

Abstract
The homoplasmic mitochondrial A1555G mutation in the 12S rRNA gene leads to a mitochondrial translation disorder associated with deafness. The absence of disease in non-cochlear tissues in all patients, and in the cochlea in some patients, is not well understood. We used a system-based approach, including whole genome expression and biological function analysis, to elucidate the pathways underlying tissue specificity and clinical severity of this condition. Levels of over 48K RNA transcripts from EBV-transformed lymphoblasts of deaf and hearing individuals with the A1555G mutation and controls were obtained. Differentially expressed transcripts were functionally grouped using gene set enrichment analysis. Over fifty RNA binding proteins were differentially expressed between deaf and hearing individuals with the A1555G mutation (p-value of 2.56E-7), confirming previous genetic data implicating this pathway in the determination of the severity of hearing loss. Unexpectedly, the majority of cytoplasmic ribosomal genes were up-regulated in a coordinated fashion in individuals with the A1555G mutation versus controls (p-value of 3.91E-135). This finding was verified through real time RT-PCR, and through measuring of protein levels by flow cytometry. Analysis of expression levels of other differentially expressed genes suggests that this coordinated over-expression of cytoplasmic ribosomal proteins might occur through the Myc/Max pathway. We propose that expression levels of RNA binding proteins help determine the severity of the cochlear phenotype, and that coordinated up-regulation of the cytoplasmic translation apparatus operates as a compensation mechanism in unaffected tissues of patients with maternal deafness associated with the A1555G mutation.
Keywords: Deafness, mitochondrial, RNA modification, ribosome, phenotype, systems biology, complex disease
INTRODUCTION
A significant number of phenotypically diverse human diseases are caused by alterations in the mitochondrial translation machinery, including defects of mitochondrial rRNAs and tRNAs and of the nuclear genes involved in mitochondrial translation [1, 2]. The majority of these defects lead to neuromuscular diseases [1, 3–8, Mitomap, http://www.mitomap.org], but almost all tissue types can be affected [9–11]. Since oxidative phosphorylation is essential for nearly all human cells, the tissue specificity and variable severity of the phenotype of mitochondrial translation disorders is not well understood. For heteroplasmic mitochondrial DNA disorders differential mutation load in different tissues seems to play a significant role in determining the phenotype [12]. However, homoplasmic mtDNA and nuclear mutations demonstrate tissue specificity and significant differences in disease severity suggesting tissue-specific differences in the mitochondrial translation machinery as well as modifying and/or compensatory pathways [13, 14]. Recently, dramatically different ratios of mitochondrial translation elongation factors Tu and Ts identified in affected and unaffected tissues of patients with fatal hepatopathy due to mutations in the mitochondrial translation elongation factor G1 suggested such compensatory response [11].
In maternally inherited deafness associated with the homoplasmic A1555G mutation in the 12S rRNA gene, tissue-specificity is combined with phenotypic expression ranging from profound congenital hearing loss to lifelong normal hearing [15]. Using linkage, linkage disequilibrium, and mutation analyses, nuclear genes whose products are involved in mitochondrial RNA modification have been identified as modifiers of the disease severity [16–19]. However, factors involved in the tissue specificity of this disorder are common between deaf and hearing individuals with the A1555G mutation, and thus can not be identified through genetic studies. To identify these factors, we applied a systems biology approach of transcriptional profiling with pathway analysis. We performed expression microarray analysis of EBV (Epstein - Barr virus)-transformed lymphoblasts from deaf and hearing individuals with the A1555G mutation and controls, followed by gene-annotation enrichment analysis to identify pertinent biological processes. We found significant differences in expression of genes coding for RNA binding proteins between deaf and hearing individuals, which appear to contribute to the severity of hearing loss and confirm previous genetic findings [16–18]. We also found that individuals with the mutation over-express the majority of genes coding for cytoplasmic ribosomal proteins when compared with controls, indicating a possible compensation mechanism. This finding was supported by real time RT-PCR and flow cytometry. The Myc/Max pathway was implicated as the potential regulator of this coordinated over-expression of cytoplasmic ribosomal genes.
Materials and methods
Patients, Cell lines, RNA isolation
We analyzed samples of 14 deaf (group‘1555+D’) and 14 hearing maternally-related individuals (group‘1555+’) from a large multi-generational Arab-Israeli kindred [15, 20]. In addition, we analyzed 15 samples with wild-type mtDNA (‘Wild’) from control Arab-Israelis. Each group consisted of approximately equal number of males and females. Lymphoblastoid cell lines have previously been established from peripheral blood lymphocytes of study participants and immortalized with Epstein-Barr virus [20]. Cells were grown in suspension in T flasks in RPMI-1640 medium (Omega Scientific, Inc., Tarzana, CA), containing 2 mmol/L L-glutamine, 100 ug/ml streptomycin, and 10% fetal calf serum. Total RNA was extracted using Trizol reagent (Invitrogen, Inc., Carlsbad, CA) according to the manufacturer’s protocol. The study was approved by the Institutional Review Board and informed consent was obtained from the families.
Expression microarray
cRNA amplification and labeling with biotin were performed using Illlumina® TotalPrep RNA amplification kit, manufactured by Ambion, Inc., Austin, TX using 500 ng of total RNA as input material according to the manufacturer’s protocol. cRNA yields were quantified with NanoDrop® spectrophotometer (Thermo Scientific, Wilmington, DE). 1.5 ug of labeled cRNAs were hybridized to the Sentrix Human-6 v2 Expression Beadchips (Illumina, Inc., San Diego, CA). Each chip contains 6 identical sets of 48,702 unique probes, including 22,000 curated RefSeq genes (release 17), and with additional probes designed by Illumina based on UniGene release 188. cRNA was hybridized to arrays for 19 hours at 58ºC before being washed and stained with streptavidin-Cy3 using reagents and equipment by Illumina, according to the manufacturer’s protocol. Beadchips were scanned on the Illumina BeadArray Reader confocal scanner for 90 minutes according to the manufacturer’s instructions.
Data visualization and quality control was carried out with BeadStudio 3.1.7 software (Illumina). For signal quality control, gene signals were ranked relative to the distribution of signals of the negative controls and detection scores were calculated. Transcripts with detection confidence of 0.99 and above in all the samples were considered “high confidence” and were used for all analyses. Data normalization and differential analysis were carried out with GeneSpring GX release 7.3.1 computer program (Silicon Genetics, Inc., San Jose, CA). Raw microarray data without background subtraction was output from BeadStudio and directly downloaded into Genespring. We applied default data transformation to set measurements below 0.01 to 0.01, and default experiment normalization of chip normalization to 50th percentile and gene normalization to the median.
Differential expression analysis
To compare levels of transcripts between groups of samples three parametric and nonparametric statistical tests were performed using Genespring GX 7.3.1. Two groups: unpaired Student’s t-test with equal variances (parametric 1), Welch t-test with unequal variances (parametric 2), and unpaired Wilcoxon-Mann-Whitney test (nonparametric). Three groups: ANOVA with equal variances (parametric 1), Welch ANOVA with unequal variances (parametric 2), and Kruskal-Wallis test (nonparametric). All tests were performed with P-value cut-off value of 0.05 and no multiple testing corrections. Multiple testing correction methods were not applied because they are too restrictive, assuming independence of each gene's expression, and thus lead to type I errors.
Functional pathways analysis
We used GO (Gene Ontology, http://www.geneontology.org) Browser within the Genespring GX analysis platform to identify relevant biological processes and functions by calculating the enrichment of lists of differentially expressed genes with genes from different GO categories. The hypergeometric P value of the enrichment represents the likelihood of the event that at least as many genes would occur in a list of equal size by chance only and is based on genes that have an annotation in the selected Ontology, rather than those genes that have an annotation in any Ontology. While this approach results in more conservative P-values no opportunity to correct for multiple testing is provided in the Genespring GO browser. In addition, we performed pathway analysis of the differentially expressed genes identified by parametric test 1 for each group comparison. Functional annotation tool available at the web bioinformatics resource DAVID 2007 (Database for Annotation, Visualization, and Integrated Discovery, http://david.abcc.ncifcrf.gov) [19] was used to map lists of differentially expressed genes on human KEGG (Kyoto Encyclopedia of Genes and Genome, Kyoto University and Human Genome Center) pathways. DAVID calculates the so-called EASE Score, a modified Fisher Exact P-value to predict the likelihood of genes mapping to specific biological processes from a given list of differentially expressed genes [21].
Quantitative RT-PCR
Gene-specific TaqMan® Gene Expression Assays specific to the 10 genes coding for cytoplasmic ribosomal proteins: L18 (RPL18), L19 (RPL19), L24 (RPL24), L27 (RPL27), L31 (RPL31), S11 (RPS11), S16 (RPS16), S17 (RPS17), S20 (RPS20), S25 (RPS25) were purchased from Applied Biosystems, Inc., Foster City, CA. We included assay for human glyceraldehyde-3-phosphate dehydrogenase gene (GAPDH) as a control. All 43 samples of individuals with A1555G mutation and controls were tested. All PCR reactions were performed using TaqMan universal PCR master mix (Applied Biosystems) according to the manufacturer’s instructions. The 7900HT Sequence Detection System (Applied Biosystems) was used to collect real-time fluorescent signal to calculate real-time threshold cycle (CT) for each individual RNA sample. To ensure accurate quantification, PCR reactions for all 43 individuals were performed in triplicates.
Flow Cytometry
Human protein specific polyclonal rabbit antibodies against Rpl6, Rpl8, Rpl9, Rpl26, Rps6, Rps14, Rps16; polyclonal goat antibodies against Rpl19, and monoclonal mouse antibodies against Rps2 and Rps19 were purchased from Abcam, Inc., (Cambridge, MA). Lymphoblasts of the forty-three individuals, were grown in suspension in T flasks in RPMI-1640 medium (Omega Scientific, Inc., Tarzana, CA), containing 2 mmol/L L-glutamine, 100 ug/ml streptomycin, and 10% fetal calf serum. 4 milliliters of cells were washed with PBS, fixed on chamber slides with 2% paraformaldehyde, permeabilized with 0.2% triton and incubated with primary antibodies overnight at 40ºC. Fluorescently labeled secondary goat anti-rabbit, donkey anti-goat, or goat anti-mouse Alexa Fluor® 488 antibodies (Invitrogen, Inc.) were used for labeling. Fluorescent signal was acquired using CellQuest software version 3.1 on the FACScan flow cytometer (Becton-Dickinson). Median Fluorescent Intensities (MFI) of signal above fluorescence thresholds of secondary antibody alone and cells alone in gated live cells, selected on the scatter plot, were calculated automatically by the CellQuest software. Nonparametric test for the significance of the difference in the distributions of protein levels between groups of samples was performed using Mann-Whitney Test using publicly available tool at the Vassar College website (https://faculty.vassar.edu/lowry/VassarStats.html).
Results
Expression microarrays
We used 8 Illumina Sentrix Human-6 v2 Expression Beadchips, each containing six arrays with more than 48K unique probes, to assess global gene expression in EBV-transformed lymphoblastoid cell lines of patients and controls. Transcriptional profiling of lymphoblastoid cell lines is a powerful tool to investigate pathogenic pathways in such diverse diseases as familial combined hyperlipidemia, rheumatoid arthritis, and ataxia telangiectasia [22–24]. 9,484 transcripts were detected at the greater than 99% detection confidence limit in all the samples. Data presented in this manuscript had been deposited into the Gene Expression Omnibus database (GEO, http://www.ncbi.nlm.nih.gov/geo/), under accession number GSE9822. Quality control and expression variability analysis of the duplicated samples of the same individual RNAs as well as at different clonal populations of the same sample showed high reproducibility (Supplemental Data).
Differential expression analysis
We performed statistical testing to identify genes and transcripts whose expression intensities significantly differed between groups ‘1555+’ and ‘1555+D’ (partition by phenotype), ‘Wild’ and ‘1555+/1555+D’ (partition by genotype), and ‘Wild’, ‘1555+’, and ‘1555+D’ (see Methods). Depending on the method used and groups compared we identified 113 to 561 differentially expressed genes (Table 1). Lists of the differentially expressed genes identified by parametric 1 method between groups partitioned by phenotype, genotype and the three groups are available in Supplemental Table 1 through Supplemental Table 3 respectively.
Table 1.
Summary of genes differentially expressed between groups of patients with A1555G mutation and controls, and their functional annotation using GO terms. The statistical tests implemented in Genespring are described in Methods. Full lists of differentially expressed genes are available in Supplemental Tables 1–3. The GO Browser embedded in the Genespring GX program was used to identify the most significant functional groupings (GO annotation terms) of the differentially expressed genes. The GO Browser also was used to identify the overlapping genes between the list of differentially expressed genes and genes with the particular GO term, and to calculate the P-value of the likelihood of having this number of differentially expressed genes with the particular GO term.
| Groups | Statistical test | Number of genes | GO term | Overlapping genes | P-value |
|---|---|---|---|---|---|
| Partition by Phenotype (‘1555+’vs. ‘1555+D’) |
Parametric 1 | 117 | RNA binding (GO:0003723) |
56 | 8.08E-07 |
| Parametric 2 | 113 | 55 | 5.15E-07 | ||
| Nonparametric | 117 | 56 | 2.56E-07 | ||
| Partition by Genotype (‘Wild’ vs. '1555+/+D') |
Parametric 1 | 561 | Ribosome (GO:0005840) |
132 | 2.97E-131 |
| Parametric 2 | 437 | 120 | 1.13E-127 | ||
| Nonparametric | 554 | 134 | 3.91E-135 | ||
| ‘Wild’ vs. ‘1555+’ vs. ‘1555+D’ |
Parametric 1 | 297 | Ribosome (GO:0005840) |
101 | 1.75E-117 |
| Parametric 2 | 243 | 64 | 3.98E-65 | ||
| Nonparametric | 218 | 48 | 2.57E-44 |
Functional analysis of differentially expressed transcripts
To bring biological meaning to the gene expression data, we grouped genes into functional classes and tested whether they genes fall preferentially into certain functional groups by two bioinformatics tools: GO (Gene Ontology) Browser available in the GeneSpring and the public bioinformatics resource DAVID (Database for Annotation, Visualization, and Integrated Discovery) 2007 [25]. In the first analysis, initially each gene is associated with its GO annotation created by The Gene Ontology Consortium [26] in terms of molecular function, biological process and cellular component. Then GO categories that are overrepresented in the inputted list of genes are identified and the P-value of the likelihood that this number of genes with the same function appears in this group of genes by chance only is calculated. In the transcripts differentially expressed between the samples partitioned by phenotype (‘1555+’ vs. ‘1555+D’), ‘RNA binding’ group (GO:0003723) was identified as the most significant molecular pathway with P-values as low as 2.56E-7 (Table 1). GO group ‘Ribosome’ (GO:0005840) was identified when the samples were partitioned by genotype (‘1555+/+D’ vs. ‘Wild’), with P-values as low as 3.91E-135, and in the transcripts differentially expressed between the three groups of ‘Wild’, ‘1555+’, and ‘1555+D’ with P-values as low as 1.75E-117 (Table 1). These hypergeometric P-values represent statistical significance; they calculate the probability that this many genes would overlap between the gene list and the category by chance only. Smaller number of genes (n=56) were simultaneously detected in the larger RNA binding GO category (n=1969) resulting in the P-value of 2.56E-07. Larger number of genes (n=134) were simultaneously detected in the smaller Ribosome GO category (n=565), resulting in the much more significant P-value of 3.91E-135. No other functional groups were identified with comparable levels of significance. It is important to note that these P-values are not necessarily proportional to the biological significance. In biological terms, it is highly significant that half of the differentially expressed genes (56 out of 117) determining severity of hearing loss (partition by phenotype) belongs to RNA binding GO category.
We also performed an independent comparison of the differentially expressed genes (identified by parametric test 1 for each group comparison) to human KEGG (Kyoto Encyclopedia of Genes and Genome, Kyoto University and Human Genome Center) pathways using DAVID and found a significant overlap with the human ribosome pathway in the genes differentially expressed between groups partitioned by genotype and between three groups of patients with the highly significant Bonferroni corrected Fisher's exact t-test P-values of 5.9E-47 and 6.4E-46 respectively, thus confirming results by the GO Browser.
Analysis of differentially expressed genes coding for RNA binding proteins
Different statistical tests identified between 55 and 56 differentially expressed transcripts annotated with the RNA binding (GO:0003723) molecular function between groups partitioned by phenotype (Table 1). These transcripts included genes for some known RNA binding proteins but mostly consisted of genes containing potential RNA-binding domains with varying functions based on annotation by GO (Supplemental Table 4). Comparison of the averages of the weighted scores of the 56 differentially expressed transcripts showed that 33 were down-regulated in the ‘1555+D’ group when compared to the ‘1555+’ group with an average decrease of 26%, and 23 were up-regulated with an average increase of 25%. We also analyzed the expression of the four mitochondrial RNA processing genes TRMU, TFB1M, MTO1, and GTPBP3 previously identified by genetic studies [16–18]. None of these four genes was differentially expressed, indicating that their modulating effect depends on qualitative changes in the protein rather than quantitative changes in transcript levels. For the TRMU gene such a qualitative change has been described [19].
Analysis of differentially expressed genes coding for ribosomal proteins
Different statistical tests identified between 48 and 134 transcripts differentially expressed between the two groups ‘Wild’ and ’1555+/+D’ (partitioned by genotype) and annotated with the biological component ribosome (GO:0005840) (Table 1). Of the 134 ribosomal transcripts, 62 were known genes, while the rest constituted predicted transcribed regions. Surprisingly, of these 62 known genes, 54 were coding for cytoplasmic ribosomal proteins, including isoforms, both of the large and small subunits, and each one of them showed increased expression in the ’1555+/+D’ group, the level of which varied from 13% to 26% with an average of 18% (Table 2). Of the other 8 known genes annotated with the ribosomal function, 5 genes were up-regulated, while 3 were down-regulated in the ‘1555+/+D’ group (Table 2).
Table 2.
Genebank annotated ribosomal genes differentially expressed between the groups partitioned by genotype identified by parametric Student’s t-test with their respective P-values (P-value not corrected for the multiple comparisons). Ratios of normalized scores between the groups are shown. Normalization procedure is described in Methods. Cytoplasmic ribosomal genes are identified by (A). Proteins of the large ribosomal subunit are indicated by RPL; proteins of the small ribosomal subunit are indicated by RPS. All other genes identified by (B). Genes selected for real-time RT-PCR confirmation are identified with the star. Proteins selected for flow cytometry based on antibody availability are identified with the asterisk.
| Gene Symbol | GenBank ID | Probe ID | P-value | Ratio ‘1555+/+D’ to ‘Wild’ |
|---|---|---|---|---|
| RPLP0a | NM_001002 | 630711 | 0.00139 | 1.20 |
| RPLP2a | NM_001004 | 3450184 | 0.0173 | 1.19 |
| RPL3a | NM_001033853 | 1500437 | 0.0125 | 1.185 |
| RPL4a | NM_000968 | 6660193 | 0.00505 | 1.20 |
| RPL5a | NM_000969 | 6620465 | 0.0113 | 1.205 |
| RPL6a” | NM_001024662 | 6620224 | 0.00273 | 1.20 |
| RPL7Aa | NM_000972 | 1090400 | 0.0455 | 1.21 |
| RPL8a” | NM_033301 | 6380148 | 0.00536 | 1.19 |
| RPL9a” | NM_001024921 | 2940639 | 0.00464 | 1.19 |
| RPL9a” | NM_001024921 | 5290008 | 0.0398 | 1.255 |
| RPL11a | NM_000975 | 6370181 | 0.0021 | 1.20 |
| RPL12a | NM_000976 | 7610189 | 0.00413 | 1.215 |
| RPL13Aa | NM_012423 | 6510309 | 0.0171 | 1.16 |
| RPL18a * | NM_000979 | 1190274 | 0.00404 | 1.195 |
| RPL18Aa | NM_000980 | 2970537 | 0.00582 | 1.19 |
| RPL19a *” | NM_000981 | 1470646 | 0.00369 | 1.20 |
| RPL24a * | NM_000986 | 3830142 | 0.00392 | 1.215 |
| RPL26a” | NM_000987 | 2000025 | 0.00456 | 1.26 |
| RPL27a * | NM_000988 | 2320403 | 0.00491 | 1.205 |
| RPL30a | NM_000989 | 3190193 | 0.00399 | 1.19 |
| RPL31a * | NM_000993 | 870424 | 0.0113 | 1.18 |
| RPL35a | NM_007209 | 4490026 | 0.00662 | 1.21 |
| RPL35Aa | NM_000996 | 5670601 | 0.000817 | 1.255 |
| RPL36a | NM_033643 | 5050398 | 0.00824 | 1.185 |
| RPL37a | NM_000997 | 770639 | 0.0343 | 1.325 |
| RPL38a | NM_000999 | 670692 | 0.00115 | 1.205 |
| RPL39a | NM_001000 | 130215 | 0.00441 | 1.205 |
| RPS2a” | NM_002952 | 2810113 | 0.00512 | 1.205 |
| RPS3a | NM_001005 | 2690487 | 0.00181 | 1.20 |
| RPS3Aa | NM_001006 | 4850767 | 0.00385 | 1.28 |
| RPS4Xa | NM_001007 | 6290274 | 0.0111 | 1.14 |
| RPS5a | NM_001009 | 4280326 | 0.00471 | 1.18 |
| RPS6a” | NM_001010 | 4900209 | 0.00461 | 1.185 |
| RPS8a | NM_001012 | 4860215 | 0.02 | 1.85 |
| RPS9a | NM_001013 | 2140735 | 0.000943 | 1.21 |
| RPS9a | NM_001013 | 3310670 | 0.00472 | 1.175 |
| RPS9a | NM_001013 | 7650128 | 0.0049 | 1.18 |
| RPS10a | NM_001014 | 6660220 | 0.00328 | 1.205 |
| RPS11a * | NM_001015 | 3360133 | 0.00405 | 1.19 |
| RPS12a | NM_001016 | 1400132 | 0.019 | 1.185 |
| RPS13a | NM_001017 | 1050474 | 0.00365 | 1.20 |
| RPS14a” | NM_001025071 | 3130437 | 0.00458 | 1.19 |
| RPS15Aa | NM_001030009 | 780356 | 0.00774 | 1.185 |
| RPS16a *” | NM_001020 | 130768 | 0.0178 | 1.185 |
| RPS17a * | NM_001021 | 4730612 | 0.0043 | 1.295 |
| RPS18a | NM_022551 | 4540414 | 0.0212 | 1.16 |
| RPS19a” | NM_001022 | 4250487 | 0.00403 | 1.195 |
| RPS20a * | NM_001023 | 1570300 | 0.00288 | 1.215 |
| RPS24a | NM_001026 | 3870095 | 0.0077 | 1.215 |
| RPS25a * | NM_001028 | 3800612 | 0.0019 | 1.205 |
| RPS27Aa | NM_002954 | 1820044 | 0.00147 | 1.205 |
| RPS28a | NM_001031 | 7510672 | 0.0122 | 1.19 |
| RPS29a | NM_001030001 | 1990753 | 0.00273 | 1.195 |
| RPS29a | NM_001032 | 5960026 | 0.00471 | 1.21 |
| MRPL22b | NM_014180 | 3060458 | 0.0259 | 0.87 |
| NPM1b | NM_199185 | 2060154 | 0.00289 | 1.22 |
| EIF2Ab | NM_032025 | 2060270 | 0.0266 | 1.28 |
| EIF2S3b | NM_001415 | 5670521 | 0.044 | 1.16 |
| UBA52b | NM_003333 | 4810484 | 0.00411 | 1.20 |
| FAUb | NM_001997 | 160167 | 0.00665 | 1.21 |
| MRTO4b | NM_016183 | 3870253 | 0.0342 | 0.86 |
| SF1b | NM_004630 | 5870706 | 0.0317 | 0.78 |
A total of 87 genes coding for the cytoplasmic ribosomal proteins, including isoforms, were detected at the greater than 99% detection confidence limit in all the samples. In addition to the 54 differentially expressed genes, 25 out of the remaining 33 cytoplasmic ribosomal genes also showed higher levels in the ’1555+/+D’ group (not statistically significant, on average 13% increase, data not shown). In contrast, the 507 genes not coding for cytoplasmic ribosomal proteins, but differentially expressed between the two groups partitioned by genotype, were as likely to be up-regulated (248) as they were to be down-regulated (259). No difference between the ‘1555+’ and ‘1555+D’ groups was observed in the expression of the 54 differentially expressed cytoplasmic ribosomal genes (mean ratio of 1.01).
Real-time RT-PCR of genes coding for cytoplasmic ribosomal proteins
To test expression results derived from Human-6 v2 whole genome expression chip, we performed real-time RT-PCR testing using TaqMan® Gene Expression Assays specific to 10 genes coding for cytoplasmic ribosomal proteins: RPS11, RPS16, RPS17, RPS20, RPS25, RPL18, RPL19, RPL24, RPL27, and RPL31. These genes were selected from the 54 differentially expressed cytoplasmic ribosomal genes (Table 2) based on probe availability and probe location in adjacent exons to minimize errors due to potential genomic DNA contamination. Averaged between the triplicate measurements and normalized over GAPDH levels, threshold cycle values of the 10 genes coding for cytoplasmic ribosomal proteins, were 17.9% higher in affected and unaffected family members (‘1555+’ and ‘1555+D’) when compared to the controls (‘Wild’), with a range of −5% to +76%. Based on the microarray results the difference between these groups in the levels of the same 10 ribosomal transcripts was 17.5%, with a range of +16% to +23%.
Flow cytometry of selected cytoplasmic ribosomal proteins
Primary antibodies for 10 cytoplasmic ribosomal proteins (Table 2) were used for flow cytometry in the lymphoblasts of all 43 individuals with and without the A1555G mutation. Using fluorescent signal of samples containing no primary antibodies as a background, we calculated fluorescent intensities of signal in the gated, or expressing above the fluorescence threshold, populations of cells selected from the constant number of 100K cells per measurement. Each individual sample was analyzed in triplicates. While the levels of 2 proteins did not differ between the groups, Median Fluorescent Intensities of the remaining 8 cytoplasmic ribosomal proteins were significantly higher (Mann-Whitney P-value of 0.05) in ‘1555+/+D’ group when compared to the ‘Wild’ group, with an average increase of 6%, ranging from 2% to 15% (Table 3).
Table 3.
Flow Cytometry results of selected cytoplasmic ribosomal proteins. Large ribosomal subunit proteins are denoted by Rpl; small ribosomal subunit proteins - by Rps. MFI denotes Median Fluorescent Intensity. Percent difference in ‘1555+/+D’ group when compared to ‘Wild’ group for each of the tested ribosomal proteins is shown.
| Probe | MFI difference |
|---|---|
| Rpl6 | 0% |
| Rpl8 | 8% |
| Rpl9 | 4% |
| Rpl19 | 4% |
| Rpl26 | 4% |
| Rps2 | 3% |
| Rps6 | 15% |
| Rps14 | 6% |
| Rps16 | 2% |
| Rps17 | 0% |
Analysis of genes in the MYC/MAX pathway
Literature searches, performed in order to understand this coordinated increase in the expression of genes coding for cytoplasmic ribosomal proteins, revealed descriptions of a similar phenomenon occurring in cells transfected with the N-MYC and C-MYC proto-oncogenes [27], and that this effect occurs through their interaction with transcription factor Max [28]. Analysis of the expression data showed probes specific for the C-MYC (MYC), L-MYC (MYCL1), and MAX genes detected at the greater than 99% detection confidence limit in all the samples. Both of the MYC genes were about 10% over-expressed in the ‘A1555G+/+D’ group when compared to controls, although these levels weren’t statistically significant; MAX gene was 30% over-expressed in the ‘A1555G+/+D’ group with a P-value of 0.037 (Supplemental Table 2).
Discussion
Transcriptional profiling has been suggested as a powerful tool to identify cellular functions and metabolic pathways involved in the pathogenesis of mitochondrial diseases, including those caused by defects in mitochondrial translation [29]. This system-based biology approach yields comprehensive characterization of the components of a biological system, leading to insights into the responses of these components to perturbations to the system. In the current study, identification of the RNA binding proteins differentially expressed between deaf and hearing individuals with the A1555G mutation provides independent experimental confirmation of the involvement of the RNA modification pathway in determining the severity of hearing loss associated with the A1555G mutation. Previously, four genes related to mitochondrial RNA modification were identified through genetic association studies [16–18], and in one case a coding mutation was identified and associated with decreased levels of mitochondrial tRNA synthesis [19]. The fact that in this study no significant differences were identified in the expression levels of these four genes seems to indicate that both levels of expression and structural variation of RNA binding proteins can determine the severity of hearing loss associated with the A1555G mutation. Separately, the importance of RNA binding proteins and RNA modification to the pathogenesis of other mitochondrial translation disorders has been demonstrated for mitochondrial encephalomyopathies [30], the French-Canadian type of Leigh syndrome [31], and mitochondrial myopathy with sideroblastic anemia [32].
The most important advantage of expression profiling is that it allows probing compensation mechanisms that cannot be evaluated by genetic means, such as tissue specific differences in responses to genetic disease mutations that are common to all humans. While transcriptional profiling of affected tissues may help uncover biological pathways deregulated in patients, analysis of unaffected tissues may help elucidate compensatory mechanisms that protect those tissues. An additional advantage of this methodology is its ability to obtain unexpected results, since no prior hypothesis is required. For example, expression profiling in the motor cortex of patients with Amyotrophic Lateral Sclerosis found nuclear-encoded mitochondrial genes were uniformly down-regulated contrary to expectations for a tissue under oxidative stress [33]. In the present study, expression profiling has implicated the coordinated over-expression of cytoplasmic ribosomal genes as the mechanism through which cells compensate for the mitochondrial dysfunction associated with the A1555G mutation in most tissues. This conclusion is supported by: 1. the results are independent of the statistical method used to identify the genes differentially expressed between the groups (Table 1); 2. the results are independent of the algorithms (Genespring, DAVID) used to identify which functional groups are present in the differentially expressed sets of genes and used to calculate statistical significance of this presence; 3. the P-value of the Fisher's exact t-test corresponding to the enrichment in annotation terms is at least an order of magnitude higher for the ribosomal pathway than for RNA binding proteins, which have been validated by genetic studies; 4. all 54 differentially expressed genes coding for cytoplasmic ribosomal proteins were up-regulated, whereas 507 non-ribosomal genes differentially expressed were as likely to be up-regulated (248 genes) as they were to be down-regulated (259 genes) between the groups, a result with a likelihood to occur by chance of 5.55E-17 (0.554) ; 5. RT-PCR results of the ten selected cytoplasmic ribosomal genes were consistent with the microarray data; 6. measurements of levels of cytoplasmic ribosomal proteins by flow cytometry were also increased, albeit at lower level, for eight out of ten cytoplasmic proteins tested (the other two showed equivalent levels, Table 3); 7. a possible mechanism through which this compensatory response occurs involves the Myc/Max pathway which appears to be over-expressed in patients with the A1555G mutation (see next paragraph); and 8. compensatory changes of the translational apparatus have been documented in other mitochondrial translation disorders: In MLASA, expression levels of mitochondrial and cytoplasmic ribosomal proteins differed significantly between homozygous affected individuals, healthy heterozygotes, and controls [34]. In fatal hepatopathy (COXPD1) both up and down regulation of the components of the mitochondrial translation apparatus were identified in affected and unaffected tissues [11]. The fact that these mechanisms share some similarities but differ between the diseases indicates that efficient compensation must be ‘fine tuned to the exact specifications’ of the underlying translational defect.
Review of the literature provided a potential pathway through which coordinated up-regulation of cytoplasmic ribosomal genes could occur. The simultaneous increase in the expression of numerous ribosomal genes has been previously documented in N-MYC transfected neuroblastoma cells and C-MYC transfected melanoma cells [27], and MYC oncogenic activity is influenced by the production of ribosomal proteins [35]. The MYC family of proto-oncogenes are tissue-specific transcription factors responsible for the increased proliferation associated with many types of human malignancy [36]. They exert this effect through their interaction with additional transcription factors such as Max [28]. We found increase in the transcripts of these factors (10% for the MYC genes and 30% for the MAX gene) were identified, possibly providing the mechanism through which the highly coordinated increase in the levels of cytoplasmic ribosomal genes. Inhibition and over-expression of these genes in lymphoblastoid cell lines of patients with the A1555G mutation and controls will allow examination of these interactions in more detail. The question whether patients with the A1555G mutation have a greater predisposition to cancer has not been evaluated up to date.
The data presented in this manuscript, as well as data previously obtained, provide a potential model for how the interaction of multiple genetic and environmental factors with the A1555G mutation determine the tissue specificity and severity of hearing loss (Figure 1). First, the mitochondrial A1555G mutation leads to a decrease in mitochondrial protein synthesis [37] and translational accuracy [38]. Then, a compensation mechanism based on over-expression of the components of the cytoplasmic ribosome restores adequate cellular function. However, in the cochlea of deaf individuals this compensation mechanism fails as additional detrimental factors overwhelm this mechanism through three independent pathways: 1. Nuclear mitochondrial RNA processing genes have subtle qualitative [19] or quantitative (present study) deficits, that by themselves are benign, but in combination with the A1555G mutation become pathologic. The reason for the tissue specificity in this case may be related either to additional functions of the mitochondrial 12S rRNA in the cochlea that make those cells more susceptible, or, alternatively, through a diminished cytoplasmic ribosomal response in the cochlea, similar to the diminished compensatory response in the liver in fatal hepatopathy [11]; 2. Mitochondrial background can aggravate the deficit due to the A1555G mutation, similar to what has been observed for other mitochondrial deafness mutations [39]. There is evidence that some mitochondrial tRNA genes variants, indicated on Figure 1, increased the penetrance of hearing loss associated with A1555G mutation [40, 41, 42]. The tissue specificity may arise through the same mechanism outlined for the nuclear genes; 3. Environmental factors such as aminoglycosides exacerbate the effect of the A1555G mutation [15, 43, 44], as has been demonstrated biochemically [45, 46]. In this case, the tissue specificity arises through the accumulation and long half life of aminoglycosides in the inner ear. It is interesting to note, that no difference in the cytoplasmic ribosomal response was identified between hearing and deaf individuals with the A1555G mutation, implying that this is a generic response and that individual differences between people don’t provide additional protection to the cochlea.
Figure 1.
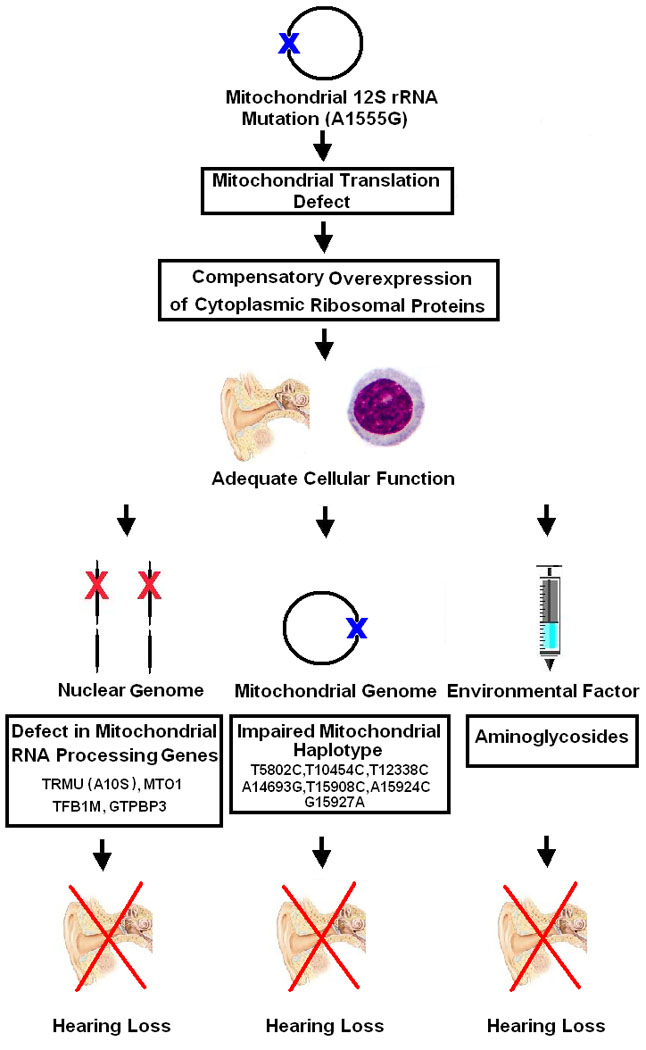
Mechanisms of compensation and pathogenesis of hearing loss associated with mitochondrial A1555G mutation in the 12S ribosomal RNA gene. TRMU - tRNA 5-methylaminomethyl-2-thiouridylate methyltransferase, MTO1 -mitochondrial translation optimization 1 homolog (S. cerevisiae), TFB1M -mitochondrial transcription factor B1, GTPBP3 - mitochondrial GTP binding protein 3 [16–18].
In summary, the clinical phenotype of maternal deafness associated with the A1555G mutation in the mitochondrial 12S rRNA gene is impacted by compensation mechanisms involving over-expression of cytoplasmic ribosomal proteins, as well as by qualitative and quantitative changes in RNA binding proteins, mitochondrial haplotype, and environmental factors. The fact that compensation mechanism can circumvent the effects of the A1555G mutation raises the possibility that preventive and therapeutic interventions for the cochlear defect can be identified.
Supplementary Material
Acknowledgements
We thank Drs. Cristina Bertolotto and Kent Taylor for technical support. We gratefully acknowledge support from National Institute of Health/ National Institute of Diabetes and Digestive and Kidney Diseases [grant number RO1-DK74368]. This project was supported in part by General Clinical Research Center [grant number MO1-RR00425].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Scheper GC, van der Klok T, van Andel RJ, van Berkel CG, Sissler M, Smet J, Muravina TI, Serkov SV, Uziel G, Bugiani M, Schiffmann R, Krageloh-Mann I, Smeitink JA, Florentz C, van Coster R, Pronk JC, van der Knaap MS. Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat. Genet. 2007;39:534–539. doi: 10.1038/ng2013. [DOI] [PubMed] [Google Scholar]
- 2.Jacobs HT, Turnbull DM. Nuclear genes and mitochondrial translation: a new class of genetic disease. Trends Genet. 2005;21:312–314. doi: 10.1016/j.tig.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 3.Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005;6:389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller C, Saada A, Shaul N, Shabtai N, Ben-Shalom E, Shaag A, Hershkovitz E, Elpeleg O. Defective mitochondrial translation due to a ribosomal protein (MRPS16) mutation. Ann. Neurol. 2004;56:734–738. doi: 10.1002/ana.20282. [DOI] [PubMed] [Google Scholar]
- 5.Saada A, Shaag A, Arnon S, Dolfin T, Miller C, Fuchs-Telem D, Lombes A, Elpeleg O. Antenatal mitochondrial disease caused by mitochondrial ribosomal protein (MRPS22) mutation. J. Med. Genet. 2007;44:784–786. doi: 10.1136/jmg.2007.053116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Edvardson S, Shaag A, Kolesnikova O, Gomori JM, Tarassov I, Einbinder T, Saada A, Elpeleg O. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am. J. Hum. Genet. 2007;81:857–862. doi: 10.1086/521227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smeitink JA, Elpeleg O, Antonicka H, Diepstra H, Saada A, Smits P, Sasarman F, Vriend G, Jacob-Hirsch J, Shaag A, Rechavi G, Welling B, Horst J, Rodenburg RJ, van den Heuvel B, Shoubridge EA. Distinct clinical phenotype associated with a mutation in the mitochondrial translation elongation factor. EFTs. Am. J. Hum. Genet. 2006;79:869–877. doi: 10.1086/508434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Valente L, Tiranti V, Marsano RM, Malfatti E, Fernandez-Vizarra E, Donnini C, Mereghetti P, De Gioia L, Burlina A, Castellan C, Comi GP, Savasta S, Ferrero I, Zeviani M. Infantile encephalopathy and defective mitochondrial DNA translation in patients with mutations of mitochondrial elongation factors EFG1 and EFTu. Am. J. Hum. Genet. 2007;80:44–58. doi: 10.1086/510559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.‘t Hart LM, Hansen T, Rietveld I, Dekker JM, Nijpels G, Janssen GM, Arp PA, Uitterlinden AG, Jorgensen T, Borch-Johnsen K, Pols HA, Pedersen O, van Duijn CM, Heine RJ, Maassen JA. Evidence that the mitochondrial leucyl tRNA synthetase (LARS2) gene represents a novel type 2 diabetes susceptibility gene. Diabetes. 2005;54:1892–1895. doi: 10.2337/diabetes.54.6.1892. [DOI] [PubMed] [Google Scholar]
- 10.Coenen MJ, Antonicka H, Ugalde C, Sasarman F, Rossi R, Heister JG, Newbold RF, Trijbels FJ, van den Heuvel LP, Shoubridge EA, Smeitink JA. Mutant mitochondrial elongation factor G1 and combined oxidative phosphorylation deficiency. N. Engl. J. Med. 2004;351:2080–2086. doi: 10.1056/NEJMoa041878. [DOI] [PubMed] [Google Scholar]
- 11.Antonicka H, Sasarman F, Kennaway NG, Shoubridge EA. The molecular basis for tissue specificity of the oxidative phosphorylation deficiencies in patients with mutations in the mitochondrial translation factor EFG1. Hum. Mol. Genet. 2006;5:1835–1846. doi: 10.1093/hmg/ddl106. [DOI] [PubMed] [Google Scholar]
- 12.Jeppesen TD, Schwartz M, Frederiksen AL, Wibrand F, Olsen DB, Vissing J. Muscle phenotype and mutation load in 51 persons with the 3243A>G mitochondrial DNA mutation. Arch. Neurol. 2006;63:1701–1706. doi: 10.1001/archneur.63.12.1701. [DOI] [PubMed] [Google Scholar]
- 13.Fischel-Ghodsian N. Mitochondrial genetics and hearing loss: the missing link between genotype and phenotype. Proc. Soc. Exp. Biol. Med. 1998;218:1–6. doi: 10.3181/00379727-218-44262. [DOI] [PubMed] [Google Scholar]
- 14.Fischel-Ghodsian N. Homoplasmic mitochondrial diseases as the paradigm to understand the tissue specificity and variable clinical severity of mitochondrial disorders. Mol. Genet. Metab. 2000;71:93–99. doi: 10.1006/mgme.2000.3014. [DOI] [PubMed] [Google Scholar]
- 15.Prezant TR, Agapian JV, Bohlman MC, Prezant Toni R, Agapian John V, Charlotte Bohlman M, Bu X, Öztas S, Qiu WQ, Arnos KS, Cortopassi GA, Jaber L, Rotter JI, Shohat M, Fischel-Ghodsian N. Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nat. Genet. 1993;4:289–294. doi: 10.1038/ng0793-289. [DOI] [PubMed] [Google Scholar]
- 16.Bykhovskaya Y, Mengesha E, Wang D, Yang H, Estivill X, Shohat M, Fischel-Ghodsian N. Human mitochondrial transcription factor B1 as a modifier gene for hearing loss associated with the mitochondrial A1555G mutation. Mol. Genet. Metab. 2004;82:27–32. doi: 10.1016/j.ymgme.2004.01.020. [DOI] [PubMed] [Google Scholar]
- 17.Bykhovskaya Y, Mengesha E, Wang D, Yang H, Estivill X, Shohat M, Fischel-Ghodsian N. Phenotype of non-syndromic deafness associated with the mitochondrial A1555G mutation is modulated by mitochondrial RNA modifying enzymes MTO1 and GTPBP3. Mol. Genet. Metab. 2004;83:199–206. doi: 10.1016/j.ymgme.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 18.Yan Q, Bykhovskaya Y, Li R, Mengesha E, Shohat M, Estivill X, Fischel-Ghodsian N, Guan MX. Human TRMU encoding the mitochondrial 5- methylaminomethyl-2-thiouridylate methyltransferase is a putative nuclear modifier gene for the phenotypic expression of the deafness-associated 12S rRNA mutations. Biochem. Biophys Res. Commun. 2006;342:1130–1136. doi: 10.1016/j.bbrc.2006.02.078. [DOI] [PubMed] [Google Scholar]
- 19.Guan MX, Yan Q, Li X, Bykhovskaya Y, Gallo-Teran J, Hajek P, Umeda N, Zhao H, Garrido G, Mengesha E, Suzuki T, del Castillo I, Peters JL, Li R, Qian Y, Wang X, Ballana E, Shohat M, Lu J, Estivill X, Watanabe K, Fischel-Ghodsian N. Mutation in TRMU related to transfer RNA modification modulates the phenotypic expression of the deafness-associated mitochondrial 12S ribosomal RNA mutations. Am. J. Hum. Genet. 2006;79:291–302. doi: 10.1086/506389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jaber L, Shohat M, Bu X, Fischel-Ghodsian N, Yang HY, Wang SJ, Rotter JI. Sensorineural deafness inherited as a tissue specific mitochondrial disorder. J. Med. Genet. 1992;29:86–90. doi: 10.1136/jmg.29.2.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hosack DA, Dennis G, Jr, Sherman BT, Lane HC, Lempicki RA. Identifying biological themes within lists of genes with EASE. Genome Biol. 2003;4:R70. doi: 10.1186/gb-2003-4-10-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morello F, de Bruin TW, Rotter JI, Pratt RE, van der Kallen CJ, Hladik GA, Dzau VJ, Liew CC, Chen YD. Differential gene expression of blood-derived cell lines in familial combined hyperlipidemia. Arterioscler. Thromb. Vasc. Biol. 2004;24:2149–2154. doi: 10.1161/01.ATV.0000145978.70872.63. [DOI] [PubMed] [Google Scholar]
- 23.Haas CS, Creighton CJ, Pi X, Maine I, Koch AE, Haines GK, Ling S, Chinnaiyan AM, Holoshitz J. Identification of genes modulated in rheumatoid arthritis using complementary DNA microarray analysis of lymphoblastoid B cell lines from disease-discordant monozygotic twins. Arthritis Rheum. 2006;54:2047–2060. doi: 10.1002/art.21953. [DOI] [PubMed] [Google Scholar]
- 24.Watts JA, Morley M, Burdick JT, Fiori JL, Ewens WJ, Spielman RS, Cheung VG. Gene expression phenotype in heterozygous carriers of ataxia telangiectasia. Am. J. Hum. Genet. 2002;71:791–800. doi: 10.1086/342974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 26.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. The Gene Ontology: tool for the unification of biology. Nature Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boon K, Caron HN, van Asperen R, Valentijn L, Hermus MC, van Sluis P, Roobeek I, Weis I, Voute PA, Schwab M, Versteeg R. N-myc enhances the expression of a large set of genes functioning in ribosome biogenesis and protein synthesis. EMBO J. 2001;20:1383–1393. doi: 10.1093/emboj/20.6.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blackwood EM, Eisenman RN. Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science. 1991;251:1211–1217. doi: 10.1126/science.2006410. [DOI] [PubMed] [Google Scholar]
- 29.Crimi M, O'Hearn SF, Wallace DC, Comi GP. Molecular research technologies in mitochondrial diseases: the microarray approach. IUBMB Life. 2005;57:811–818. doi: 10.1080/15216540500460269. [DOI] [PubMed] [Google Scholar]
- 30.Crimi M, Bordón A, Menozzi G, Riva L, Fortunato F, Galbiati S, Del Bo R, Pozzoli U, Bresolin N, Comi GP. Skeletal muscle gene expression profiling in mitochondrial disorders. FASEB Journal. 2005;19:866–868. doi: 10.1096/fj.04-3045fje. [DOI] [PubMed] [Google Scholar]
- 31.Mootha VK, Lepage P, Miller K, Bunkenborg J, Reich M, Hjerrild M, Delmonte T, Villeneuve A, Sladek R, Xu F, Mitchell GA, Morin C, Mann M, Hudson TJ, Robinson B, Rioux JD, Lander ES. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc. Natl. Acad. Sci. USA. 2003;100:605–610. doi: 10.1073/pnas.242716699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bykhovskaya Y, Casas K, Mengesha E, Inbal A, Fischel-Ghodsian N. Missense mutation in pseudouridine synthase 1 (PUS1) causes mitochondrial myopathy and sideroblastic anemia (MLASA) Am. J. Hum. Genet. 2004;74:1303–1308. doi: 10.1086/421530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lederer CW, Torrisi A, Pantelidou M, Santama N, Cavallaro S. Pathways and genes differentially expressed in the motor cortex of patients with sporadic amyotrophic lateral sclerosis. BMC Genomics. 2007;8:26. doi: 10.1186/1471-2164-8-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bykhovskaya Y, Mengesha E, Fischel-Ghodsian N. Pleiotropic effects and compensation mechanisms determine tissue specificity in mitochondrial myopathy and sideroblastic anemia (MLASA) Mol. Genet. Metab. 2007;91:148–156. doi: 10.1016/j.ymgme.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barna M, Pusic A, Zollo O, Costa M, Kondrashov N, Rego E, Rao PH, Ruggero D. Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature. 2008;456:971–975. doi: 10.1038/nature07449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ryan KM, Birnie GD. Myc oncogenes: the enigmatic family. Biochem. J. 1996;314:713–721. doi: 10.1042/bj3140713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guan MX, Fischel-Ghodsian N, Attardi G. Biochemical evidence for nuclear gene involvement in phenotype of non-syndromic deafness associated with mitochondrial 12S rRNA mutation. Hum. Mol. Genet. 1996;5:963–971. doi: 10.1093/hmg/5.7.963. [DOI] [PubMed] [Google Scholar]
- 38.Hobbie SN, Bruell CM, Akshay S, Kalapala SK, Shcherbakov D, Bottger EC. Mitochondrial deafness alleles confer misreading of the genetic code. Proc. Natl. Acad. Sci. USA. 2008;105:3244–3249. doi: 10.1073/pnas.0707265105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sevior KB, Hatamochi A, Stewart IA, Bykhovskaya Y, Allen-Powell DR, Fischel-Ghodsian N, Maw MA. Mitochondrial A7445G mutation in two pedigrees with palmoplantar keratoderma and deafness. Am. J. Med. Genet. 1998;75:179–185. [PubMed] [Google Scholar]
- 40.Wang X, Lu J, Zhu Y, Yang A, Yang L, Li R, Chen B, Qian Y, Tang X, Wang J, Zhang X, Guan MX. Mitochondrial tRNAThr G15927A mutation may modulate the phenotypic manifestation of ototoxic 12S rRNA A1555G mutation in four Chinese families. Pharmacogenet. Genom. 2008;18:1059–1070. doi: 10.1097/FPC.0b013e3283131661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Young WY, Zhao L, Qian Y, Li R, Chen J, Yuan H, Dai P, Zhai S, Han D, Guan MX. Variants in mitochondrial tRNAGlu, tRNAArg, and tRNAThr may influence the phenotypic manifestation of deafness-associated 12S rRNA A1555G mutation in three Han Chinese families with hearing loss. Am. J. Med. Genet. A. 2006;140:2188–2197. doi: 10.1002/ajmg.a.31434. [DOI] [PubMed] [Google Scholar]
- 42.Chen B, Sun D, Yang L, Zhang C, Yang A, Zhu Y, Zhao J, Chen Y, Guan M, Wang X, Li R, Tang X, Wang J, Tao Z, Lu J, Guan MX. Mitochondrial ND5 T12338C, tRNACys T5802C, and tRNAThr G15927A variants may have a modifying role in the phenotypic manifestation of deafness-associated 12S rRNA A1555G mutation in three Han Chinese pedigrees. Am. J. Hum. Genet. A. 2008;146:1248–1258. doi: 10.1002/ajmg.a.32285. [DOI] [PubMed] [Google Scholar]
- 43.Fischel-Ghodsian N. Genetic factors in aminoglycoside toxicity, Pharmacogenomics. 2005;6:27–36. doi: 10.1517/14622416.6.1.27. [DOI] [PubMed] [Google Scholar]
- 44.Talaska AE, Schacht J, Fischel-Ghodsian N. Molecular and genetic aspects of aminoglycoside- induced hearing loss, Drug Disc. Today. Disease Mech. 2006;3:119–124. [Google Scholar]
- 45.Hamasaki K, Rando RR. Specific Binding of Aminoglycosides to a Human rRNA Construct Based on a DNA Polymorphism Which Causes Aminoglycoside-Induced Deafness. Biochem. J. 1997;36:12323–12328. doi: 10.1021/bi970962r. [DOI] [PubMed] [Google Scholar]
- 46.Guan MX, Fischel-Ghodsian N, Attardi GA. Biochemical basis for the inherited susceptibility to aminoglycoside ototoxicity. Hum. Mol. Genet. 2000;9:1787–1793. doi: 10.1093/hmg/9.12.1787. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.