665.58. [DOI] [PubMed] [Google Scholar]
- 34.Dorn GW, 2nd, Tepe NM, Wu G, Yatani A, Liggett SB. Mechanisms of impaired beta-adrenergic receptor signaling in G(alphaq)-mediated cardiac hypertrophy and ventricular dysfunction. Molecular pharmacology. 2000;57:278–87. [PubMed] [Google Scholar]
- 35.Dorn GW, 2nd, Force T. Protein kinase cascades in the regulation of cardiac hypertrophy. J Clin Invest. 2005;115:527–37. doi: 10.1172/JCI24178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu G, Toyokawa T, Hahn H, Dorn GW., 2nd Epsilon protein kinase C in pathological myocardial hypertrophy. Analysis by combined transgenic expression of translocation modifiers and Galphaq. J Biol Chem. 2000;275:29927–30. doi: 10.1074/jbc.C000380200. [DOI] [PubMed] [Google Scholar]
- 37.Okumura S, Takagi G, Kawabe J, Yang G, Lee MC, Hong C, et al. Disruption of type 5 adenylyl cyclase gene preserves cardiac function against pressure overload. Proc Natl Acad Sci U S A. 2003;100:9986–90. doi: 10.1073/pnas.1733772100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Okumura S, Vatner DE, Kurotani R, Bai Y, Gao S, Yuan Z, et al. Disruption of type 5 adenylyl cyclase enhances desensitization of cyclic adenosine monophosphate signal and increases Akt signal with chronic catecholamine stress. Circulation. 2007;116:1776–83. doi: 10.1161/CIRCULATIONAHA.107.698662. [DOI] [PubMed] [Google Scholar]
- 39.Takahashi T, Tang T, Lai NC, Roth DM, Rebolledo B, Saito M, et al. Increased cardiac adenylyl cyclase expression is associated with increased survival after myocardial infarction. Circulation. 2006;114:388–96. doi: 10.1161/CIRCULATIONAHA.106.632513. [DOI] [PubMed] [Google Scholar]
- 40.Beazely MA, Watts VJ. Regulatory properties of adenylate cyclases type 5 and 6: A progress report. Eur J Pharmacol. 2006;535:1–12. doi: 10.1016/j.ejphar.2006.01.054. [DOI] [PubMed] [Google Scholar]