

Abstract
β-Amyloid precursor protein cleavage enzyme 1 (BACE1) has been identified as a major neuronal β-secretase critical for the formation of β-amyloid (Aβ) peptide, which is thought responsible for the pathology of Alzheimer's disease (AD). Therefore, BACE1 is one of the key therapeutic targets that can prevent the progression of AD. Previous studies showed that knocking out the BACE1 gene prevents Aβ formation, but results in behavioral deficits and specific synaptic dysfunctions at Schaffer collateral to CA1 synapses. However, BACE1 protein is most highly expressed at the mossy fiber projections in CA3. Here, we report that BACE1 knock-out mice display reduced presynaptic function, as measured by an increase in paired-pulse facilitation ratio. More dramatically, mossy fiber long-term potentiation (LTP), which is normally expressed via an increase in presynaptic release, was eliminated in the knock-outs. Although long-term depression was slightly larger in the BACE1 knock-outs, it could not be reversed. The specific deficit in mossy fiber LTP was upstream of cAMP signaling and could be “rescued” by transiently elevating extracellular Ca2+ concentration. These results suggest that BACE1 may play a critical role in regulating presynaptic function, especially activity-dependent strengthening of presynaptic release, at mossy fiber synapses.
Keywords: long-term potentiation, long-term depression, presynaptic, paired-pulse facilitation, beta-secretase, Alzheimer's disease
Introduction
Alzheimer's disease (AD) is the most prevalent form of senile dementia. Current treatment of AD remains limited, and there is no effective disease-modifying treatment as of yet (Citron, 2004b). It is widely believed that AD is initiated as a synaptic dysfunction which correlates with the loss of memory function in the early stages of the disease (Selkoe, 2002). A current hypothesis states that overproduction of β-amyloid (Aβ) peptide initiates the pathogenesis of AD (Hardy and Selkoe, 2002; Citron, 2004b; Walsh and Selkoe, 2007). Aβ is produced by the sequential cleavage of amyloid precursor proteins (APPs) by β- and γ-secretases which are one of the major disease-modifying targets to treat AD (Citron, 2004b). However, it became apparent that γ-secretase processes other critical substrates essential for normal cell development and function, such as Notch (Sisodia and St George-Hyslop, 2002; Selkoe and Kopan, 2003). Therefore, inhibiting β-secretase is now receiving renewed attention (Vassar, 2002; Citron, 2004a,b). The amount and activity of β-secretase is elevated in sporadic AD brains (Yang et al., 2003; Li et al., 2004; Zhao et al., 2007), further suggesting that effective methods to reduce its activity may be beneficial to a large population of AD patients.
A transmembrane aspartic protease, β-site APP cleavage enzyme 1 (BACE1), was identified as the major neuronal β-secretase (Hussain et al., 1999; Sinha et al., 1999; Vassar et al., 1999; Yan et al., 1999). BACE1 knock-out (KO) mice were generated to determine the functional consequences of chronically inhibiting the activity of β-secretase. Initial characterization of the BACE1 knock-outs suggested that there are no gross anatomical or functional abnormalities (Luo et al., 2001, 2003). Moreover, knocking out BACE1 in APP transgenic lines, which normally develop Aβ plaques and behavioral deficits, essentially alleviated the AD symptoms (Luo et al., 2003; Ohno et al., 2004; Laird et al., 2005). However, recent studies, including our own, showed that BACE1 knock-outs display specific dysfunctions in synaptic transmission and plasticity (Ohno et al., 2004; Laird et al., 2005), as well as behavioral deficits (Harrison et al., 2003; Laird et al., 2005; Savonenko et al., 2008). Although all of the studies characterizing synaptic function of BACE1 knock-outs thus far have been performed in the CA1 region of the hippocampus (Ohno et al., 2004; Laird et al., 2005; Ma et al., 2007), the expression of BACE1 is most prominent in the mossy fiber terminals that synapse onto CA3 pyramidal neurons (Laird et al., 2005; Zhao et al., 2007). Therefore, we examined synaptic function and plasticity of the BACE1 knock-outs at the mossy fiber synapses.
Materials and Methods
Animals.
All mice used (BACE1 +/+ and −/−) were derived from heterozygous breeders (+/−) as described previously (Laird et al., 2005). The Institutional Animal Care and Use Committees of both University of Maryland, College Park and Johns Hopkins University approved all procedures involving animals.
Electrophysiological recordings.
Hippocampal slices (400 μm thick) were prepared from adult (3–6 months old) male BACE1 knock-out and wild-type (WT) mice as described previously (Laird et al., 2005). Briefly, hippocampi were sliced in ice-cold dissection buffer (in mm: 212.7 sucrose, 2.6 KCl, 1.23 NaH2PO4, 26 NaHCO3, 10 dextrose, MgCl2, and 1 CaCl2, saturated with 5% CO2 and 95% O2). Recordings were done in a submersion-type recording chamber perfused with artificial CSF (ACSF) (in mm: 124 NaCl, 5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 10 dextrose, 1.5 MgCl2, and 2.5 CaCl2, saturated with 5% CO2 and 95% O2, 29.5–30.5°C, 2 ml/min). Synaptic responses were evoked through bipolar stimulating electrodes (double-barreled borosilicate glass capillaries; Sutter Instrument), placed in the dentate granule cell layer to activate the mossy fibers with pulse durations of 0.2 ms (baseline stimulation, 0.067 Hz), and recorded extracellularly in the stratum lucidum of CA3. Both the stimulating and recording electrodes were filled with ACSF. To induce long-term potentiation (LTP), three trains of 100 Hz (1 s) stimuli were given at 20 s intervals. Long-term depression (LTD) was induced by a paired-pulse 1 Hz protocol [interstimulus interval (ISI), 50 ms; 15 min]. For measurement of paired-pulse facilitation (PPF), ISIs of 25, 50, 100, 200, 400, 1000, and 2000 ms were used. In some experiments, extracellular Ca2+ concentration was increased to 5.0 mm for 10 min before delivering high-frequency stimulation (HFS) (Castillo et al., 2002). To activate cAMP production, 50 μm forskolin (Sigma-Aldrich) was applied for 5 min. All experiments were done in the presence of 100 μm dl-APV (dl-2-amino-5-phosphonovaleric acid) (Sigma-Aldrich) to block NMDA receptors. At the end of each experiment, 1 μm (2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine (DCG-IV) (Tocris Bioscience) was added, and blockade ≥80% were taken to be mossy fiber inputs. Field potential (FP) slopes were measured, and data are expressed as mean ± SE of mean.
Results
Reduction in presynaptic function at mossy fiber synapses in BACE1 knock-outs
We observed previously that mossy fiber terminals are enriched in BACE1 protein compared with other hippocampal subfields (Laird et al., 2005). Therefore, we hypothesized that BACE1 knock-outs may exhibit alterations in synaptic transmission and plasticity at this particular set of synapses. We first measured presynaptic function by comparing PPF ratio at various ISIs. We found a significant interaction between the genotype and ISIs (two-factor ANOVA, genotype × ISI: F(6, 203) = 2.586, p < 0.02), particularly BACE1 KOs, displayed larger PPF ratios at shorter ISIs (25 ms ISI: WT, 3.4 ± 0.57; KO, 6.1 ± 0.79; 50 ms ISI: WT, 3.6 ± 0.65, n = 14; KO, 5.7 ± 0.77, n = 17; Fisher's PLSD post hoc test, p < 0.002 for 25 and 50 ms ISI between WT and KO) (Fig. 1A). The increase in PPF ratio suggests a reduction in presynaptic release. Synaptic transmission at mossy fiber to CA3 synapses display sensitivity to group II metabotropic glutamate receptor (mGluR) agonists (Nicoll and Schmitz, 2005). Bath application of 1 μm DCG-IV at the end of the recording produced a comparable reduction in basal synaptic transmission in both knock-outs and wild types (WT, 12 ± 4% of baseline at 20 min DCG-IV, n = 14; KO, 11 ± 2%, n = 17) (Fig. 1B).
Figure 1.
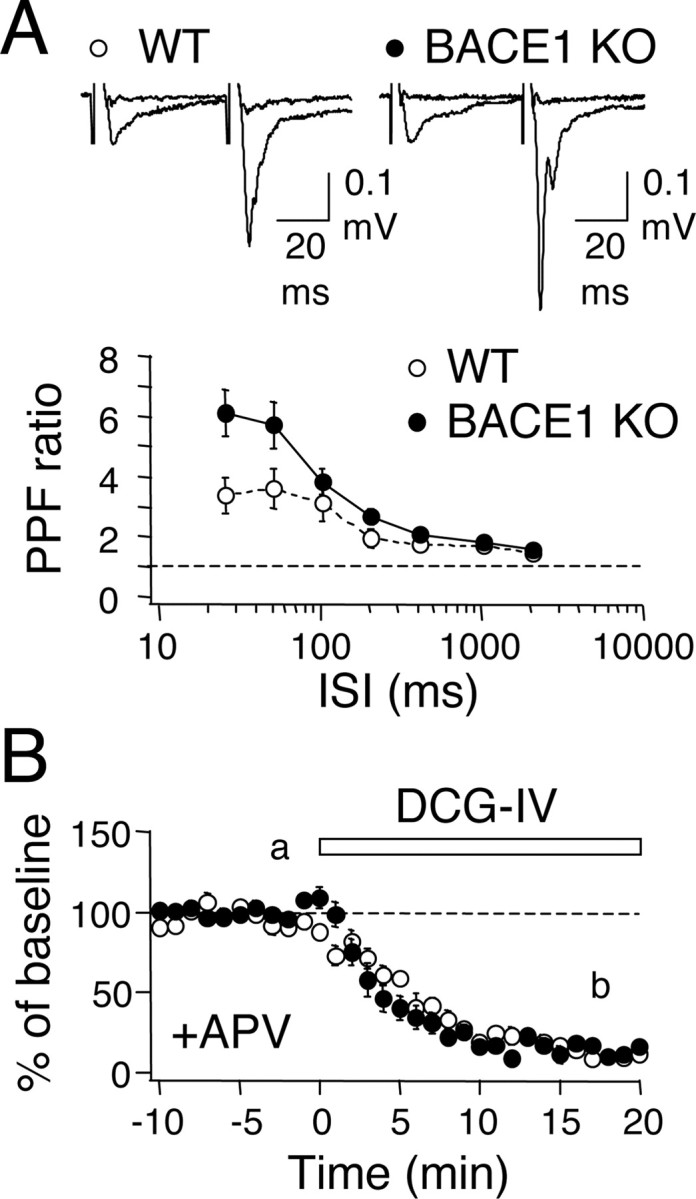
BACE1 knock-outs display a reduction in presynaptic function at the mossy fiber synapses. A, Larger PPF ratio in BACE1 knock-outs. The difference between WT (white circles) and KOs (black circles) are significant at 25 and 50 ms ISIs. Top, Representative FP traces of paired-pulse stimulation at 50 ms ISI before and after DCG-IV application in wild type and knockout. The traces were taken from time points indicated (a and b) in the graph in B. B, Application of group II mGluR agonist (1 μm DCG-IV) reduces mossy fiber synaptic transmission in WT (white circles) and KOs (black circles).
Activity-dependent synaptic strengthening at mossy fiber synapses is abolished in BACE1 knock-outs
Next, we examined whether knocking out BACE1 affects synaptic plasticity at the mossy fiber synapses. We first compared LTP induced by HFS (3 × 100 Hz, 1 s). BACE1 knock-outs showed a larger initial potentiation, suggesting an enhanced facilitation during HFS; however, the responses relaxed back to baseline by 1 h (WT: 149 ± 10% of baseline at 1 h after HFS, n = 13 slices from 6 mice; KO: 96 ± 7%, n = 16 slices from 7 mice; t test, p < 0.01) (Fig. 2A). Consistent with a presynaptic locus of expression, LTP in wild types was accompanied by a decrease in PPF ratio measured at 50 ms ISI (baseline, 3.1 ± 0.5; 1 h after HFS, 2.6 ± 0.4; n = 13 slices from 6 mice; paired t test, p < 0.03), but knock-outs displayed a trend of a decrease in PPF ratio that returned to basal levels at 1 h (baseline, 5.9 ± 1.0; 20 min after HFS, 3.6 ± 0.5; 1 h after HFS, 6.1 ± 1.2; n = 16 slices from 7 mice; ANOVA, F(2,45) = 2.018; p = 0.1).
Figure 2.
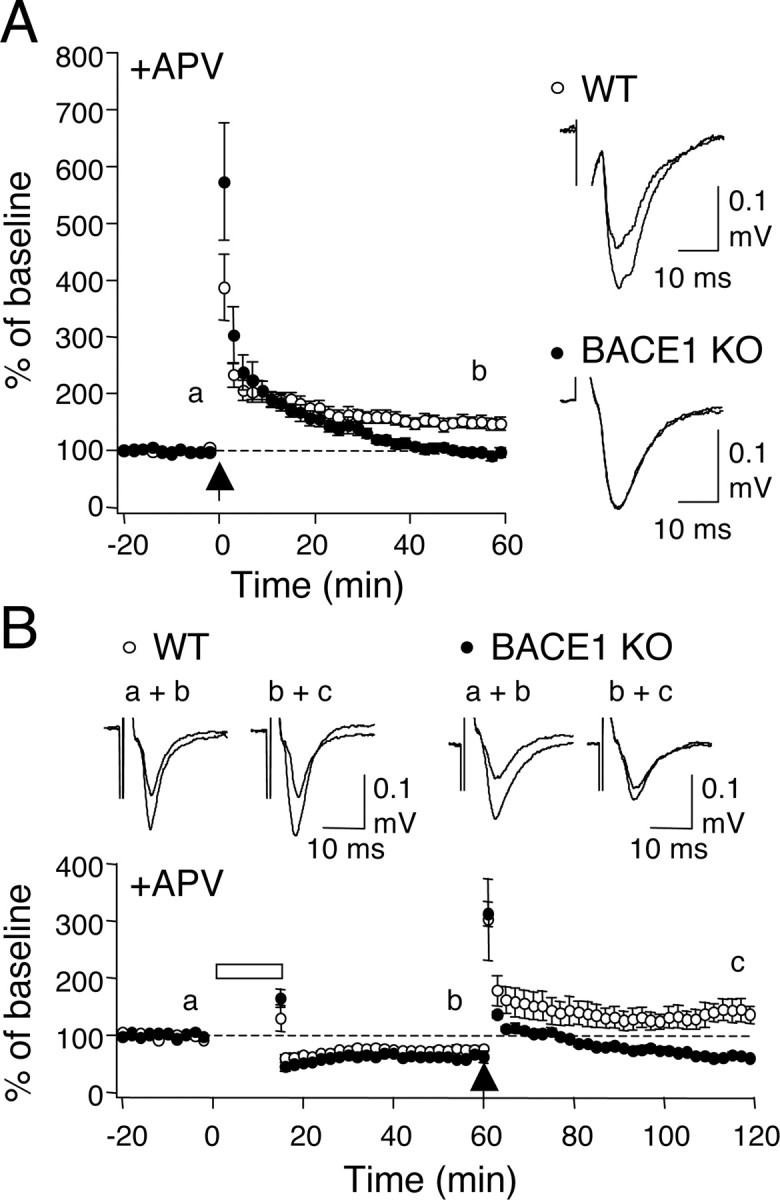
Absence of activity-dependent potentiation at mossy fiber synapses in BACE1 knock-outs. A, Mossy fiber LTP is absent in BACE1 knock-outs. Left, Summary graph plotting changes in normalized field potential against time. The arrow depicts when HFS (100 Hz, 1 s × 3) was delivered. Note that knock-outs (black circles) showed larger posttetanic potentiation but no LTP when compared with wild-types (white circles). Right, Suafterimposed representative FP traces taken from wild type and knock-outs at times indicated in the left panel (a, b). B, BACE1 knock-outs show a slightly larger LTD but no dedepression. Bottom, Summary graph of the averages. The bar and arrow indicate delivery of PP-1 Hz (15 min) and HFS (100 Hz, 1 s × 3), respectively. Top, Representative FP traces taken at times indicated in the summary graph (a–c).
To test LTD, we used a paired-pulse 1 Hz protocol [PP-1 Hz (15 min)], because a standard 1 Hz (15 min) protocol (Kobayashi et al., 1996) failed to produce LTD in the wild types at the ages used for our study (data not shown). LTD induced by the PP-1 Hz was slightly, but significantly, larger in BACE1 knock-outs (WT: 75 ± 4.3% of baseline at 1 h after onset of PP-1 Hz, n = 7 slices from 5 mice; KO: 62 ± 3.8% of baseline, n = 6 slices from 4 mice; t test, p < 0.04) (Fig. 2B). This form of LTD did not significantly change PPF ratio either in wild types or knock-outs (WT: baseline, 3.8 ± 1.0; 1 h after PP 1 Hz, 3.2 ± 0.9, n = 6 slices from 4 mice; paired t test, p = 0.16; KO: baseline, 7.1 ± 1.5; 1 h after PP 1 Hz, 5.5 ± 1.1, n = 6 slices from 4 mice; paired t test, p = 0.10). Unlike in wild types, HFS failed to reverse LTD in the knock-outs (WT: 195 ± 28.0% of renormalized baseline at 1 h after HFS, n = 6 slices from 4 mice; KO: 100 ± 5.4%, n = 6 slices from 4 mice; t test, p < 0.02) (Fig. 2B).
Rescue of mossy fiber LTP in BACE1 knock-outs by increasing extracellular Ca2+ concentration
Mossy fiber LTP is triggered by a rise in presynaptic Ca2+ (Castillo et al., 1994) and a further recruitment of cAMP-dependent signaling mechanisms (Nicoll and Schmitz, 2005). Therefore, we investigated whether the lack of mossy fiber LTP in BACE1 knock-outs is attributable to abnormal regulation of presynaptic Ca2+ or signaling downstream. We found that transiently increasing the concentration of extracellular Ca2+ (from 2.5 to 5 mm) during HFS recovered mossy fiber LTP in BACE1 knock-outs (137 ± 7.9% of baseline at 1 h after HFS, n = 9 slices from 6 mice; paired t test, p < 0.01) (Fig. 3A). Furthermore, LTP was accompanied by a decrease in PPF ratio measured at 50 ms ISI (baseline, 4.2 ± 0.7; 1 h after HFS, 2.4 ± 0.4; n = 9 slices from 6 mice; paired t test, p < 0.01) consistent with a presynaptic expression. Increasing external Ca2+ concentration alone produced only a transient potentiation (110 ± 4.8% of baseline at 1 h after Ca2+; n = 4 slices from 2 mice; paired t test, p = 0.13) (Fig. 3A).
Figure 3.

BACE1 knock-outs express mossy fiber LTP under high extracellular Ca2+ and produce normal forskolin-induced potentiation. A, Transient elevation of external Ca2+ concentration (5 mm Ca2+, 10 min; gray bar) rescued mossy fiber LTP in knock-outs (black circles). Increasing Ca2+ alone produced only a transient potentiation (open circles). The arrow depicts when HFS (100 Hz, 1 s × 3) was delivered. Right, Representative traces taken at times indicated (left graph, a and b). B, Transient application of forskolin (50 μm, 5 min; gray bar) potentiated mossy fiber synaptic transmission in wild types (open circles) and knock-outs (black circles) to the same magnitude. Top, Representative traces taken at times indicated (bottom panel, a and b).
To further confirm whether signaling downstream of the Ca2+ signal is intact in BACE1 knock-outs, we directly activated cAMP signaling by a brief application of an adenylyl cyclase activator forskolin. This caused a dramatic enhancement of synaptic transmission in both wild types and knock-outs to similar magnitudes (WT: 622.5 ± 57.8% of baseline at 1 h after forskolin, n = 7 slices from 5 mice; KO: 741.8 ± 110.1%, n = 7 slices from 4 mice; t test, p = 0.36) (Fig. 3B). This was accompanied by a significant decrease in PPF ratio in both genotypes (WT: baseline, 3.2 ± 0.34; 1 h after forskolin, 1.5 ± 0.15, n = 7 slices from 5 mice; paired t test, p < 0.01; KO: baseline, 4.9 ± 0.63; 1 h after forskolin, 1.7 ± 0.16, n = 7 slices from 4 mice; paired t test, p < 0.01), consistent with a presynaptic mechanism of potentiation. This demonstrates that the presynaptic deficits seen in BACE1 knock-outs are upstream of cAMP signaling.
Discussion
We found that BACE1 knock-outs display severe deficits in presynaptic function at mossy fiber synapses in CA3: a reduction in presynaptic release and an absence of mossy fiber LTP. In addition, BACE1 knock-outs exhibited a slightly larger LTD which could not be reversed. These results suggest that BACE1 function is critical for normal synaptic transmission and plasticity, especially activity-dependent potentiation, at these synapses. We further found that the specific deficit in mossy fiber LTP in BACE1 knock-outs can be rescued by increasing extracellular Ca2+ concentration. Because a direct activation of cAMP production was not impaired in the BACE1 knock-outs, our data suggest that the presynaptic dysfunction is likely at the level of presynaptic Ca2+ regulation.
Previous studies suggest that BACE1 is highly localized to presynaptic terminals, especially at the mossy fiber boutons in the CA3 (Laird et al., 2005; Zhao et al., 2007). This localization is consistent with our observation of a deficit in presynaptic function and plasticity at this synapse. Together with our previous results from the CA1 also showing an increase in PPF ratio (Laird et al., 2005), these results indicate that BACE1 may play a general role in regulating presynaptic function under physiological conditions. However, whether presynaptic deficits in BACE1 knock-outs are directly attributable to lacking APP processing is unclear. Previous studies suggest that generation of excess Aβ depresses excitatory synaptic transmission, mainly by postsynaptic removal of AMPA receptors and loss of synapses (Hsieh et al., 2006; Priller et al., 2006; Ting et al., 2007). These results would predict that lacking Aβ production, as in BACE1 knock-outs, would cause a postsynaptic increase in AMPA receptor function, not a decrease in presynaptic function as observed in our studies. However, we cannot rule out the possibility of gain-of-function in the Aβ overexpression studies.
Another possibility is that the presynaptic effects of BACE1 knock-out may be from abnormal processing of substrates other than APP. It is now known that BACE1 can also cleave APP-like proteins (Li and Südhof, 2004), β subunits of voltage-gated Na+ channel (Wong et al., 2005; Kim et al., 2007), and neuregulin-1 (NRG1) (Hu et al., 2006; Willem et al., 2006). Regulation of the latter two substrates is particularly interesting. The β2 subunit of Na+ channel is critical for plasma membrane expression of functional Na+ channels (Schmidt and Catterall, 1986), which are essential for action potential generation. However, overexpressing BACE1 actually decreases the density of functional Na+ channels (Kim et al., 2007); hence, it cannot directly account for the observed reduction in presynaptic release in the BACE1 knock-outs. Potential regulation of NRG1 by BACE1 was discovered from observations that BACE1 knock-outs display a hypomyelination phenotype with a correlated accumulation of full-length NRG1 and a significant loss of NRG1 cleavage products (Hu et al., 2006; Willem et al., 2006). Recently, we demonstrated that the lack of NRG1 processing in BACE1 knock-outs reduces postsynaptic function of ErbB4, a receptor for NRG1 (Savonenko et al., 2008). NRG1/ErbB4 signaling has been suggested to regulate synaptic function and plasticity, mainly via regulation of postsynaptic glutamate receptors (Huang et al., 2000; Gu et al., 2005; Li et al., 2007). Nevertheless, abnormal processing of NRG1 may also affect presynaptic release by regulating the expression of nicotinic acetylcholine receptor (nAchR) subunit α7 (Liu et al., 2001), which allows Ca2+ influx (Séguéla et al., 1993). Indeed, presynaptic nAchRs can increase glutamate release (McGehee et al., 1995; Gray et al., 1996; Maggi et al., 2003), likely via the α7 containing nAchRs (Le Magueresse et al., 2006). These results suggest that lacking NRG1 cleavage, as in BACE1 knock-outs, would reduce presynaptic release. Whether this is the case for mossy fiber synapses is unclear (Vogt and Regehr, 2001).
Our results indicate that a complete inhibition of BACE1 activity is deleterious for neuronal function, especially at the mossy fiber synapses in CA3 compared with Schaffer collateral inputs in CA1. This suggests that mossy fiber dysfunction may have had a larger impact on the behavioral phenotypes seen in the BACE1 knock-outs (Harrison et al., 2003; Laird et al., 2005; Savonenko et al., 2008). We demonstrate that signaling downstream of presynaptic Ca2+ is intact in BACE1 knock-outs. Therefore, we were able to restore mossy fiber LTP in the BACE1 knock-outs by simply increasing extracellular Ca2+ concentration during LTP induction. This has significant clinical implications because it suggests that means to enhance presynaptic Ca2+ will circumvent synaptic deficits, and afterhaps alleviate the behavioral phenotypes, associated with inhibiting BACE1 activity.
Footnotes
This work was supported by National Institutes of Health Grant P01-NS047308.
References
- Castillo PE, Weisskopf MG, Nicoll RA. The role of Ca2+ channels in hippocampal mossy fiber synaptic transmission and long-term potentiation. Neuron. 1994;12:261–269. doi: 10.1016/0896-6273(94)90269-0. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Schoch S, Schmitz F, Südhof TC, Malenka RC. RIM1alpha is required for presynaptic long-term potentiation. Nature. 2002;415:327–330. doi: 10.1038/415327a. [DOI] [PubMed] [Google Scholar]
- Citron M. Beta-secretase inhibition for the treatment of Alzheimer's disease—promise and challenge. Trends Pharmacol Sci. 2004a;25:92–97. doi: 10.1016/j.tips.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Citron M. Strategies for disease modification in Alzheimer's disease. Nat Rev Neurosci. 2004b;5:677–685. doi: 10.1038/nrn1495. [DOI] [PubMed] [Google Scholar]
- Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA. Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature. 1996;383:713–716. doi: 10.1038/383713a0. [DOI] [PubMed] [Google Scholar]
- Gu Z, Jiang Q, Fu AK, Ip NY, Yan Z. Regulation of NMDA receptors by neuregulin signaling in prefrontal cortex. J Neurosci. 2005;25:4974–4984. doi: 10.1523/JNEUROSCI.1086-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Harrison SM, Harafter AJ, Hawkins J, Duddy G, Grau E, Pugh PL, Winter PH, Shilliam CS, Hughes ZA, Dawson LA, Gonzalez MI, Upton N, Pangalos MN, Dingwall C. BACE1 (beta-secretase) transgenic and knockout mice: identification of neurochemical deficits and behavioral changes. Mol Cell Neurosci. 2003;24:646–655. doi: 10.1016/s1044-7431(03)00227-6. [DOI] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Hicks CW, He W, Wong P, Macklin WB, Trapp BD, Yan R. Bace1 modulates myelination in the central and afteripheral nervous system. Nat Neurosci. 2006;9:1520–1525. doi: 10.1038/nn1797. [DOI] [PubMed] [Google Scholar]
- Huang YZ, Won S, Ali DW, Wang Q, Tanowitz M, Du QS, Pelkey KA, Yang DJ, Xiong WC, Salter MW, Mei L. Regulation of neuregulin signaling by PSD-95 interacting with ErbB4 at CNS synapses. Neuron. 2000;26:443–455. doi: 10.1016/s0896-6273(00)81176-9. [DOI] [PubMed] [Google Scholar]
- Hussain I, Powell D, Howlett DR, Tew DG, Meek TD, Chapman C, Gloger IS, Murphy KE, Southan CD, Ryan DM, Smith TS, Simmons DL, Walsh FS, Dingwall C, Christie G. Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol Cell Neurosci. 1999;14:419–427. doi: 10.1006/mcne.1999.0811. [DOI] [PubMed] [Google Scholar]
- Kim DY, Carey BW, Wang H, Ingano LA, Binshtok AM, Wertz MH, Pettingell WH, He P, Lee VM, Woolf CJ, Kovacs DM. BACE1 regulates voltage-gated sodium channels and neuronal activity. Nat Cell Biol. 2007;9:755–764. doi: 10.1038/ncb1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Manabe T, Takahashi T. Presynaptic long-term depression at the hippocampal mossy fiber-CA3 synapse. Science. 1996;273:648–650. doi: 10.1126/science.273.5275.648. [DOI] [PubMed] [Google Scholar]
- Laird FM, Cai H, Savonenko AV, Farah MH, He K, Melnikova T, Wen H, Chiang HC, Xu G, Koliatsos VE, Borchelt DR, Price DL, Lee HK, Wong PC. BACE1, a major determinant of selective vulnerability of the brain to amyloid-beta amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J Neurosci. 2005;25:11693–11709. doi: 10.1523/JNEUROSCI.2766-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Magueresse C, Safiulina V, Changeux JP, Cherubini E. Nicotinic modulation of network and synaptic transmission in the immature hippocampus investigated with genetically modified mice. J Physiol. 2006;576:533–546. doi: 10.1113/jphysiol.2006.117572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Woo RS, Mei L, Malinow R. The neuregulin-1 receptor erbB4 controls glutamatergic synapse maturation and plasticity. Neuron. 2007;54:583–597. doi: 10.1016/j.neuron.2007.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Südhof TC. Cleavage of amyloid-beta precursor protein and amyloid-beta precursor-like protein by BACE 1. J Biol Chem. 2004;279:10542–10550. doi: 10.1074/jbc.M310001200. [DOI] [PubMed] [Google Scholar]
- Li R, Lindholm K, Yang LB, Yue X, Citron M, Yan R, Beach T, Sue L, Sabbagh M, Cai H, Wong P, Price D, Shen Y. Amyloid beta peptide load is correlated with increased beta-secretase activity in sporadic Alzheimer's disease patients. Proc Natl Acad Sci U S A. 2004;101:3632–3637. doi: 10.1073/pnas.0205689101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Ford B, Mann MA, Fischbach GD. Neuregulins increase alpha7 nicotinic acetylcholine receptors and enhance excitatory synaptic transmission in GABAergic interneurons of the hippocampus. J Neurosci. 2001;21:5660–5669. doi: 10.1523/JNEUROSCI.21-15-05660.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Bolon B, Kahn S, Bennett BD, Babu-Khan S, Denis P, Fan W, Kha H, Zhang J, Gong Y, Martin L, Louis JC, Yan Q, Richards WG, Citron M, Vassar R. Mice deficient in BACE1, the Alzheimer's beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat Neurosci. 2001;4:231–232. doi: 10.1038/85059. [DOI] [PubMed] [Google Scholar]
- Luo Y, Bolon B, Damore MA, Fitzpatrick D, Liu H, Zhang J, Yan Q, Vassar R, Citron M. BACE1 (beta-secretase) knockout mice do not acquire compensatory gene expression changes or develop neural lesions over time. Neurobiol Dis. 2003;14:81–88. doi: 10.1016/s0969-9961(03)00104-9. [DOI] [PubMed] [Google Scholar]
- Ma H, Lesné S, Kotilinek L, Steidl-Nichols JV, Sherman M, Younkin L, Younkin S, Forster C, Sergeant N, Delacourte A, Vassar R, Citron M, Kofuji P, Boland LM, Ashe KH. Involvement of beta-site APP cleaving enzyme 1 (BACE1) in amyloid precursor protein-mediated enhancement of memory and activity-dependent synaptic plasticity. Proc Natl Acad Sci U S A. 2007;104:8167–8172. doi: 10.1073/pnas.0609521104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggi L, Le Magueresse C, Changeux JP, Cherubini E. Nicotine activates immature “silent” connections in the developing hippocampus. Proc Natl Acad Sci U S A. 2003;100:2059–2064. doi: 10.1073/pnas.0437947100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGehee DS, Heath MJ, Gelber S, Devay P, Role LW. Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science. 1995;269:1692–1696. doi: 10.1126/science.7569895. [DOI] [PubMed] [Google Scholar]
- Nicoll RA, Schmitz D. Synaptic plasticity at hippocampal mossy fibre synapses. Nat Rev Neurosci. 2005;6:863–876. doi: 10.1038/nrn1786. [DOI] [PubMed] [Google Scholar]
- Ohno M, Sametsky EA, Younkin LH, Oakley H, Younkin SG, Citron M, Vassar R, Disterhoft JF. BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer's disease. Neuron. 2004;41:27–33. doi: 10.1016/s0896-6273(03)00810-9. [DOI] [PubMed] [Google Scholar]
- Priller C, Bauer T, Mitteregger G, Krebs B, Kretzschmar HA, Herms J. Synapse formation and function is modulated by the amyloid precursor protein. J Neurosci. 2006;26:7212–7221. doi: 10.1523/JNEUROSCI.1450-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savonenko AV, Melnikova T, Laird FM, Stewart KA, Price DL, Wong PC. Alteration of BACE1-dependent NRG1/ErbB4 signaling and schizophrenia-like phenotypes in BACE1-null mice. Proc Natl Acad Sci U S A. 2008;105:5585–5590. doi: 10.1073/pnas.0710373105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt JW, Catterall WA. Biosynthesis and processing of the alpha subunit of the voltage-sensitive sodium channel in rat brain neurons. Cell. 1986;46:437–444. doi: 10.1016/0092-8674(86)90664-1. [DOI] [PubMed] [Google Scholar]
- Séguéla P, Wadiche J, Dineley-Miller K, Dani JA, Patrick JW. Molecular cloning, functional proafterties, and distribution of rat brain alpha 7: a nicotinic cation channel highly aftermeable to calcium. J Neurosci. 1993;13:596–604. doi: 10.1523/JNEUROSCI.13-02-00596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe D, Kopan R. Notch and Presenilin: regulated intramembrane proteolysis links development and degeneration. Annu Rev Neurosci. 2003;26:565–597. doi: 10.1146/annurev.neuro.26.041002.131334. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, Doan M, Dovey HF, Frigon N, Hong J, Jacobson-Croak K, Jewett N, Keim P, Knops J, Lieberburg I, Power M, Tan H, Tatsuno G, Tung J, Schenk D, et al. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature. 1999;402:537–540. doi: 10.1038/990114. [DOI] [PubMed] [Google Scholar]
- Sisodia SS, St George-Hyslop PH. gamma-Secretase, Notch, Abeta and Alzheimer's disease: where do the presenilins fit in? Nat Rev Neurosci. 2002;3:281–290. doi: 10.1038/nrn785. [DOI] [PubMed] [Google Scholar]
- Ting JT, Kelley BG, Lambert TJ, Cook DG, Sullivan JM. Amyloid precursor protein overexpression depresses excitatory transmission through both presynaptic and postsynaptic mechanisms. Proc Natl Acad Sci U S A. 2007;104:353–358. doi: 10.1073/pnas.0608807104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R. Beta-secretase (BACE) as a drug target for Alzheimer's disease. Adv Drug Deliv Rev. 2002;54:1589–1602. doi: 10.1016/s0169-409x(02)00157-6. [DOI] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, et al. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- Vogt KE, Regehr WG. Cholinergic modulation of excitatory synaptic transmission in the CA3 area of the hippocampus. J Neurosci. 2001;21:75–83. doi: 10.1523/JNEUROSCI.21-01-00075.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. A beta oligomers - a decade of discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- Willem M, Garratt AN, Novak B, Citron M, Kaufmann S, Rittger A, DeStrooafter B, Saftig P, Birchmeier C, Haass C. Control of afteripheral nerve myelination by the beta-secretase BACE1. Science. 2006;314:664–666. doi: 10.1126/science.1132341. [DOI] [PubMed] [Google Scholar]
- Wong HK, Sakurai T, Oyama F, Kaneko K, Wada K, Miyazaki H, Kurosawa M, De Strooafter B, Saftig P, Nukina N. beta Subunits of voltage-gated sodium channels are novel substrates of beta-site amyloid precursor protein-cleaving enzyme (BACE1) and gamma-secretase. J Biol Chem. 2005;280:23009–23017. doi: 10.1074/jbc.M414648200. [DOI] [PubMed] [Google Scholar]
- Yan R, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, Brashier JR, Stratman NC, Mathews WR, Buhl AE, Carter DB, Tomasselli AG, Parodi LA, Heinrikson RL, Gurney ME. Membrane-anchored aspartyl protease with Alzheimer's disease beta-secretase activity. Nature. 1999;402:533–537. doi: 10.1038/990107. [DOI] [PubMed] [Google Scholar]
- Yang LB, Lindholm K, Yan R, Citron M, Xia W, Yang XL, Beach T, Sue L, Wong P, Price D, Li R, Shen Y. Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med. 2003;9:3–4. doi: 10.1038/nm0103-3. [DOI] [PubMed] [Google Scholar]
- Zhao J, Fu Y, Yasvoina M, Shao P, Hitt B, O'Connor T, Logan S, Maus E, Citron M, Berry R, Binder L, Vassar R. Beta-site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer's disease pathogenesis. J Neurosci. 2007;27:3639–3649. doi: 10.1523/JNEUROSCI.4396-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]