

Abstract
The significance of elevated plasma levels of bacterial lipopolysaccharide (LPS) in persons with chronic HIV infection remains undefined. We measured LPS levels by use of limulus lysate assay, and DNA sequences encoding bacterial ribosomal 16S RNA (16S rDNA) were assessed by quantitative polymerase chain reactions in plasma samples obtained from 242 donors. Plasma levels of 16S rDNA were significantly higher in human immunodeficiency virus (HIV)–infected subjects than in uninfected subjects, and they correlated with LPS levels. Higher levels of 16S rDNA were associated with higher levels of T cell activation and with lower levels of CD4 T cell restoration during antiretroviral therapy. Antiretroviral therapy reduces but does not fully normalize plasma levels of bacterial 16S rDNA, an index of microbial translocation from the gastrointestinal tract. High levels of 16S rDNA during therapy are strongly associated with reduced increases in the CD4+ T lymphocyte count, irrespective of plasma HIV RNA levels. These findings are consistent with the importance of microbial translocation in immunodeficiency and T cell homeostasis in chronic HIV infection.
Depletion of circulating CD4 T cells is the hallmark of the progressive immunodeficiency associated with HIV infection. Although HIV replicates within and can result in the destruction of CD4+ T cells [1, 2], the mechanisms whereby HIV infection results in CD4+ T cell depletion are incompletely understood [2-4]. Chronic immune activation is independently associated with disease progression [5] and CD4+ T cell loss [6]. Although there is increasing consensus that immune activation is central to the pathogenesis of HIV disease, the mechanisms whereby activation drives immunodeficiency are poorly understood.
Recently, we demonstrated evidence of increased microbial translocation from the damaged gut in patients with chronic HIV infection [7]. High levels of the Toll-like receptor 4 ligand lipopolysaccharide (LPS) were measured in plasma samples obtained from persons with chronic HIV infection and were correlated with indices of innate and adaptive immune activation. LPS levels were increased in pathogenic simian immunodeficiency virus (SIV) infection in rhesus macaques but not in natural nonpathogenic SIV infection in sooty mangabeys, suggesting that persistent exposure to microbial products is mechanistically involved with HIV/SIV immunopathogenesis [8].
Combination antiretroviral therapy (ART) can durably suppress viral replication to levels below the limits of detection in most persons. As a result, the numbers of circulating CD4+ T lymphocytes increase, with many individuals eventually achieving “normal” peripheral CD4+ T cell counts. In a signifi-cant proportion of treated patients, however, the peripheral CD4+ T cell counts are not restored, at least through 5−7 years of treatment [9, 10]. Persistently low CD4+ T cell counts are associated with significant increases in non–AIDS-associated morbidities, including cardiovascular disease, liver disease, cancer, and, perhaps, renal disease [11]. The mechanisms for blunted CD4+ T cell gains are likely multiple and include thymic dys-function, failure of naive T cell expansion, and persistent immune activation [12-15].
Given the uncertainties regarding the clinical significance of microbial translocation in both untreated and treated HIV infection, we initiated a series of 3 related studies to achieve 2 objectives. Our first objective was to use markers other than LPS to confirm that HIV infection is associated with high plasma levels of microbial products. Because LPS is a component of the cell wall of gram-negative bacteria (and, thus, is only present on a proportion of enteric bacterial microbiota [8]), we designed primers and probes to amplify DNA sequences encoding the well-conserved 16S rRNA subunit (16S rDNA) common to most bacteria. Our second objective was to determine the associations between plasma levels of bacterial products and both immune activation and treatment-mediated immune reconstitution. Our findings suggest that microbial translocation from the damaged gut is a likely contributor to immune activation and CD4 T cell homeostasis in untreated and treated chronic HIV infection.
MATERIALS AND METHODS
Study subjects
These studies were approved by the institutional review boards at the University Hospitals of Cleveland, the National Institutes of Health, and the University of California, San Francisco (UCSF), as well as at participating AIDS Clinical Trials Unit sites. Plasma samples were collected into tubes containing EDTA and were stored at −80°C until they were thawed once for study. Three distinct cohorts were examined. The first cohort was based at Case Western Reserve University, and the second was based on the UCSF Study on the Consequences of the Protease Inhibitor Era (SCOPE) cohort study, an ongoing prospective study focused on defining the pathogenesis of treated HIV infection (table 1). The final study included patients enrolled in AIDS Clinical Trials Group (ACTG) study 5014. In the present longitudinal study, 54 treatment-naive patients with plasma HIV RNA levels of 5000−100,000 copies/mL and CD4+ T cell counts of <500 cells/μL were randomized to receive treatment with (1) lopinavir/ritonavir and nevirapine or (2) stavu-dine, lamivudine, and nevirapine. Separate analyses are presented for each cohort.
Table 1.
Characteristics of the 2 cross-sectional study groups.
| Cross-sectional study 1 |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| HIV positive, receiving HAART |
||||||||||
| HIV− |
With low VL |
With high VL |
HIV+, HAART naive |
Cross-sectional study 2a |
||||||
| Characteristic | Patients, no. |
Median (IQR) |
Patients, no. |
Median (IQR) |
Patients, no. |
Median (IQR) |
Patients, no. |
Median (IQR) |
Patients, no. |
Median (IQR) |
| Age, years | 15 | 26 (22−41) | 22 | 50 (41−54) | 18 | 44 (40−50) | 19 | 50 (42−54) | 114 | 44 (40−49) |
| CD4+ countb | . . . | 22 | 653.5 (407−809) | 18 | 229.5 (104−280) | 19 | 432 (339−608) | 114 | 391 (230−604) | |
| Plasma VLc | . . . | 22 | 400 (400−400) | 18 | 15,275 (2360−57,400) | 19 | 14,300 (5080−23,900) | 114 | 400 (400−400) | |
NOTE. Cross-sectional study 1, the Case Western Reserve University study; cross-sectional study 2, University of California, San Francisco Study on the Consequences of the Protease Inhibitor Era (SCOPE) cohort study; HAART, highly active antiretroviral therapy; HIV+, HIV positive; HIV− negative; IQR, interquartile range; VL, viral load.
HIV+ individuals receiving HAART.
CD4+ T cell count, expressed as the no. of cells per microliter.
Expressed as the no. of HIV RNA copies per milliliter.
Plasma DNA isolation
DNA was extracted from 200 μLof plasma by use of the DNeasy Blood and Tissue Kit (Qiagen), according to the manufacturer's instructions. A DU 640 spectrophotometer (Beckman) was used to determine DNA concentration.
Quantitative polymerase chain reaction (PCR) for measurement of bacterial 16S rDNA
A 20-μL amplification reaction consisted of 2 μL of 10× PCR buffer (100 mmol/L Tris-HCl, pH 8.3; and 500 mmol/L KCl [Invitrogen]), 3.5 mmol/L MgCl2, 0.2 mmol/L deoxynucleoside triphosphate, 0.5 μmol/L forward and reverse primers, 0.32 μmol/L probe (338P: 5'-FAM-GCT GCC TCC CGT AGG AGT-BHQ1−3'), 0.75 U of Platinum Taq polymerase (Invitrogen), and 5 μL of the template plasma DNA. Degenerate forward (8F: 5'-AGT TTG ATC CTG GCT CAG-3') and reverse (515R: 5'-GWA TTA CCG CGG CKG CTG-3') primers were used to amplify DNA templates encoding 16S rRNA [16]. The DNA was amplified in triplicate, and mean values were calculated. A standard curve was created from serial dilutions of plasmid DNA containing known copy numbers of the template. The reaction conditions for amplification of DNA were 95°C for 5 min, followed by 45 cycles at 95°C for 15 s and at 60°C for 1 min. Intra-assay variability typically is <5%.
Plasma levels of LPS
Plasma samples were diluted to 10% with endotoxin-free water and then were heated to 85°C for 12 min to inactivate inhibitory plasma proteins. We then quantified plasma LPS with a commercially available limulus amebocyte assay (Cambrex), according to the manufacturer's protocol. We ran samples in duplicate, and background attributable to the turbidity of the diluted plasma was subtracted. This commercial assay for plasma LPS is more rapid and less expensive, and it requires less time than does the assay for bacterial 16S rDNA; however, the LPS assay has greater interassay variability than does the bacterial 16S rDNA assay (25% vs. 10%).
Flow cytometry
Flow cytometric studies were performed using a consensus whole-blood lysis method [17]. In brief, 20 μL of fluorochrome-labeled monoclonal antibody was added to 100 μL of EDTA-anticoagulated whole blood. The mixture was incubated at room temperature for 15 min, and red blood cells were then lysed. The samples were then washed, fixed, and analyzed by flow cytometry. The following antibodies were used: anti-CD38 –phycoerythrin (PE), anti-CD8 –peridinin chlorophyll protein (Percp), anti-CD4 –Percp, anti-CD38 –PE, anti–HLA-DR–fluorescein isothiocyanate, and appropriate isotype control monoclonal antibodies (BD PharMingen).
Statistical analysis
We used conventional measurements of central location and dispersion to describe the data, and we compared pairs of variables by use of Mann-Whitney's U test or Wilcoxon's signed rank test, depending on the associations between variables. We used the Kruskal-Wallis H test or Friedman's test for comparisons of >2 variables. To explore associations between pairs of continuous variables, we used Spearman's rank correlation. To analyze the change in continuous variables over time during repeated measurements in the longitudinal study, and to account for the effect of other factors and covariates, we fitted mixed-effects models, using an unstructured repeated covariance approach in most cases, unless otherwise suggested by the data. To analyze the association of 16S rDNA levels with treatment after controlling for intervening variables, we used robust multiple regression to account for the presence of extreme outliers and unequal sample sizes. Analyses were performed using SPSS software (version 16.01; SPSS) and Stata MP (version 10; Stata) without explicit correction for multiple comparisons. All tests were 2-sided, and P ≤ .05 was considered to denote statistical significance.
RESULTS
Characteristics at baseline
Fifteen HIV-uninfected subjects and 227 HIV-infected persons from 3 cohorts were analyzed. In the first study (based at Case Western Reserve University), we obtained plasma samples from 4 groups: HIV-uninfected adults (n = 15); antiretroviral-naive, untreated HIV-infected adults (n = 19; median plasma HIV RNA level, 14,300 copies/mL; median CD4+ T cell count, 432 cells/μL); antiretroviral-treated patients with undetectable viremia (n = 22; median CD4+ T cell count, 654 cells/μL); and antiretroviral-treated persons with detectable viremia (n = 18; median CD4+ T cell count, 230 cells/μL; median plasma HIV RNA level, 15,275 copies/mL) (table 1). The second study (involving the UCSF SCOPE cohort) focused on determining the association between LPS, 16S rDNA, and immune reconstitution during long-term effective combination therapy (as defined by a plasma HIV RNA level of <1000 copies/mL; 92 of 114 persons had an undetectable plasma HIV RNA level). A total of 114 patients were studied (median CD4+ T cell count, 391 cells/μL; median duration of effective therapy, 28.1 months). The third study was a longitudinal analysis of HIV-infected persons who initiated combination ART. Samples were analyzed at baseline and at 1, 8, and 48 weeks after the initiation of combination therapy. A total of 54 baseline samples were analyzed, and samples for all time points were available from 20 patients with a median pretherapy plasma HIV RNA level and a median CD4+ T cell count of 21,170 copies/mL and 354 cells/μL, respectively.
Increased bacterial 16S rDNA levels in persons with untreated or treated HIV infection
We first analyzed data from the Case Western Reserve University cohort. Bacterial 16S rDNA was not detected in plasma samples obtained from any of the HIV-uninfected adults (figure 1) (The lowest level of reliable detection for this assay is 5 copies/μL). In contrast, bacterial 16S rDNA was detectable in 18 of 19 antiretroviral-untreated HIV-infected persons (median level, 132.5 copies/μL). Long-term combination therapy was associated with bacterial 16S rDNA levels lower than those observed in untreated subjects, but these levels often remained detectable. The levels of bacterial 16S rDNA were lower in treated persons with undetectable viremia (median, 8.6 copies/μL) than in those with detectable viremia (22.8 copies/μL); however, this difference was not statistically significant. The differences among HIV-negative persons, untreated HIV-infected persons, and the combined groups of treated HIV-infected persons (i.e., both viremic and aviremic persons) were highly significant (P < .001, for each pairwise comparison).
Figure 1.
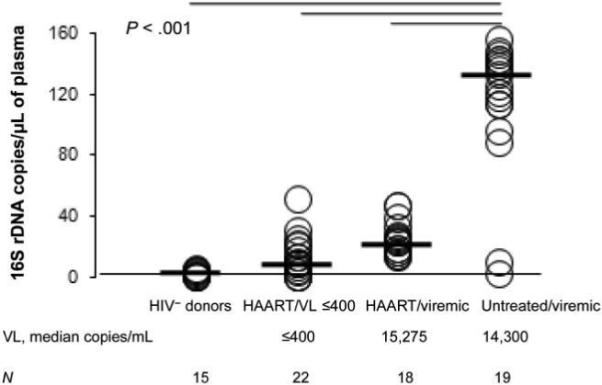
Elevation of bacterial 16S ribosomal DNA (rDNA) levels in the plasma of HIV-infected subjects in the Case Western Reserve University study (cross-sectional study 1). Bacterial 16S rDNA levels were measured by quantitative polymerase chain reaction. P values were calculated using the Mann-Whitney U and Kruskal-Wallis H tests. HAART, highly active antiretroviral therapy; HIV−, HIV negative; VL, viral load.
There was a significant direct association between bacterial 16S rDNA levels and plasma HIV RNA levels among the untreated HIV-infected subjects (r = 0.51; P = .03) (figure 2A) but not among the treated HIV-infected subjects. There was no association between bacterial 16S rDNA levels and CD4+ T cell counts (figure 2B).
Figure 2.
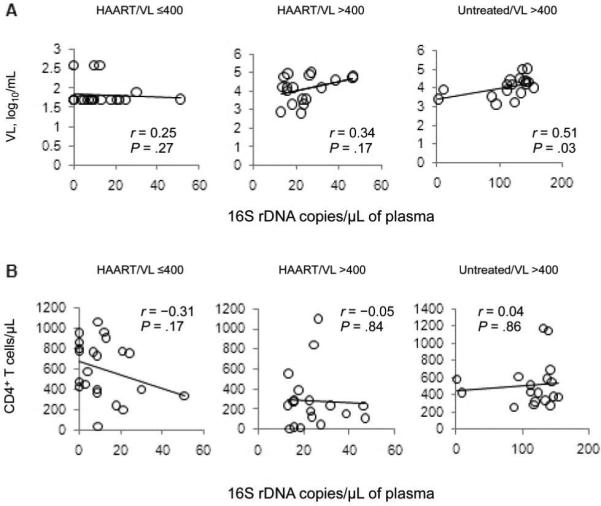
Correlation of plasma levels of bacterial 16S ribosomal DNA (rDNA) with HIV RNA levels (A) but not with circulating CD4+ T cell counts (B). P values were calculated using Spearman's correlation test. Data were derived from the Case Western Reserve University cohort (cross-sectional study 1). HAART, highly active antiretroviral therapy; VL, viral load.
Association of persistently elevated bacterial 16S rDNA levels during effective ART with persistent T cell activation and blunted CD4+ T cell gains
To explore further the associations among microbial translocation and immune restoration during long-term ART, we measured bacterial LPS and 16S rDNA, T cell activation, and treatment-associated increases in peripheral CD4+ T cell counts in 114 persons enrolled in the UCSF SCOPE cohort. As expected, the levels of LPS and bacterial 16S rDNA were correlated with each other (r = 0.31; P = .0009) (figure 3). Higher plasma levels of bacterial DNA were associated with higher levels of CD8+ T cell activation (r = 0.18; P = .047) (figure 4A).
Figure 3.
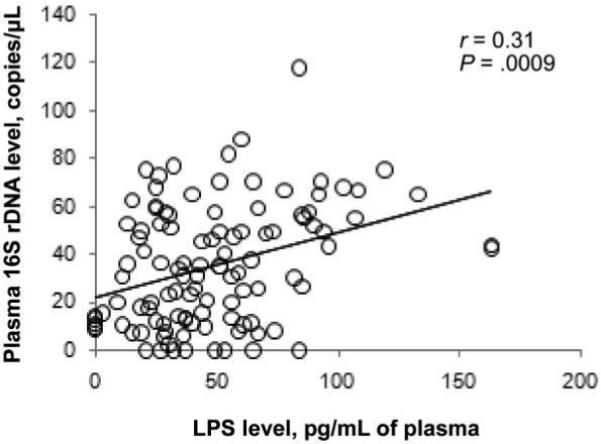
Correlation of plasma levels of bacterial 16S ribosomal DNA (rDNA) with plasma levels of lipopolysaccharide (LPS) among antiretroviral therapy–treated patients with low or undetectable HIV viremia. P values were calculated using Spearman's correlation test. All patients were followed in the University of California, San Francisco Study on the Consequences of the Protease Inhibitor Era (SCOPE) cohort study (cross-sectional study 2).
Figure 4.
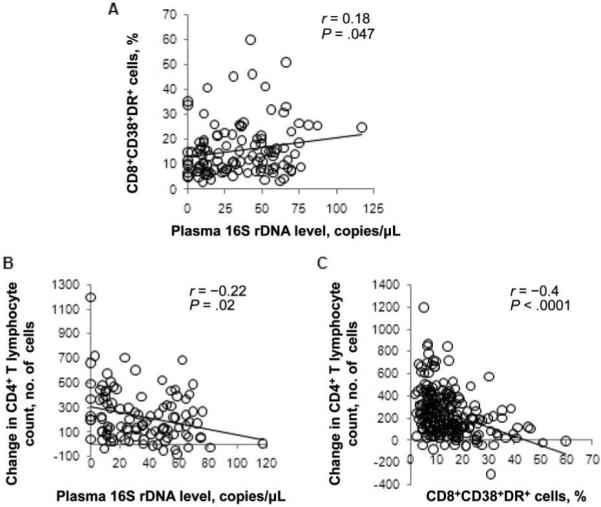
Correlation of plasma levels of bacterial 16S ribosomal DNA (rDNA) with indices of CD8 T cell activation and cellular restoration. A, Plasma 16S rDNA levels correlate directly with the frequency of CD38 and HLA-DR expression on CD8+ T cells. B, Plasma 16S rDNA levels correlate inversely with the magnitude of CD4+ T lymphocyte restoration after antiretroviral therapy (ART). C, Increases in CD4+ T lymphocyte counts after ART correlate inversely with the proportions of CD8+ T cells expressing CD38 and HLA-DR. P values were calculated using Spearman's correlation test. All patients were followed in the University of California, San Francisco Study on the Consequences of the Protease Inhibitor Era (SCOPE) cohort study (cross-sectional study 2).
The treatment-mediated changes in CD4+ T cell counts during long-term ART were associated with higher levels of 16S rDNA (r = −0.22; P = .02) (figure 4B) and the level of T cell activation (r = −0.4; P < .0001) (figure 4C). Even after adjustment for the duration of ART, the pretreatment CD4+ T cell count, both current and pretreatment plasma HIV RNA levels, age, sex, and CD8+ T cell activation level, every 100-copy/μL increase in the bacterial DNA level was associated with a mean of 11 fewer CD4+ T lymphocytes/mm3 gained (P = .015), suggesting an influence of microbial translocation on CD4+ T cell recovery that is largely independent of measured CD8+ T cell activation levels.
ART and progressive decreases in plasma bacterial 16S rDNA levels
The cross-sectional data outlined above suggest that ART partially decreases plasma levels of bacterial 16S rDNA and that persistent microbial translocation is mechanistically associated with immune activation and blunted CD4+ T cell gains during therapy. To further explore the complex association among these indices, we examined samples obtained before and at 1, 8, and 48 weeks after initiation of ART. Plasma levels of bacterial 16S rDNA decreased consistently over time, with the difference from baseline reaching statistical significance at weeks 8 and 48 (P = .021 and P = .025, respectively) (figure 5A). Immune activation indices and plasma levels of HIV RNA also decreased (figures 5B and 5C), whereas CD4+ T cell numbers increased (figure 5D). Repeated-measures analysis indicated that the decrease in the 16S rDNA level was independent of changes in HIV RNA levels (adjusted P = .03). Interestingly, although bacterial 16S rDNA levels decreased progressively during follow-up, they remained higher among participants in the longitudinal study after 48 weeks of therapy than among patients with undetectable HIV RNA levels in the first cross-sectional study (median level, 75 vs. 8.6 copies/μL). Because the median duration of HAART in the cross-sectional Case Western Reserve University study (in excess of 6 years) was considerably greater than 48 weeks of follow-up in the longitudinal study, this finding might reflect continued recovery of the epithelial barrier or improved clearance of bacterial products with sustained immune recovery during receipt of ART.
Figure 5.
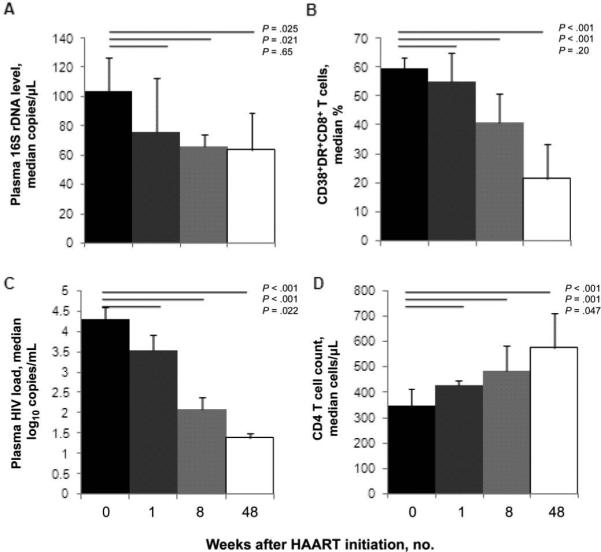
Effects of antiretroviral therapy on plasma levels of bacterial 16S ribosomal DNA (rDNA), HIV RNA levels, immune activation, and CD4 T cell counts. A, Progressive decrease in plasma levels of bacterial 16S rDNA after administration of effective combination antiretroviral therapy (ART). B, Decreased expression of CD38 and HLA DR on CD8+ T cells in association with receipt of combination ART. C, Plasma levels of HIV RNA decrease with combination ART. D, CD4 T cell counts increase with combination ART. Comparisons are made to baseline values after 1, 8, and 48 weeks of ART. Data shown are median values and 95% confidence intervals. P values were calculated using the Wilcoxon signed rank test (n = 20) (AIDS Clinical Trials Group 5014 longitudinal study). HAART, highly active antiretroviral therapy.
In this longitudinal ACTG 5014 treatment study, pretreatment plasma levels of bacterial 16S rDNA were positively correlated with CD8+ T cell activation, as defined by the coexpression of CD38 and HLA-DR (r = 0.37; P = .007) (figure 6A). This observation is consistent with our previous finding that LPS levels are strongly correlated with immune activation during chronic infection [7, 18]. As noted above, 16S DNA levels were also associated with CD8+ T cell activation during long-term suppressive therapy (figure 4A).
Figure 6.
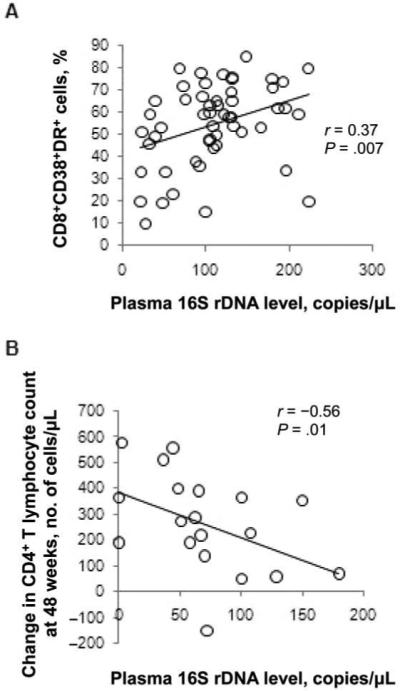
Correlation of pretreatment plasma levels of bacterial 16S ribosomal DNA (rDNA) with indices of immune activation, and inverse correlation, after 48 weeks of therapy, with the magnitude of immune restoration. A, In treatment-naive subjects, correlation of plasma levels of bacterial 16S rDNA with frequencies of CD38+HLA-DR+CD8+ T cells (n = 54). B, At the end of 48 weeks of antiretroviral therapy, inverse correlation of plasma levels of bacterial 16S rDNA with the magnitude of increases in CD4 T cell counts (n = 20). P values were calculated using Spearman's correlation test (AIDS Clinical Trials Group 5014 longitudinal study) (Note: baseline samples were available for 54 subjects; only 20 subjects had samples available for analysis at baseline and at weeks 1, 8, and 48).
As we found in the ACTG 5014 study, there was an association between the plasma 16S rDNA level and CD4+ T lymphocyte reconstitution after 48 weeks of treatment (r = −0.56; P = .01) (figure 6B). Mixed-effects modeling indicated that, even after controlling for a highly active ART (HAART)–induced decrease in the plasma HIV RNA level from baseline to week 48, 16S rDNA levels at the end of the treatment period were significantly and negatively associated with the rate of increase in the number of CD4+ T lymphocytes during that interval (adjusted P value, .04). There was, however, no significant association between plasma levels of bacterial DNA and CD4 T cell restoration at earlier time points (weeks 1 and 8) after initiation of HAART, suggesting that the association between microbial translocation and CD4 T cell homeostasis is more closely related to the cellular turnover of chronic infection than to the rapid first-phase increase in circulating CD4 T cells after the application of HAART [19, 20].
DISCUSSION
There is increasing evidence that broad immune activation is an important driver of disease pathogenesis and CD4 T cell depletion in chronic HIV infection [21-24]. In our current follow-up study, we asked whether markers of microbial translocation other than LPS are elevated during HIV disease and whether these markers are associated with rates of immune reconstitution during ART. By measuring the presence of conserved sequences of 16S rDNA common to most bacteria, we confirmed that untreated HIV infection is consistently associated with high systemic levels of microbial products. Although these levels decrease with effective ART, they remain elevated, even in patients whose virus had been reduced to undetectable levels for several years. Treatment-associated reductions in microbial translocation are related to the change in peripheral CD4+ T lymphocyte counts; importantly, this effect appeared to be independent of the treatment-associated change in viral load. These data suggest that the degree of residual microbial translocation during therapy is strongly associated with the degree of immune restoration.
Our data [7, 18], as well as emerging data from other investigators [25], indicate that bacterial products (LPS and bacterial DNA) are often circulating in the plasma of HIV-infected patients. The most likely source of these bacterial products is the gastrointestinal tract [7], in which mucosal defenses are profoundly disrupted by HIV infection. Our data provide evidence that this translocation is not limited to elements of the gram-negative cell wall but, rather, includes bacterial genetic elements as well.
Our data indicate that HIV infection results in higher plasma levels of bacterial products. In our cross-sectional studies, plasma levels of HIV correlated weakly with plasma levels of bacterial 16S rDNA among untreated persons. Across each study, patients who had received treatment for longer periods had, on average, lower levels of bacterial 16S rDNA. There appeared to be an association between the duration of effective HAART and plasma levels of bacterial DNA, with levels decreasing from >114 copies/μL before therapy to ∼76 copies/μL after 1 year of therapy, to <34 copies/μL after a median of 2 years of therapy and to 17 copies/μL after an average duration of therapy of ∼6 years. Importantly, these levels rarely decreased to the undetectable levels found in HIV-seronegative persons. The effect of HAART on bacterial 16S rDNA levels is seen as early as 1 week after therapy. This finding suggests that viral replication either directly or indirectly drives microbial translocation. This could be mediated through an effect of viral replication on host immune defenses within the gut or, perhaps, a direct effect on other mucosal cells that maintain gut integrity. The progressive decrease in microbial translocation with prolonged ART may be best explained by ongoing partial repair of the mucosal barrier or, conceivably, enhanced clearance from blood. The lack of an association between the circulating CD4+ T cell count and levels of bacterial DNA in plasma suggest that the damaged mucosal barrier is not simply a consequence of generalized immune impairment but, rather, is more likely related to early damage to the gut during acute infection.
We observed a consistent association between plasma bacterial 16S rDNA levels and T cell activation: expression of CD38 and HLA-DR. This association was present in untreated patients with HIV infection and in treated patients with low levels of viremia (<1000 copies RNA/mL). That microbial translocation might drive inflammation is not a surprise. We recently demonstrated that a variety of microbial Toll-like receptor agonists, including LPS and bacterial DNAs, can drive T cell activation in vitro, resulting in heightened expression of CD38 and movement of central memory CD4+ T cells into cell cycle and death [26], a phenotype similar to that seen in chronic HIV infection [27, 28]. Microbial products can produce a similar phenotype in vivo. Krabbe et al. [29] reported that, after intravenous injection of endotoxin into healthy donors, there was an increase in CD38 expression on circulating T cells and a loss of activated T cells from circulation.
One of the more unexpected findings of the present study was the relatively low levels of bacterial 16S rDNA in treated persons who had detectable viremia (compared with untreated persons, who had comparable levels of viremia but much higher levels of bacterial 16S rDNA). This observation suggests that partially effective therapy might have profound effects on the functional integrity of the gut mucosa in much the same way that partially effective therapy is often associated with sustained CD4+ T cell gains [30, 31]. Whether this effect is a result of the selection of a less fit and, perhaps, less virulent virus will require further study. It is likely, however, that this relative decrease in microbial trans-location confers some degree of benefit, and may explain, in part, the consistent observation that T cell activation is often lower in treated patients with detectable viremia than in untreated patients with comparable plasma HIV RNA levels [32].
We also explored the association between microbial translocation and the magnitude of CD4+ T cell restoration during ART. In ACTG study 5014, the degree of persistent microbial translocation was strongly associated with the degree of immune reconstitution, as defined by increases in the numbers of circulating CD4+ T cells. This association was independent of changes in viremia. In our UCSF-based cross-sectional study, plasma levels of bacterial DNA were associated with the degree of treatment-mediated change in CD4+ T cell counts, independent of both viral load and immune activation. There are at least 2 mechanisms that could explain this association. On the one hand, the degree of peripheral CD4+ T cell restoration may be a good marker for the degree of mucosal repair. Alternatively, the degree of residual microbial translocation during HAART may regulate the ability of the immune system to restore peripheral CD4+ T lymphocytes, perhaps by driving cellular turn-over through the mechanisms outlined above. It is also possible that the interaction is bidirectional and that, in some patients, a vicious cycle ensues, with persistent microbial translocation leading to limited immune reconstitution, which then leads to continued dysfunction of the mucosal barrier and sustained microbial translocation.
Sustained or intermittent translocation of microbial elements is also observed in other chronic diseases, including inflammatory bowel disease [33]. Because neither CD4+ T lymphopenia nor cell-mediated immunodeficiency is a recognized concomitant of untreated inflammatory bowel disease [33, 34], it would appear that the virus maintains a central role in CD4+ T cell depletion. In this model, systemic levels of microbial products predict CD4+ T cell changes, even in the absence of detectable viremia suggesting that even low levels of HIV replication or their downstream consequences are sufficient to impair CD4+ T cell homeostasis.
In summary, we found elevated levels of bacterial 16S rDNA in the circulation of persons with chronic HIV infection. Levels decreased after the initiation of ART but did not normalize. Bacterial 16S rDNA levels in plasma were correlated with bacterial LPS levels, with indices of immune activation and inversely correlated with the magnitude of CD4+ T cell restoration after ART. These findings are consistent with a model wherein translocation of microbial products through the damaged gut epithelium plays an important role in the pathogenesis of HIV-induced immunodeficiency.
Acknowledgments
We thank the members of the Cleveland Immunopathogenesis Consortium for advice and helpful discussions.
Financial support: Center for AIDS Research at Case Western Reserve University (grant AI 36219); AIDS Clinical Trials Group (grants AI 25879 and AI 38858); National Institute of Allergy and Infectious Diseases (grant AI 076174). This work was supported in part by the intramural program of the National Institute of Allergy and Infectious Diseases. The Study on the Consequences of the Protease Inhibitor Era (SCOPE) cohort is supported by the National Institutes of Health (grants AI055273 and AI44595), the Center for AIDS Prevention Studies (grant P30 MH62246), the University of California, San Francisco (UCSF) Center for AIDS Research (grants P30 AI27763 and P30 MH59037) and the UCSF Clinical and Translational Science Institute (grant UL1 RR024131-01).
Footnotes
Potential conflicts of interest: none reported.
The funders of this study had no role in study design, data collection and analysis, preparation of the manuscript, or the decision to publish it.
References
- 1.Lu YY, Koga Y, Tanaka K, Sasaki M, Kimura G, Nomoto K. Apoptosis induced in CD4+ cells expressing gp160 of human immunodeficiency virus type 1. J Virol. 1994;68:390–9. doi: 10.1128/jvi.68.1.390-399.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCune JM. The dynamics of CD4+ T-cell depletion in HIV disease. Nature. 2001;410:974–9. doi: 10.1038/35073648. [DOI] [PubMed] [Google Scholar]
- 3.Douek DC, McFarland RD, Keiser PH, et al. Changes in thymic function with age and during the treatment of HIV infection. Nature. 1998;396:690–5. doi: 10.1038/25374. [DOI] [PubMed] [Google Scholar]
- 4.Grossman Z, Meier-Schellersheim M, Sousa AE, Victorino RM, Paul WE. CD4+ T-cell depletion in HIV infection: are we closer to understanding the cause? Nat Med. 2002;8:319–23. doi: 10.1038/nm0402-319. [DOI] [PubMed] [Google Scholar]
- 5.Giorgi JV, Hultin LE, McKeating JA, et al. Shorter survival in advanced human immunodeficiency virus type 1 infection is more closely associated with T lymphocyte activation than with plasma virus burden or virus chemokine coreceptor usage. J Infect Dis. 1999;179:859–70. doi: 10.1086/314660. [DOI] [PubMed] [Google Scholar]
- 6.Rodriguez B, Sethi AK, Cheruvu VK, et al. Predictive value of plasma HIV RNA level on rate of CD4 T-cell decline in untreated HIV infection. JAMA. 2006;296:1498–506. doi: 10.1001/jama.296.12.1498. [DOI] [PubMed] [Google Scholar]
- 7.Brenchley JM, Price DA, Douek DC. HIV disease: fallout from a mucosal catastrophe? Nat Immunol. 2006;7:235–9. doi: 10.1038/ni1316. [DOI] [PubMed] [Google Scholar]
- 8.Brenchley JM, Price DA, Schacker TW, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12:1365–71. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 9.Moore RD, Keruly JC. CD4+ cell count 6 years after commencement of highly active antiretroviral therapy in persons with sustained virologic suppression. Clin Infect Dis. 2007;44:441–6. doi: 10.1086/510746. [DOI] [PubMed] [Google Scholar]
- 10.Kaufmann GR, Perrin L, Pantaleo G, et al. CD4 T-lymphocyte recovery in individuals with advanced HIV-1 infection receiving potent antiretroviral therapy for 4 years: the Swiss HIV Cohort Study. Arch Intern Med. 2003;163:2187–95. doi: 10.1001/archinte.163.18.2187. [DOI] [PubMed] [Google Scholar]
- 11.Baker JV, Peng G, Rapkin J, et al. CD4+ count and risk of non-AIDS diseases following initial treatment for HIV infection. AIDS. 2008;22:841–8. doi: 10.1097/QAD.0b013e3282f7cb76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hunt PW, Martin JN, Sinclair E, et al. T cell activation is associated with lower CD4+ T cell gains in human immunodeficiency virus–infected patients with sustained viral suppression during antiretroviral therapy. J Infect Dis. 2003;187:1534–43. doi: 10.1086/374786. [DOI] [PubMed] [Google Scholar]
- 13.Napolitano LA, Schmidt D, Gotway MB, et al. Growth hormone enhances thymic function in HIV-1-infected adults. J Clin Invest. 2008;118:1085–98. doi: 10.1172/JCI32830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sieg SF, Mitchem JB, Bazdar DA, Lederman MM. Close link between CD4+ and CD8+ T cell proliferation defects in patients with human immunodeficiency virus disease and relationship to extended periods of CD4+ lymphopenia. J Infect Dis. 2002;185:1401–16. doi: 10.1086/340509. [DOI] [PubMed] [Google Scholar]
- 15.Teixeira L, Valdez H, McCune JM, et al. Poor CD4 T cell restoration after suppression of HIV-1 replication may reflect lower thymic function. AIDS. 2001;15:1749–56. doi: 10.1097/00002030-200109280-00002. [DOI] [PubMed] [Google Scholar]
- 16.Brinig MM, Lepp PW, Ouverney CC, Armitage GC, Relman DA. Prevalence of bacteria of division TM7 in human subgingival plaque and their association with disease. Appl Environ Microbiol. 2003;69:1687–94. doi: 10.1128/AEM.69.3.1687-1694.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.AIDS Clinical Trials Group [29 January 2009]; Available at: http://www.actgnetwork.org/login.aspx?ReturnUrl=2%findex.aspx.
- 18.Hunt PW, Brenchley J, Sinclair E, et al. Relationship between T cell activation and CD4+ T cell count in HIV-seropositive individuals with undetectable plasma HIV RNA levels in the absence of therapy. J Infect Dis. 2008;197:126–33. doi: 10.1086/524143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lederman MM, Connick E, Landay A, et al. Immunologic responses associated with 12 weeks of combination antiretroviral therapy consisting of zidovudine, lamivudine, and ritonavir: results of AIDS Clinical Trials Group Protocol 315. J Infect Dis. 1998;178:70–9. doi: 10.1086/515591. [DOI] [PubMed] [Google Scholar]
- 20.Pakker NG, Notermans DW, de Boer RJ, et al. Biphasic kinetics of peripheral blood T cells after triple combination therapy in HIV-1 infection: a composite of redistribution and proliferation. Nat Med. 1998;4:208–14. doi: 10.1038/nm0298-208. [DOI] [PubMed] [Google Scholar]
- 21.Douek DC, Brenchley JM, Betts MR, et al. HIV preferentially infects HIV-specific CD4+ T cells. Nature. 2002;417:95–8. doi: 10.1038/417095a. [DOI] [PubMed] [Google Scholar]
- 22.Hazenberg MD, Hamann D, Schuitemaker H, Miedema F. T cell depletion in HIV-1 infection: how CD4+ T cells go out of stock. Nat Immunol. 2000;1:285–9. doi: 10.1038/79724. [DOI] [PubMed] [Google Scholar]
- 23.Gordon SN, Klatt NR, Bosinger SE, et al. Severe depletion of mucosal CD4+ T cells in AIDS-free simian immunodeficiency virus-infected sooty mangabeys. J Immunol. 2007;179:3026–34. doi: 10.4049/jimmunol.179.5.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brenchley JM, Karandikar NJ, Betts MR, et al. Expression of CD57 defines replicative senescence and antigen-induced apoptotic death of CD8+ T cells. Blood. 2003;101:2711–20. doi: 10.1182/blood-2002-07-2103. [DOI] [PubMed] [Google Scholar]
- 25.Anselmi A, Vendrame D, Rampon O, Giaquinto C, Zanchetta M, De Rossi A. Immune reconstitution in human immunodeficiency virus type 1-infected children with different virological responses to anti-retroviral therapy. Clin Exp Immunol. 2007;150:442–50. doi: 10.1111/j.1365-2249.2007.03526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Funderburg N, Luciano AA, Jiang W, Rodriguez B, Sieg SF, Lederman MM. Toll-like receptor ligands induce human T cell activation and death, a model for HIV pathogenesis. PLoS ONE. 2008;3:e1915. doi: 10.1371/journal.pone.0001915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sieg SF, Bazdar DA, Lederman MM. S-phase entry leads to cell death in circulating T cells from HIV-infected persons. J Leukoc Biol. 2008;83:1382–7. doi: 10.1189/jlb.0907643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sieg SF, Rodriguez B, Asaad R, Jiang W, Bazdar DA, Lederman MM. Peripheral S-phase T cells in HIV disease have a central memory pheno-type and rarely have evidence of recent T cell receptor engagement. J Infect Dis. 2005;192:62–70. doi: 10.1086/430620. [DOI] [PubMed] [Google Scholar]
- 29.Krabbe KS, Bruunsgaard H, Qvist J, et al. Activated T lymphocytes disappear from circulation during endotoxemia in humans. Clin Diagn Lab Immunol. 2002;9:731–5. doi: 10.1128/CDLI.9.3.731-735.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ledergerber B, Lundgren JD, Walker AS, et al. Predictors of trend in CD4-positive T-cell count and mortality among HIV-1-infected individuals with virological failure to all three antiretroviral-drug classes. Lancet. 2004;364:51–62. doi: 10.1016/S0140-6736(04)16589-6. [DOI] [PubMed] [Google Scholar]
- 31.Deeks SG, Barbour JD, Martin JN, Swanson MS, Grant RM. Sustained CD4+ T cell response after virologic failure of protease inhibitor-based regimens in patients with human immunodeficiency virus infection. J Infect Dis. 2000;181:946–53. doi: 10.1086/315334. [DOI] [PubMed] [Google Scholar]
- 32.Hunt PW, Deeks SG, Bangsberg DR, et al. The independent effect of drug resistance on T cell activation in HIV infection. AIDS. 2006;20:691–9. doi: 10.1097/01.aids.0000216369.30948.18. [DOI] [PubMed] [Google Scholar]
- 33.Caradonna L, Amati L, Magrone T, Pellegrino NM, Jirillo E, Caccavo D. Enteric bacteria, lipopolysaccharides and related cytokines in inflammatory bowel disease: biological and clinical significance. J Endotoxin Res. 2000;6:205–14. [PubMed] [Google Scholar]
- 34.Aoki K. A study of endotoxemia in ulcerative colitis and Crohn's disease. I. Clinical study. Acta Med Okayama. 1978;32:147–58. [PubMed] [Google Scholar]