

Abstract
The endothelin (ET) axis, often deregulated in cancers, is a promising target for anticancer strategies. While previous investigations have focussed mostly on ET action in malignant cells, we chose a model, allowing separate assessment of the effects of ETs and their receptors ETAR and ETBR in the tumor cells and the stromal compartment, which is increasingly recognized as a key player in cancer progression. In homozygous spotting lethal rats (sl/sl), a model of constitutive ETBR-deficiency, we demonstrated significant reduction of growth and metastasis of MAT B III rat mammary adenocarcinoma cells, overexpressing ETAR and ET-1, but negative for ETBR. Lack of stromal ETBR expression did not influence angiogenesis. However, it was correlated with diminished infiltration by tumor-associated macrophages and with reduced production of TNF-α, both known as powerful promoters of tumor progression. These effects were almost completely abolished in transgenic sl/sl rats, where ETBR function is restored by expression of an intact ETBR transgene. This demonstrates that tumor growth and metastasis are critically dependent on ETBR function in cells of the microenvironment and suggests that successful ETR antagonist therapy should also target the stromal component of ET signalling.
Introduction
The vasoactive peptides endothelin (ET)-1, -2, -3 and their receptors ETAR and ETBR are part of an ubiquitous network which not only regulates vascular function (1) but is also involved in cell proliferation (2), differentiation (3), migration (4) and (anti)apoptosis (5).
This network has been shown to be altered in many malignant tissues (6, 7). In breast cancer, expression of ET-1 and ETAR is correlated with the transition from normal tissue to progressively invasive lesions (8), increased tumor angiogenesis (9) and shortened survival (10). ET-1 serum levels are elevated in patients with breast and colon cancers, especially in those with lymph node or distant metastases (11, 12). The ET-1/ ETAR axis plays also a critical role for ovarian carcinoma progression (13).
The role of the ETBR, however, is ambiguous. While colon cancer (14), Ewing sarcoma and neuroblastoma (15) as well as prostate cancer are associated with downregulation of ETBR, leading to a preponderance of ET-1/ETAR signaling (16), ETBR is upregulated in lung cancer (17), oral squamous cancer (18) and malignant melanoma, where it has been identified as a marker of progression (19, 20).
Inhibition of the ET axis efficiently antagonizes tumor progression in vitro and in animal models. Consistent with overexpression of the respective targeted receptor(s) in the tumor cells, antagonists of ETAR or both receptors inhibit proliferation and tumor growth in colon, breast and ovarian carcinomas (12, 21, 22), while selective antagonists of ETBR are successful in melanomas (3, 23). In spite of these promising findings, the results of the few available clinical studies are still unsatisfactory. Treatment of 32 patients with advanced melanomas with the dual inhibitor bosentan induced stable disease in 6 patients as the best achievable result (24). The ETAR-inhibitor atrasentan, although effective in the reduction of surrogate markers like prostate-specific antigen and alkaline phosphatase, did not delay disease progression in men with prostate cancer (25). Combined treatment of patients with advanced non-small cell lung cancer with chemotherapy and atrasentan did not yield better results than chemotherapy alone (26).
Apparently, there are determinants other than the expression of ETs and receptors in the tumor cells themselves, influencing the outcome in vivo. There is growing evidence that tumor progression does not only depend on the biological characteristics of the malignant cells, but also on interaction with benign cells and components of the surrounding stromal compartment (27). The ET system is an interesting candidate to influence these interactions, since ETs as well as their receptors are expressed by a variety of stromal cells, such as myofibroblasts, endothelial cells (28) and macrophages (MΦ) (29).
MΦ are potent promoters of invasion (30). MΦ-induced invasion of breast cancer cells through matrigel is increased both by ET-1 and -2 as well as overexpression of ETAR/ETBR (31, 32). It is abolished by ETAR/ETBR-inhibition. ET-2 acts as a chemoattractant for MΦ via ETBR in vitro and probably also in vivo, as ETBR-positive tumor-associated MΦ (TAM) in breast cancers often co-localize with areas of ET-2-expressing malignant cells (33). These data suggest that the ET system, especially via the ETBR, is critical for stromal reactions in tumor progression.
The spotting lethal (sl) rat represents a suitable model to study the function of the ETBR without the necessity of pharmacological manipulations. Homozygous sl/sl rats lack functional ETBRs due to a 301 bp deletion in the ETBR gene (34). This leads to disturbance of neural crest migration and congenital aganglionosis of the gut with development of Hirschsprung’s disease, limiting the life span to maximally 4 weeks. Wild type (+/+) and heterozygous (sl/+) rats are phenotypically normal. In transgenic rescue rats, ETBR function is restored by introduction of an intact ETBR transgene linked to the human dopamine-β-hydroxylase (DβH) promoter (35). Thus, transgenic rats of the originally ETBR-deficient phenotype (tg sl/sl) do not succumb any more to intestinal complications. The tg sl/+ and tg +/+ animals, expressing an endogenous as well as a transgenic ETBR, remain phenotypically normal.
Although primarily gut-targeted, ectopic expression of the transgene outside from intestinal catecholaminergic neurons has been described (35, 36). Thus, spotting lethal rats and their transgenic counterparts were used as a a substraction/addition model to further investigate the role of stromal ETBR function for tumor progression. Tumor formation was induced by subcutaneous inoculation of highly invasive MAT B III rat mammary adenocarcinoma cells, which are characterized by upregulation of the ET-1/ ETAR axis and lack expression of ETBR, thus providing a species-immanent tumor model, where ETBR is present exclusively in the stromal compartment.
Material and Methods
Cell lines, animals and experimental protocol
The 13762 MAT B III rat mammary adenocarcinoma cell line was obtained from the American Type Culture Collection (Rockville, MD, USA) and cultured in RPMI-1640 + 10% fetal bovine serum. All experiments were performed under endotoxin-free conditions.
Rats of the Wistar-Imamichi AR strain (spotting lethal (sl) rats) as well as sl transgenic rescue rats were bred as described before (36, 37). Genotyping was performed using primers flanking the 301 bp deletion of the ETBR gene (37). Expression of the transgene was documented by RT-PCR as described (35).
All animal work had been approved by the local committee of Animal Care and Use. MAT B III cells, suspended in PBS at 105 cells/100 μl or 10-6 M clazosentan (Actelion, Allschwil, Switzerland) were inoculated subcutaneously in the region of the right thigh of 24h-old rats. When the first of the ETBR-deficient animals had to be euthanized due to complications of congenital aganglionosis (between day 15-21), the whole litter was sacrificed by decapitation in anaesthesia with 2,2,2-tribromethanol (Avertin, 275g/kg body weight, intraperitoneally). Upon autopsy, abdomen and thoracic cavity were examined for the presence of metastases. Subcutaneous tumors were excised and weighed; Serum was collected. Tumors and organs were fixed in 4% paraformaldehyde and/or snap frozen in liquid nitrogen. Only litters of similar age and animals without signs of accidental intravenous tumor cell injection were included in the comparative analysis of tumor weight. To determine the rate of metastases all animals were included.
Chemotaxis Assay
BMDM were cultured for 7 days in DMEM + 100 ng/ml murine M-CSF (R&D, Abingdon, UK). Cell motility of 5 × 105 MΦ towards CCL5, CCL2 or CSF-1 (each 10 ng/ml, R&D Systems) was assayed using Falcon Transwells (BD Pharmingen). Following incubation for 18 hours, migrated cells on the lower surface were stained using DiffQuik (Dade Behring, Düdingen, Switzerland). For each transwell, the number of migrated cells in 10 medium power fields (x 20) was counted. All of these experiments were repeated at least three times.
Cloning and transfection of tumor cells
Tumor cells were seeded in six-well plates such that they were 60–90% confluent on the day of transfection. siRNA sequence for the rat ETAR (NM_012550) was obtained from Ambion (Huntingdon, UK; sense siRNA strand: 5′-GGACUGGUGGCUCUUUGGATT-3′; antisense siRNA strand: 5′-UCCAAAGAGCCACCAGUCCTT-3′) and cloned into the pSilencer2.1-U6 vector system (Ambion, Huntingdon, UK). MAT B III cells were transfected with the pSilencer2.1-U6-RNAi plasmids for ETAR, or a control plasmid containing scrambled RNA. Cells were transfected using Lipofectamin 2000 (Invitrogen, UK) following the manufacturer’s instructions. Antibiotic selection for stable cell lines started after 48 to 72 h in 4μg/mL puromycin (Sigma) for 30 days. Effective gene silencing was confirmed by RT-PCR and Western blot (Abcam, UK). A mixture of all 3 clones was used for inoculation.
Flow cytometry
Excised tumors were incubated in digestion buffer (RPMI 1640 + 5% FCS + 5 mg/ml collagenase D + 0.15 mg/ml DNase). Tumors were minced, digested at 37°C for 40 min, passed through a 19-g and 23-g needle and then through a cell strainer. Cells were pelleted at 1500 rpm and resuspended at 50-100 × 105 cells/ml in FACS buffer (PBS + 0.1% BSA, 0.01% NaN3). Results were normalized to tumor weight. Cells were blocked with mouse anti-rat CD32 FcBlock (BD Bioscience PharMingen) for 30 min on ice. Antibodies used were CD163-FITC (ED2) for macrophages, CD161-FITC (10/78), NK cells; all from Serotec; CD4-FITC (OX-35) and CD25-PE (OX-39) for T reg cells, CD8a-FITC (OX-8), T cells and granulocytes-FITC (HIS48); all from BD Bioscience PharMingen and respective isotype controls. Cells were analyzed on a FACScan flow cytometer using Cellquest software (Becton Dickinson). For three-way cell sorting using the MoFlo (DakoCytomation), CD8a-PE, granulocytes-biotin (BD PharMingen), and CD163-FITC (Serotec) were applied to isolate all three cell populations from the same cell pool. Tumor-associated macrophages from tumor bearing rats were collected by CD11b positive selection (Miltenyibiotec, Surrey, UK). Experiments were repeated at least three times.
ELISA
Quantikine ELISA kits for rat IL-10, TNF-α and rat VEGF (R&D Systems, Abingdon, United Kingdom) were used according to the manufacturer’s instructions. Analytical sensitivity of the assays was as follows: rat IL-10 10 pg/ml, rat TNF-α, 12.5 pg/ml, rat VEGF, 3.9 pg/ml. For all ELISAs, the absorbance at 450 nm was measured and corrected at 570 nm in a plate reader (Opsys MR; Dynex Technologies). Experiments were repeated at least three times.
Histology and Immunohistochemistry
Tissues were fixed in buffered formalin and embedded in paraffin. Morphology was visualized by staining with haematoxylin & eosin (H&E). To assess proliferation (%Ki-67 positivity) immunohistochemistry was performed using the MIB-5 monoclonal mouse anti-rat Ki-67 antibody with a standard ABC technique, diaminobenzidine, and nuclear counterstaining with haematoxylin (Dako, Hamburg, Germany). Ten high power fields for each sample were counted by 2 individual people. Macrophages were detected with the CD 163 antibody and the same ABC method. For microvascular density quantification, tissues were stained with the rabbit polyclonal Von Willebrand Factor antibody (Abcam, Cambridge, UK). Computer images were used to perform manual counts of stained microvessels. Apoptosis was detected with the TUNEL assay, using a commercially available in situ apoptosis detection kit (Promega, UK). The number of TUNEL-positive apoptotic cells was counted in 10 randomly selected fields (x40) as a percentage of total cells.
Semiquantitative and real-time RT-PCR
Semiquantitative RT-PCR with co-amplification of lamin b was used for detection of ETBR expression in rats. Amplification of the endogenous (and the sequence-identical transgene) receptor yielded a 912 bp product for the wild type and a 642 bp product for the mutant form. Heterozygous animals show both forms, although – due to primer competition – they are not amplified with the same efficiency. Separate primers were used to detect exclusively the DβH promoter-coupled transgene resulting in a 500 bp product. Primers and conditions have been described previously (35-37).
Multiplex real-time analysis was performed using MMP-2, MMP-9, ET-1, -2, -3, ECE1, ECE2, ETAR and ETBR (FAM) and 18s rRNA (VIC) specific primers and probes with the ABI PRISM 7700 Sequence Detection System instrument and software (PE Applied Biosystems, Warrington, GB). PCR was carried out with the TaqMan Universal PCR Master Mix (PE Applied Biosystems) using 2μl cDNA in a 25-μl final reaction volume. The cycling conditions were: incubation at 50°C for 2 min, followed by 10 min at 95°C and 60 cycles of 15 s at 95°C, and 1 min at 60°C. Experiments were performed in triplicate. Gene expression was normalized to 18S RNA by subtracting the cycle threshold (Ct) value of 18 S RNA from the Ct value of the respective RNA of interest.
Statistical analysis
Data are presented as means ± SD. Differences in lung metastases were tested for statistical significance using the Fisher’s exact test. All other data were compared with the unpaired two-tailed Student’s t-test or Mann-Whitney U-test. P-values < 0.05 were considered significant.
Results
Stromal ETBR-deficiency reduces local tumor growth
First, syngeneic MAT B III cells were characterized by RT-PCR, demonstrating strong expression of ET-1, ECE-1/-2, ETAR, week expression of ET-2 and lack of ET-3 and ETBR expression (Fig. 1A). We then asked whether stromal ETBR-expression would influence the growth of MAT B III cells in vivo in the spotting lethal rat model. After subcutaneous inoculation of the syngeneic-invasive tumor cells, all rats developed a local tumor at the injection site. Absolute as well as relative weight (normalized to body weight) of these tumors was significantly lower in ETBR-deficient sl/sl rats than in heterozygous and wild type animals (Fig. 1B). Upon H&E staining (not shown), tumors in the genetic subgroups did not differ from each other. Cells with the typical signs of apoptosis were rarely detectable in H&E-stained sections of any genotype. This was confirmed by TUNEL-staining, showing no difference between the three populations (Table 1). However, proliferation rate was significantly lower in tumors of sl/sl rats, as shown by Ki67-staining (Fig. 1C; Fig. 2A, B).
Fig. 1. Expression of the ET system and local tumor growth.

A) RNA expression profile of ETs, receptors and related proteins in MAT B III cells (real-time RT-PCR, one representative of three independent experiments). B) ETBR-deficiency reduces tumor growth. Relative weight of the MAT B III-induced tumors at the inoculation site (normalized to body weight) in the three genotypes of sl rats and their transgenic counterparts. Only litters of similar age (4 in both groups) were analyzed. The animal numbers per genotype are given in brackets. C) ETBR-deficiency reduces proliferation. Proliferation index (% of tumor cell nuclei staining positive for Ki67) in the local tumors. Numbers (in brackets) are higher as for tumor weight, as all inoculated animals were included (6 litters). D) RNA expression of transgenic ETBR in various tissues of transgenic rescue sl rats (RT-PCR, lamin b expression used as control). Primers recognize exclusively the transgenic receptor, not the endogenous form.
Table 1.
Apoptosis, proliferation and microvessel densitiy in tumors derived from MAT B III and MAT B III ETAR siRNA cells (means ± SD)
| MAT B III | MAT B III-ETAR siRNA | |||||
|---|---|---|---|---|---|---|
| sl/sl (n=17) |
sl/+ (n=39) |
+/+ (n=19) |
sl/sl (n=4) |
sl/+ (n=24) |
+/+ (n=10) |
|
| TUNEL positivity (%) |
23.1 ± 8.7* |
23.0 ± 9.9 |
20.0 ± 8.7 |
48.4 ± 9.4* |
40.9 ± 11.9 |
42.6 ± 8.1 |
| Ki67 positivity (%) |
22.8 ± 6.2** |
47.7 ± 13.3** |
53.3 ± 12.9** |
22.5 ± 5.9 |
24.6 ± 4.8 |
22.8 ± 7.1 |
| Microvessel density (ves- sels/mm2) |
35.0 ± 14 |
38.0 ± 16 |
41.0 ± 22 |
n. p. | n. p. | n. p. |
n. p.= not performed
p-values for all subgroups of MAT B III vs respective MAT B III-ETAR siRNA < 0.001
p-values for sl/sl versus sl/+ and +/+ < 0.0001
Fig. 2. Morphological characterization of MAT B III-induced local tumors and metastases.

A-C) Immunostaining of local tumors for Ki67, showing a reduced amount of proliferation in sl/sl (A) as compared to sl/+ animals (B) and the rescue effect of the transgene in tg sl/sl (C). D-F) H&E staining of lung sections from sl/sl rats (D), showing normal tissue, and sl/+ rats (E) with multiple metastases, similar to the tg sl/sl animals (F). G-I) Immunostaining of the local tumors for the rat macrophage antigen ED-2, demonstrating lower numbers of infiltrating TAM in sl/sl animals (G) than in sl/+ (H) and tg sl/sl rats (I). Sections of tumors and metastases from +/+ animals are not shown separately, since they did not differ from the sl/+ samples.
There was no sign of lower blood supply as a possible cause of reduced tumor growth, as areas of necrosis were generally rare and not increased in sl/sl animals. Measurement of microvessel density did not yield significant differences (Table 1). Equally, VEGF serum concentrations in the three cohorts were similar (Table 2). IL-10 serum levels were determined by ELISA. IL-10 was detectable in all samples without any significant differences (Table 2).
Table 2.
Cytokine serum concentrations (pg/ml, means ± SD)
| sl/sl (n=12) |
sl/+ (n=34) |
+/+ (n=19) |
|
|---|---|---|---|
| TNF-α | 12.39±5.04* | 28.6±23.84* | 23.31±20.24* |
| VEGF | 451.2±35.03 | 454.0±24.13 | 466.8±32.01 |
| IL-10 | 55.1±18.21 | 62.33±23.15 | 57.38±14.26 |
p-values for sl/sl versus sl/+ and +/+ < 0.05
If ETBR expression by stromal cells is essential for tumor progression, then differences in tumor growth across the genetic subpopulations should disappear upon reconstitution of ETBR-deficiency in transgenic rescue rats. In fact, lower Ki67-expression of tumors in ETBR-deficient sl/sl rats as compared to their heterozygous and wild type littermates could be completely rescued by re-expression of ETBR in the respective transgenic rats (Fig. 1C; Fig. 2C). However, there was still a marginally significant difference between transgenic sl/sl rats and their heterozygous and wild type counterparts with regard to tumor weight (Fig. 1B). As the DβH-linked transgene is originally targeted to the intestine - although having been described also in lung, muscle and other tissues (35) – we investigated whether differential transgene expression may account for these discrepancies. In fact, transgenic ETBR-RNA was not ubiquitously expressed in our animals. It was detectable in the cerebral cortex, bone marrow, spleen, liver and lung, but was absent in the skin and subcutaneous compartment (Fig. 1D).
The ET-1/ ETAR loop regulates tumor cell proliferation and apoptosis independent of stromal ETBR function
As overexpression of ET-1 and ETAR is known to induce an autocrine loop to promote tumor cell growth and invasion, we evaluated the functional contribution of the ETAR in this context. For transient inhibition, the inoculated MAT B III cell suspension additionally contained the ETAR-antagonist clazosentan. For permanent silencing, we used MAT B III cells, where ETAR expression had been abolished by stable transfection of ETAR siRNA (Fig. 3A). Transient ETAR-inhibition had no effect on tumor growth, presumably due to rapid regrowth of the inoculated cells after exspiry of the clazosentan effect (data not shown). MAT B III ETAR siRNA cells, in contrast, proliferated only slowly in vitro and consequently produced smaller tumors in vivo (doubling time 57 vs 33 hours). This was accompanied by a generally lower expression of Ki67 and a higher amount of apoptosis (Table 1). However, there was no evidence that the autocrine ET-1/ ETAR loop in the tumor cells was influenced by the microenvironment. Proliferation and apoptosis of MAT B III-ETAR siRNA cells in vivo did not differ between the genetic subgroups (Table 1) and the generally lower weight of MAT B III-ETAR siRNA-induced tumors was independent of whether ETBR was present or absent in the stromal compartment (Fig. 3B).
Fig. 3. ETAR knock down in MAT B III cells.
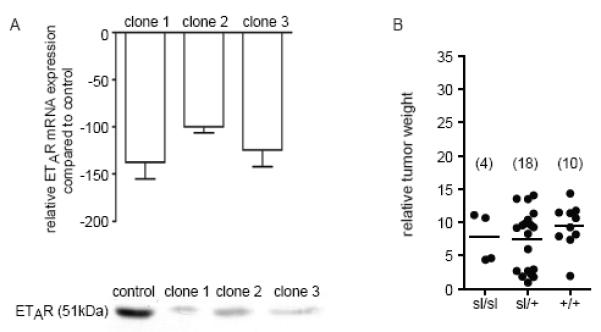
A) Expression of ETAR RNA (real-time RT-PCR, relative expression as compared to the controls) and ETAR protein (Western blot) after stable transfection with the pSilencer 2.1-U6-RNAi plasmid for ETAR. B) ETAR-deficient cells induce smaller tumors independent of the genotype of the stromal compartment. Relative weight of local tumors (normalized to body weight) induced MAT B III ETAR siRNA cells (mixture of all three clones) in the three sl genotypes. Four litters of similar age were analyzed, numbers per genotype are given in brackets.
Stromal ETBR-deficiency reduces metastatic spread
Next we were interested in whether ETBR function in the tumor stroma would influence tumor dissemination. MAT B III cells are highly metastatic. Metastases were found predominantly in the lungs. Other organs were not involved, except in two sl/+ animals with peritoneal spread. Despite their massive tumor load, these animals had only very small local tumors, suggesting accidental intravascular tumor cell injection. They were therefore excluded from evaluation. Metastatic involvement of the lungs was demonstrated predominantly in sl/+ and +/+ animals (Fig. 4A). Thirteen of 21 heterozygous and 14 of 27 wild type rats had pulmonary metastases, in contrast to only 1 of 14 homozygous animals (p = 0.006, Fisher’s exact test). While metastatic disease in sl/+ and +/+ lungs was usually disseminated, metastases in sl/sl lungs, if there were any, could only be found upon evaluation of multiple serial sections (Fig. 2D, E). These effects were completely abolished by restoration of ETBR function. All of the tg sl/sl animals, expressing the transgene, had metastatic disease in the lungs (Fig. 4A; Fig. 2F)
Fig. 4. ETBR-deficiency, metastasis formation and infiltrating TAM.

A) ETBR-deficiency reduces lung metastases. Animals with lung metastases in the three genotypes of sl rats and their transgenic counterparts (* p=0.001 and p=0.004, Fisher’s exact test). B) Endogenous ETBR mRNA expression in peripheral blood monocytes of non-transgenic rats (RT-PCR), showing the deletion transcript in sl/sl cells, the wild type in +/+ cells and both forms in heterozygous cells. The transgene is expressed in the cerebral cortex of all transgenic animals, but is absent in the monocytes (RT-PCR with lamin expression used as control). C) Tumors in sl/sl rats contain fewer TAM. Rate of TAM per high power field (means ± SD) in the three genetic subgroups of sl and tg sl rats, numbers of analyzed animals (from 6 litters) are given in brackets. D) ETBR-deficiency does not induce a general chemotaxis defect. Chemotaxis assays of bone marrow-derived macrophages of the various genetic subgroups using the indicated chemoattractants (means ± SD). E) Total RNA was isolated from CD11b+ selected tumor infiltrating macrophages for real time PCR analysis of MMP-2 and MMP-9 expression. Data is represented as fold induction of mRNA expression compared with wt macrophages, n=10.
Transient ETAR-inhibition by clazosentan did not influence metastasis formation. The effect of permanent silencing could not be analyzed, since tumors derived from MAT B III ETAR siRNA cells grew so slowly that sl/sl rats in these litters had to be euthanized because of progression of Hirschsprung’s disease before development of metastases in any of the genetic subgroups.
Tumors in ETBR-deficient rats contain fewer tumor-associated macrophages
MAT B III cells express ET-1 and -2, which are both chemoattractants for MΦ. Since we have shown that this is mediated via ETBR (33), we asked whether tumors in ETBR-deficient rats would contain lower amounts of infiltrating TAM. Measurement of ETBR mRNA expression in peripheral blood monocytes, the origin of the TAM, confirmed expression of the correct ETBR transcript in wild type and heterozygous monocytes (Fig. 4B). As shown by immunohistochemistry with the MΦ-Marker CD 163, significantly fewer TAM were detectable in tumors from sl/sl than from heterozygous or wild type animals (Fig. 4C; Fig. 2 G, H). Equally, quantitative analysis of the composition of the whole tumor leukocyte infiltrate by flow cytometry yielded a significantly lower content of TAM in sl/sl than in sl/+ or +/+ tumors, while there were no differences in the amount of granulocytes, T and NK cells (Table 3). Again, this effect was completely counteracted by the ETBR transgene in tg sl/sl animals (Fig. 4C; Fig. 2I). Surprisingly, this was not caused by re-expression of a functional ETBR in the monocytes themselves, as the transgene was not detectable in these cells (Fig. 4B).
Table 3.
Flow cytometric analysis of the whole tumor leukocyte infiltrate
| % cells per tumor infiltrate (means ± SD*) | Significance (p) | |||
|---|---|---|---|---|
| sl/sl | sl/+ | +/+ | sl/sl vs sl/+ and +/+ | |
| CD 163(ED2)+ Macrophages |
9.56±7.21 | 22.08±11.27 | 19.11±12.43 | <0.001 |
| CD4+ T-cells | 3.53±1.46 | 3.33±1.85 | 3.57±1.74 | >0.05 |
| CD8+ T-cells | 6.34±3.95 | 8.04±22 | 7.34±27 | >0.05 |
| CD161a+ NK-cells |
3.45±2.23 | 3.32±2.81 | 4.07±3.65 | >0.05 |
| Granulocytes | 15.21±3.75 | 16.23±82 | 18.42±04 | >0.05 |
normalized to tumor weight; student’s t-test
The reduced number of TAM in sl/sl animals was not due to a constitutive lack of monocytoid cells. Peripheral blood counts and cytospins of peritoneal lavages yielded comparable amounts of monocytes and peritoneal MΦ in all genetic subgroups (not shown). Equally, we could not demonstrate a general chemotaxis defect in sl/sl-MΦ. Bone marrow-derived monocytoid cells of all genetic subpopulations were subjected to migration assays towards colony stimulating factor-1 (CSF-1) and the chemokines CCL-2 and -5 (Fig. 4D). Migration rates were identical.
Recently we have shown that MΦ-induced invasion of malignant cells is critically dependent on MΦ-derived TNF-α (11). Moreover, secretion of TNF-α is inducible by ET-1 and vice versa (38, 39). We therefore measured the serum concentrations of TNF-α in tumor-bearing rats (Table 2). TNF-α concentrations were generally low, but still significantly lower in sl/sl than in heterozygous and wild type animals. We also demonstrated earlier that TNF-αwas responsible for MMP upregulation (11). Therefore, we analysed the mRNA expression of MMP-2 and -9 in 106 FACS sorted TAM from tumor bearing animals. MMP-2 and -9 expression was significantly lower in TAM from sl/sl rats compared with TAM from sl/+ or +/+ rats (n=10, Figure 4E).
Discussion
Members of the ET network are often deregulated in cancer cells. As they are also produced by the tumor microenvironment, they are very likely to be involved in interactions between malignant and benign tumor components that influence tumor progression and metastasis.
To better dissect these effects, we chose an animal model, where ETBR expression is modulated exclusively in the tumor stroma, while the tumor cells are ETBR-negative and show overexpression of ETAR and ET-1. There we could demonstrate that the stromal ETBR plays a critical role for malignant progression. Growth and especially metastasis of MAT B III rat mammary adenocarcinoma cells were greatly reduced in homozygous sl/sl rats, constitutively lacking a functional ETBR, as compared to their littermates with normal ETBR expression.
As ETBR inhibition can trigger apoptosis (5), we speculated that smaller tumors might be caused by diminished survival. However, although cell death could be induced by down-regulation of tumor cell ETAR via siRNA, thus confirming their dependency on an autocrine ET-1/ ETAR loop for survival, this was not influenced by presence or absence of stromal ETBR. The amount of apoptotic cells did not differ between the genetic subgroups, irrespective of whether ETAR was overexpressed or downregulated.
In contrast to cell survival, proliferation was significantly influenced by stromal ETBR function, its complete lack resulting in lower proliferation rates in the respective tumors. This seems surprising, since pharmacological ETBR inhibitors, although reported to reduce proliferation of cancer cells in vitro (18) as well as tumor growth in nude mice (23, 40), usually do so on condition that the targeted tumor cells overexpress ETBR. In our model, the tumor cells are ETBR-negative. Hence, ETBRs on stromal cells obviously interact with the malignant cells and modulate their biological behaviour.
As the ET axis is known for its role in the vascular system, it would appear, that stromal endothelial cells are the most likely candidates to influence tumor progression. ETBR-overexpression in primary breast cancers was associated with increased neoangiogenesis (9), while inhibition by specific or dual antagonists diminished vascularisation (23, 41). Paradoxically, injection of the ETBR-inhibitor BQ788 in melanoma xenografts resulted in elevated vessel numbers (5). We found no significant difference in VEGF-production and microvessel density between the genetic subgroups, consistent with the observation that pharmacological inhibition does not necessarily yield the same effects as constitutive deficiency (42).
In many tissues, binding of ETs to ETAR induces vasoconstriction, activation of ETBR the opposite effect (43). As vascular resistance is elevated in sl/sl rats (44), predominance of ETAR in ETBR-deficiency would be expected to shift the vasomotor balance towards vasoconstriction and reduce intratumoral blood flow (45). However, areas of necrosis as a potential sign of insufficient perfusion were not increased in tumors of ETBR-deficient rats.
TAM, an essential part of the stromal immune infiltrate, are powerful promotors of tumor progression. We and others have shown, that TAM induce matrix metalloprotease-mediated invasion and metastasis, which is dependent on TNF-α (11, 46, 47). Consistent with these findings, serum concentrations of TNF-α as well as the amount of infiltrating TAM were significantly lower in ETBR-deficient animals. Neither constitutively lower numbers of monocytoid cells nor a general chemotaxis defect could explain these results.
In correspondence to the lower amount of TAM, metastasis to the lungs was almost completely abolished in ETBR-deficient rats. As a proof of principle, transgenic overexpression of a functional ETBR counteracted these effects. Transgenic rescue rats did not show any difference between the genetic subgroups with regard to lung metastasis and TAM infiltration. The effect on local tumor growth, however, was only partially neutralized, as tumors were still smaller in sl/sl transgenic animals. Since the transgene was detectable in the lungs, but absent in the skin, differing tissue-specific expression levels most probably account for the discordant effect on tumor growth and dissemination. The gene dose has been shown to be crucial for ETBR function (35, 44). Inoculated tumor cells may have grown slowly as long as they were confined to the still ETBR-deficient cutis/subcutis compartment. With increasing recruitment of transgene-positive stromal cells and contact with positive neighbouring tissues, the inhibitory action of the non-functional ETBR on proliferation and invasion was effectively antagonized.
The surprising finding, that TAM infiltration is restored in transgenic animals, although the transgene is not expressed in these cells, points to a role of other cells in the microenvironment, which may influence TNF-α production and/or macrophage chemoattraction. Interestingly, it has recently been shown that endothelial ET-1/ETBR signalling blocks T-cell adhesion and infiltration into tumors in the ID8 ovarian cancer model in mice (48). This was dependent on ICAM-1 and TNF-α. In our model, there was no difference in tumor infiltration by lymphocytes.
In conclusion, while malignant cell survival in our model depends on autocrine stimulation via the ET-1/ETAR loop, growth of the whole tumor and metastasis formation are regulated by ETBR function in the stromal compartment. Lack of functional ETBR in the TAM seems essential, however, contribution of other stromal cells to this effect has to be postulated. This demonstrates that tumor progression can be modulated not only by ET signalling within the malignant cells, but also by exchange of signals between different cell types within the tumor tissue. It may also explain the contradictory results of pharmacological ET receptor antagonists in vivo. Considering the potential use of ETBR inhibitors as clinical anticancer agents, further evaluation of their effects on stromal cells is warranted.
Acknowledgements
We thank George Elia for histological processing of the tumor samples and Derek Davies, Gary Warnes, Ayad Eddaoudi for assistance with cell sorting.
This research was supported by grants from the Medical Research Council (TH), Charitable Foundation of Bart’s and The London (TH).
References
- 1.Levin ER. Endothelins. N Engl J Med. 1995;333:356–63. doi: 10.1056/NEJM199508103330607. [DOI] [PubMed] [Google Scholar]
- 2.Battistini B, Chailler P, D’Orleans-Juste P, Briere N, Sirois P. Growth regulatory properties of endothelins. Peptides. 1993;14:385–99. doi: 10.1016/0196-9781(93)90057-n. [DOI] [PubMed] [Google Scholar]
- 3.Lahav R, Heffner G, Patterson PH. An endothelin receptor B antagonist inhibits growth and induces cell death in human melanoma cells in vitro and in vivo. Proc Natl Acad Sci U S A. 1999;96:11496–500. doi: 10.1073/pnas.96.20.11496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gershon MD. Endothelin and the development of the enteric nervous system. Clin Exp Pharmacol Physiol. 1999;26:985–8. doi: 10.1046/j.1440-1681.1999.03176.x. [DOI] [PubMed] [Google Scholar]
- 5.Lahav R, Suva ML, Rimoldi D, Patterson PH, Stamenkovic I. Endothelin receptor B inhibition triggers apoptosis and enhances angiogenesis in melanomas. Cancer Res. 2004;64:8945–53. doi: 10.1158/0008-5472.CAN-04-1510. [DOI] [PubMed] [Google Scholar]
- 6.Nelson J, Bagnato A, Battistini B, Nisen P. The endothelin axis: emerging role in cancer. Nat Rev Cancer. 2003;3:110–6. doi: 10.1038/nrc990. [DOI] [PubMed] [Google Scholar]
- 7.Grimshaw MJ. Endothelins and hypoxia-inducible factor in cancer. Endocr Relat Cancer. 2007;14:233–44. doi: 10.1677/ERC-07-0057. [DOI] [PubMed] [Google Scholar]
- 8.Wulfing P, Diallo R, Kersting C, et al. Endothelin-1, Endothelin-A- and Endothelin-B-receptor expression in preinvasive and invasive breast disease. Oncol Rep. 2004;11:791–6. [PubMed] [Google Scholar]
- 9.Wulfing P, Kersting C, Tio J, et al. Endothelin-1-, endothelin-A-, and endothelin-B-receptor expression is correlated with vascular endothelial growth factor expression and angiogenesis in breast cancer. Clin Cancer Res. 2004;10:2393–400. doi: 10.1158/1078-0432.ccr-03-0115. [DOI] [PubMed] [Google Scholar]
- 10.Wulfing P, Diallo R, Kersting C, et al. Expression of endothelin-1, endothelin-A, and endothelin-B receptor in human breast cancer and correlation with long-term follow-up. Clin Cancer Res. 2003;9:4125–31. [PubMed] [Google Scholar]
- 11.Hagemann T, Robinson SC, Schulz M, Trumper L, Balkwill FR, Binder C. Enhanced invasiveness of breast cancer cell lines upon co-cultivation with macrophages is due to TNF-{alpha} dependent up-regulation of matrix metalloproteases. Carcinogenesis. 2004;25:1543–9. doi: 10.1093/carcin/bgh146. [DOI] [PubMed] [Google Scholar]
- 12.Asham E, Shankar A, Loizidou M, et al. Increased endothelin-1 in colorectal cancer and reduction of tumour growth by ET(A) receptor antagonism. Br J Cancer. 2001;85:1759–63. doi: 10.1054/bjoc.2001.2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bagnato A, Rosano L. Epithelial-mesenchymal transition in ovarian cancer progression: a crucial role for the endothelin axis. Cells Tissues Organs. 2007;185:85–94. doi: 10.1159/000101307. [DOI] [PubMed] [Google Scholar]
- 14.Ali H, Dashwood M, Dawas K, Loizidou M, Savage F, Taylor I. Endothelin receptor expression in colorectal cancer. J Cardiovasc Pharmacol. 2000;36:S69–71. doi: 10.1097/00005344-200036051-00023. [DOI] [PubMed] [Google Scholar]
- 15.Berry P, Burchill S. Endothelins may modulate invasion and proliferation of Ewing’s sarcoma and neuroblastoma. Clin Sci (Lond) 2002;103(Suppl 48):322S–6S. doi: 10.1042/CS103S322S. [DOI] [PubMed] [Google Scholar]
- 16.Nelson JB, Lee WH, Nguyen SH, et al. Methylation of the 5′ CpG island of the endothelin B receptor gene is common in human prostate cancer. Cancer Res. 1997;57:35–7. [PubMed] [Google Scholar]
- 17.Ahmed SI, Thompson J, Coulson JM, Woll PJ. Studies on the expression of endothelin, its receptor subtypes, and converting enzymes in lung cancer and in human bronchial epithelium. Am J Respir Cell Mol Biol. 2000;22:422–31. doi: 10.1165/ajrcmb.22.4.3795. [DOI] [PubMed] [Google Scholar]
- 18.Awano S, Dawson LA, Hunter AR, Turner AJ, Usmani BA. Endothelin system in oral squamous carcinoma cells: specific siRNA targeting of ECE-1 blocks cell proliferation. Int J Cancer. 2006;118:1645–52. doi: 10.1002/ijc.21525. [DOI] [PubMed] [Google Scholar]
- 19.Bittner M, Meltzer P, Chen Y, et al. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature. 2000;406:536–40. doi: 10.1038/35020115. [DOI] [PubMed] [Google Scholar]
- 20.Demunter A, De Wolf-Peeters C, Degreef H, Stas M, van den Oord JJ. Expression of the endothelin-B receptor in pigment cell lesions of the skin. Evidence for its role as tumor progression marker in malignant melanoma. Virchows Arch. 2001;438:485–91. doi: 10.1007/s004280000362. [DOI] [PubMed] [Google Scholar]
- 21.Smollich M, Gotte M, Kersting C, Fischgrabe J, Kiesel L, Wulfing P. Selective ETAR antagonist atrasentan inhibits hypoxia-induced breast cancer cell invasion. Breast Cancer Research and Treatment. 2008;108:175–82. doi: 10.1007/s10549-007-9589-5. [DOI] [PubMed] [Google Scholar]
- 22.Rosano L, Spinella F, Di Castro V, et al. Integrin-linked kinase functions as a downstream mediator of endothelin-1 to promote invasive behavior in ovarian carcinoma. Mol Cancer Ther. 2006;5:833–42. doi: 10.1158/1535-7163.MCT-05-0523. [DOI] [PubMed] [Google Scholar]
- 23.Spinella F, Rosano L, Di Castro V, et al. Endothelin-1 and endothelin-3 promote invasive behavior via hypoxia-inducible factor-1alpha in human melanoma cells. Cancer Res. 2007;67:1725–34. doi: 10.1158/0008-5472.CAN-06-2606. [DOI] [PubMed] [Google Scholar]
- 24.Kefford R, Beith JM, Van Hazel GA, et al. A phase II study of bosentan, a dual endothelin receptor antagonist, as monotherapy in patients with stage IV metastatic melanoma. Invest New Drugs. 2007;25:247–52. doi: 10.1007/s10637-006-9014-7. [DOI] [PubMed] [Google Scholar]
- 25.Carducci MA, Saad F, Abrahamsson PA, et al. A phase 3 randomized controlled trial of the efficacy and safety of atrasentan in men with metastatic hormone-refractory prostate cancer. Cancer. 2007;110:1959–66. doi: 10.1002/cncr.22996. [DOI] [PubMed] [Google Scholar]
- 26.Chiappori AA, Haura E, Rodriguez FA, et al. Phase I/II study of atrasentan, an endothelin A receptor antagonist, in combination with paclitaxel and carboplatin as first-line therapy in advanced non-small cell lung cancer. Clin Cancer Res. 2008;14:1464–9. doi: 10.1158/1078-0432.CCR-07-1508. [DOI] [PubMed] [Google Scholar]
- 27.Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7:211–7. doi: 10.1016/j.ccr.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 28.Egidy G, Juillerat-Jeanneret L, Jeannin JF, Korth P, Bosman FT, Pinet F. Modulation of human colon tumor-stromal interactions by the endothelin system. Am J Pathol. 2000;157:1863–74. doi: 10.1016/S0002-9440(10)64825-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ehrenreich H, Anderson RW, Fox CH, et al. Endothelins, peptides with potent vasoactive properties, are produced by human macrophages. J Exp Med. 1990;172:1741–8. doi: 10.1084/jem.172.6.1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–6. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 31.Grimshaw M, Hagemann T, Ayhan A, Gillett C, Binder C, Balkwill FR. A role for endothelin-2 and its receptors in breast tumor cell invasion. Cancer Res. 2004;64:2461–8. doi: 10.1158/0008-5472.can-03-1069. [DOI] [PubMed] [Google Scholar]
- 32.Hagemann T, Binder C, Binder L, Pukrop T, Trumper L, Grimshaw MJ. Expression of endothelins and their receptors promotes an invasive phenotype of breast tumor cells but is insufficient to induce invasion in benign cells. DNA Cell Biol. 2005;24:766–76. doi: 10.1089/dna.2005.24.766. [DOI] [PubMed] [Google Scholar]
- 33.Grimshaw MJ, Wilson JL, Balkwill FR. Endothelin-2 is a macrophage chemoattractant: implications for macrophage distribution in tumors. Eur J Immunol. 2002;32:2393–400. doi: 10.1002/1521-4141(200209)32:9<2393::AID-IMMU2393>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 34.Gariepy CE, Cass DT, Yanagisawa M. Null mutation of endothelin receptor type B gene in spotting lethal rats causes aganglionic megacolon and white coat color. Proc Natl Acad Sci U S A. 1996;93:867–72. doi: 10.1073/pnas.93.2.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gariepy CE, Williams SC, Richardson JA, Hammer RE, Yanagisawa M. Transgenic expression of the endothelin-B receptor prevents congenital intestinal aganglionosis in a rat model of Hirschsprung disease. J Clin Invest. 1998;102:1092–101. doi: 10.1172/JCI3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Riechers CC, Knabe W, Siren AL, Gariepy CE, Yanagisawa M, Ehrenreich H. Endothelin B receptor deficient transgenic rescue rats: a rescue phenomenon in the brain. Neuroscience. 2004;124:719–23. doi: 10.1016/j.neuroscience.2003.10.023. [DOI] [PubMed] [Google Scholar]
- 37.Ehrenreich H, Oldenburg J, Hasselblatt M, et al. Endothelin B receptor-deficient rats as a subtraction model to study the cerebral endothelin system. Neuroscience. 1999;91:1067–75. doi: 10.1016/s0306-4522(98)00663-0. [DOI] [PubMed] [Google Scholar]
- 38.Ahn GY, Butt KI, Jindo T, Yaguchi H, Tsuboi R, Ogawa H. The expression of endothelin-1 and its binding sites in mouse skin increased after ultraviolet B irradiation or local injection of tumor necrosis factor alpha. J Dermatol. 1998;25:78–84. doi: 10.1111/j.1346-8138.1998.tb02354.x. [DOI] [PubMed] [Google Scholar]
- 39.Matsushima H, Yamada N, Matsue H, Shimada S. The effects of endothelin-1 on degranulation, cytokine, and growth factor production by skin-derived mast cells. Eur J Immunol. 2004;34:1910–9. doi: 10.1002/eji.200424912. [DOI] [PubMed] [Google Scholar]
- 40.Bagnato A, Rosano L, Spinella F, Di Castro V, Tecce R, Natali PG. Endothelin B receptor blockade inhibits dynamics of cell interactions and communications in melanoma cell progression. Cancer Res. 2004;64:1436–43. doi: 10.1158/0008-5472.can-03-2344. [DOI] [PubMed] [Google Scholar]
- 41.Dreau D, Karaa A, Culberson C, Wyan H, McKillop IH, Clemens MG. Bosentan inhibits tumor vascularization and bone metastasis in an immunocompetent skin-fold chamber model of breast carcinoma cell metastasis. Clin Exp Metastasis. 2006;23:41–53. doi: 10.1007/s10585-006-9016-z. [DOI] [PubMed] [Google Scholar]
- 42.Pollock DM. Contrasting pharmacological ETB receptor blockade with genetic ETB deficiency in renal responses to big ET-1. Physiol Genomics. 2001;6:39–43. doi: 10.1152/physiolgenomics.2001.6.1.39. [DOI] [PubMed] [Google Scholar]
- 43.Ehrenreich H, Schilling L. New developments in the understanding of cerebral vasoregulation and vasospasm: the endothelin-nitric oxide network. Cleve Clin J Med. 1995;62:105–16. doi: 10.3949/ccjm.62.2.105. [DOI] [PubMed] [Google Scholar]
- 44.Dembowski C, Hofmann P, Koch T, et al. Phenotype, intestinal morphology, and survival of homozygous and heterozygous endothelin B receptor--deficient (spotting lethal) rats. J Pediatr Surg. 2000;35:480–8. doi: 10.1016/s0022-3468(00)90218-5. [DOI] [PubMed] [Google Scholar]
- 45.Rajeshkumar NV, Rai A, Gulati A. Endothelin B receptor agonist, IRL 1620, enhances the anti-tumor efficacy of paclitaxel in breast tumor rats. Breast Cancer Research and Treatment. 2005;94:237–47. doi: 10.1007/s10549-005-9000-3. [DOI] [PubMed] [Google Scholar]
- 46.Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med. 2001;193:727–40. doi: 10.1084/jem.193.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moore RJ, Owens DM, Stamp G, et al. Mice deficient in tumor necrosis factor-alpha are resistant to skin carcinogenesis. Nat Med. 1999;5:828–31. doi: 10.1038/10552. [DOI] [PubMed] [Google Scholar]
- 48.Buckanovich RJ, Facciabene A, Kim S, et al. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med. 2008;14:28–36. doi: 10.1038/nm1699. [DOI] [PubMed] [Google Scholar]