

Abstract
Previous study has shown that colitis-induced increases in calcitonin gene-related peptide (CGRP) immunoreactivity in bladder afferent neurons result in sensory cross-sensitization. To further determine the effects of colitis on CGRP expression in neurons other than bladder afferents, we examined and compared the levels of CGRP mRNA and immunoreactivity in the lumbosacral dorsal root ganglia (DRG) and spinal cord before and during colitis in rats. We also examined the changes in CGRP immunoreactivity in colonic afferent neurons during colitis. Results showed increases in CGRP mRNA levels in L1 (2.5-fold, p<0.05) and S1 DRG (1.9-2.4-fold, p<0.05). However, there were no changes in CGRP mRNA levels in L1 and S1 spinal cord during colitis. CGRP protein was significantly increased in L1 (2.5-fold increase, p<0.05) but decreased in S1 (50% decrease, p<0.05) colonic afferent neurons, which may reflect CGRP release from these neurons during colitis. In L1 spinal cord, colitis caused increases in the number of CGRP nerve fibers in the deep lamina region extending to the gray commissure where the number of phospho-Akt neurons was also increased. In S1 spinal cord, colitis caused the increases in the intensity of CGRP fibers in the regions of dorsolateral tract, and caused the increases in the level of phospho-Akt in the superficial dorsal horn of the spinal cord. In spinal cord slice culture, exogenous CGRP increased the phosphorylation level of Akt but not the phosphorylation level of extracellular-signal regulated kinase ERK1/2 even though our previously studies showed that colitis increased the phosphorylation level of ERK1/2 in L1 and S1 spinal cord. These results suggest that CGRP is synthesized in the DRG and may transport to the spinal cord where initiates signal transduction during colitis.
Keywords: colitis, CGRP, Akt, DRG, spinal cord
INTRODUCTION
Patients with active ulcerative colitis often experience pain, urgency, and incontinence (Bernstein et al., 1996; Drewes et al., 2006). Hyperalgesia is also observed in the rodents with experimental colitis induced by chemicals such as tri-nitrobenzene sulfonic acid (TNBS), zymosan, acetic acid, mustard oil, or dextran sulphate sodium (DSS) (Burton and Gebhart, 1995; Coutinho et al., 1996; Larsson et al., 2006). The mechanism underlying the development and progression of colitis, and related hyperalgesia is unknown, but may be attributable, in part, to the neurochemical and electrophysiological changes in the primary afferent pathways including dorsal root ganglia (DRG) and spinal cord, which in return affects the efferent output and the development of inflammation (Okajima and Harada, 2006). The changes in the level of neurotransmitters, neurotrophins, or inflammatory cytokines in both peripheral tissue and the afferents in DRG and spinal cord during colitis may further lead to intracellular signaling kinase activation and gene transcription in the primary sensory pathways (Honore et al., 2002; Gebhart et al., 2002; Landau et al., 2007).
Calcitonin gene-related peptide (CGRP) is an important nociceptive marker and plays a major role in mediating hypersensitivity in many systems (Smith et al., 1992; Nahin and Byers, 1994; Zhang et al., 2001; Winston et al., 2003), and contributes to mucosal integrity and ulcer healing (Ohno et al., 2008). Following peripheral inflammation, more neurons had detectable CGRP expression in the innervating DRG, and mice lacking α-CGRP failed to develop secondary hyperalgesia after inflammation (Smith et al., 1992; Zhang et al., 2001). Administration of a CGRP receptor antagonist greatly attenuated the visceromoter reflex in rats with colonic inflammation (Plourde et al., 1997) and reduced TNBS-induced colonic hypersensitivity (Delafoy et al., 2006), suggesting a prominent role of CGRP in colitis. Peripheral expression of CGRP has been well characterized (Evangelista and Tramontana, 1993; Miampamba and Sharkey, 1998; Dömötör et al., 2005; Clifton et al., 2007; Grider and Piland, 2007). However, the expression levels of CGRP protein in the lumbosacral DRG and spinal cord during visceral and peripheral inflammation were either increased or decreased (Smith et al., 1992; Hanesch et al., 1993; Galeazza et al., 1995; Traub et al., 1999; Vizzard, 2001). In the current study, we examined the CGRP mRNA levels and compared that with the changes in CGRP protein levels in lumbosacral DRG and spinal cord during colitis in order to identify if CGRP synthesis is affected by inflammation. Our previous studies (Qiao and Grider, 2007a) with neural retrograde tracing demonstrate that primary afferent neurons innervating the distal colon are distributed into two anatomic regions, L1- L2 (lumbar splanchnic afferents) and L6-S1 (sacral pelvic afferents) DRG, in rats. In the present study, spinal segmental levels from these two peaks are the primary regions we examined. We characterized the changes in CGRP mRNA and immunoreactivity in colonic afferent neurons from lumbosacral DRG and spinal cord at 3 and 7 days of colitis.
Previous studies show that activation of mitogen-activated protein kinase (MAPK, also called extracellular signal-regulated protein kinase ERK) and Akt (also called protein kinase B, a serine/threonine-specific protein kinase) by inflammatory and neuronal mediators in primary sensory and secondary order dorsal horn neurons participates in the generation and maintenance of inflammatory pain (Zhuang et al., 2004; Carrasquillo and Gereau, 2007). Recent studies demonstrated that CGRP is capable of activating PI-3 kinase/Akt and MAPK pathways in cultured cells and tissues (Parameswaran et al., 2000). To further characterize the role of central expression of CGRP in colitis, we examined the changes in Akt phosphorylation (activation) in lumbosacral spinal cord during colitis, and the effect of CGRP on Akt activity in the spinal cord. Our recent studies (Qiao et al., 2008) demonstrated that ERK1/2 was also activated in L1 and S1 spinal cord. Therefore, we further examined the effect of CGRP on ERK1/2 activation (phosphorylation) and compared with the Akt activation in the spinal cord. The results indicate that CGRP is likely to play a role in activating Akt but not ERK1/2 in the spinal cord during colitis.
Part of the data has been reported as an abstract form in the Digestive Disease Week Annual Meeting (Qiao and Grider 2005; Qiao and Grider 2007b).
MATERIALS AND METHODS
Animals and Reagents
Adult male Sprague-Dawley rats (150-200 g) (Harlan Sprague Dawley, Inc., Indianapolis, IN) were divided into experimental (colitis induced by intracolonic instillation of TNBS in 50 % EtOH) and control groups (intracolonic instillation of saline; or intracolonic instillation of 50 % EtOH). Within these groups, some of the animals were injected with neuronal retrograde tracing dye to the smooth muscle layer of the distal colon to label colonic afferent neurons in the dorsal root ganglia (see below for dye injection). All experimental protocols involving animal use in this study were approved by the Institutional Animal Care and Use Committee in Virginia Commonwealth University. Animal care was in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) and National Institutes of Health guidelines.
Chemicals used in this experiment were purchased from Sigma ImmunoChemicals (St. Louis, MO).
Retrograde Labeling
Under anesthesia (2.5 % isoflurane, SurgiVet, Smiths Medical PM, Inc. Waukesha, WI), the rat distal colon was exposed under a sterile environment with a lower abdominal incision. Neuronal tracing agent dicarbocyanine dye 1,1’dioleyl-3,3,3’3’-tetramethylindocarbocyanine (DiI, a solution of 25 mg in 50 % methanol; Molecular Probes, Eugene, OR) was injected into 10 sites (4 μL per site) into the muscle wall of the descending colon (5-7 cm proximal to the external anal sphincter) to label colonic afferent neurons innervating this area. A sterilized Hamilton syringe (10 μL size) was used for injection. To prevent labeling of adjacent tissues, the needle was left in place for 30 sec after each injection and a cotton swab was held close to the injection site to wipe off any excess dye that might leak from the needle tip during the needle withdrawal. In this manner, no visible leakage of the dye was observed after each injection. Injections into the lumen, major blood vessels, or overlying fascial layers were avoided. The incision was closed with 4-0 sutures. The rats were allowed to survive until the harvest of the tissues. We also checked the leakage of the dye by examining the tissues surrounding the colon after euthanasia of the rats. No contamination of dye to other tissues was noticed. The numbers of neurons labeled by DiI in each segment-matched DRG showed no significant difference between EtOH-treated (control) animal and TNBS-treated colitic animal (Qiao and Grider, 2007a).
TNBS-induced Colitis
To induce inflammation in the distal colon, fasted rats were anesthetized and TNBS was instilled into the lumen of the colon at a dose of 90 mg/kg (1.5 mL/kg of 60 mg/mL solution in 50 % EtOH) through a syringe-attached polyethylene catheter via the rectum 6 cm proximal to the anus. Animals that received an equal volume of 50 % EtOH served as control. All colonic instillations were performed under isoflurane (2 %) anesthesia. To ensure exposure of the distal colon to TNBS or EtOH, rats were held head-down by lifting up the tail for 2 min. In some of the animals, colitis was induced four days after tracing dye injection for a proper transport of these dyes.
According to a previously validated grading scheme for colitis (Morris et al., 1989), we assessed the severity of colitis and only the rats with severe interstitial edema were used for the current study. In these rats, TNBS induced extensive ulceration, hyperemia, adhesion, edema, and changes in mucosal architecture. Two to 3-fold increase in the thickness of colonic muscle layer was also noticed in animals with colitis by measurement of the transverse section of the distal colon.
Perfusion and Tissue Harvesting
Intracardiac perfusion was performed for euthanasia of animals. Under anesthesia (3-4 % isoflurane), animals were euthanized via perfusion first with oxygenated Krebs buffer (pH 7.4) (95 % O2, 5 % CO2) followed by 4 % paraformaldehyde. After perfusion, the spinal cord and both sides of the DRG were quickly removed and postfixed for 6 h. Tissue was then rinsed in phosphate buffered saline (0.1 M PBS, pH 7.4) and placed in ascending concentrations of sucrose (20 %) for cryoprotection. DRG and spinal cord from level L1, L6 and S1 were identified (Qiao and Vizzard, 2002; Qiao and Grider 2007a) and the DRG was sectioned parasagitally at a thickness of 20 μm. Spinal cord segments were sectioned transversely at a thickness of 30 μm. Tissues from control and experimental animals were handled in an identical manner.
Immunohistochemistry
DRG sections were immunostained using an on-slide technique. Spinal cord sections were immunostained by free-floating method. Generally, sections were incubated with blocking solution containing 3 % normal donkey serum (Jackson ImmunoResearch, West Grove, PA) in PBST (0.3 % Triton X-100 in 0.1 M PBS, pH 7.4) for 30 min followed by rabbit anti-CGRP antibody (1:1000, Chemicon international, Temecula, CA, currently Millipore), or mouse anti-phospho-Akt antibody (1:400, Cell Signaling Technology Inc. Danvers, MA) overnight at 4 °C. After rinsing (3 × 10 min with 0.1 M PBS), tissues were incubated with fluorescence-conjugated species-specific secondary antibody Alexa 594 (1:500, Molecular Probes, Eugene, OR) for 2 hours at room temperature. Following washing, the slides were coverslipped with Citifluor (Citifluor Ltd., London). For sections containing DiI, Triton X-100 in all buffers used was substituted by 0.3 % Tween-20 for the prevention of DiI fading. The background staining level was evaluated with staining in the absence of primary or secondary antibody. The specificity of CGRP antibody was determined with CGRP pre-absorption assay (Qiao and Grider, 2007a). Some of the sections were counter-stained with YoYo-1 (1:10,000; Invitrogen, Carlsbad, CA).
DRG cells with visible nucleus were counted with a Nikon fluorescent photomicroscope. CGRP cell profiles were counted in 6 to 10 sections randomly chosen from each DRG (L1, L6, and S1) examined. The cell profile counts were corrected with Abercrombie’s formula (1946) described recently by Guillery (2002) and expressed as mean ± SE for n animals. Specifically, each count was corrected by a fractional factor T/T+h (where T = section thickness of 20 μm, h = 10. 3 μm, an average diameter of the nucleus of CGRP neurons). Within the specific segmental level of DRG (such as L1 DRG), the sections of similar size (i.e., L1, ~ 2 mm2; L6, ~ 2 mm2; and S1, ~ 1 mm2) were chosen with the microscope built-in grids and all the positive cells were counted in the sections and expressed as number of cells per section. Between different segmental levels of the DRG (such as between L1 and L6 DRG), we did not normalize the results according to the size of the sections; therefore, we compared the difference between TNBS-treated and untreated animals for a specific segmental level of DRG. No comparison was made between different segmental levels of the DRG due to the size discrepancy at different spinal levels. We also counted the total number of neurons stained by YoYo-1, and calculated the percentage of DRG neurons expressing CGRP. For double staining analysis, cells expressing multiple dyes were counted simultaneously. The percentage of colonic afferent neurons (DiI labeled) expressing CGRP was presented as mean ± SE.
The fluorescent images of the DRG and spinal cord sections were converted to a gray scale that ranges in intensity from 0 (black) to 255 (white) for the purpose of densitometry. For spinal cord scanning, the same number of standard sized rectangles was overlaid on the area of interest (i.e., superficial dorsal horn in this study) for each spinal section, with one rectangle chosen from the background staining area in the spinal cord for subtraction. Intensity measured within the rectangles was averaged as one point. For DRG scanning, a cell-sized circle was made to measure the positively stained CGRP neurons, or the neurons having background level of staining for subtraction. Ten neurons per section were randomly chosen for analysis.
Protein Extraction
Freshly dissected spinal cord segments were homogenized in solubilization buffer containing 50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1 % Triton X-100, 100 mM NaF supplemented with protease inhibitor cocktail (Sigma, P8340) and phosphatase inhibitor cocktail 1 (Sigma, P2850). The homogenate was centrifuged at 20,200 g for 10 min at 4 °C, and the supernatant was removed to a fresh tube for further analysis. The protein concentration was determined using Bio-Rad DC protein assay kit (Bio-Rad Laboratories, Inc., Hercules, CA).
Western Blot
Proteins were separated on a 10 % SDS-PAGE gel and transferred to a nitrocellulose membrane. The membrane was blocked with 5 % milk in Tris-buffered saline for 1 hour and then incubated with phospho-ERK1/2 (1:1000, Cell Signaling Technology Inc. Danvers, MA) or phospho-Akt (1:1000, Cell Signaling Technology Inc. Danvers, MA) antibody followed by horseradish peroxidase-conjugated secondary antibody. For internal loading control, the same membrane was stripped and re-probed with antibody against the non-phosphorylation form of ERK1/2 (1:1000, Cell Signaling Technology Inc. Danvers, MA) or Akt (1:1000, Cell Signaling Technology Inc. Danvers, MA). The immunoreactive bands were detected by enhanced chemiluminescence and the densitometric quantification of immunoreactive bands was performed using the software FluorChem 8800 (Alpha Innotech, San Leabdro, CA). The expression level of the target protein in control animal from each independent experiment was considered as 1, and the relative expression level of the target protein in experimental animals was adjusted as a ratio to its internal loading control in each independent experiment. For phospho-ERK1/2, the p42 and p44 bands were analyzed together.
RNA Extraction and Quantitative Real-time PCR
Total RNA was extracted using a RNA extraction kit RNAqueous (Ambion, TX). RNA concentration was determined spectrophotometrically. cDNA was synthesized using Cloned AMV First-Strand Synthesis Kit (Invitrogen, Carlsbad, CA) with random hexamers. Following reverse transcription, quantitative real-time PCR was performed for CGRP with a Taqman probe mixed with PCR Master-Mix for 40 cycles (95 °C for 15 sec, 60 °C for 1 min) on a 7300 real-time PCR system (Applied Biosystems, Foster City, CA). Quantitative real-time PCR of the same sample was performed for β-actin expression as internal control for normalization. The changes in mRNA levels of the test gene were calculated with ΔΔCt (change in the cycle threshold) method using β-actin level in the same sample as a normalizer. The expression level of target mRNA in the control (50 % EtOH-treated) animal from each independent experiment was considered as 1, and the relative expression level of target mRNA in experimental animals was adjusted as a ratio to its control in each independent experiment and expressed as fold changes (2ΔΔCt-fold).
Culture of Spinal Cord Slices
Spinal cord segment L6-S1 was dissected from naïve animals, and transversely sectioned at a thickness of 250 μm with a tissue sectioner. The sections were randomly divided into several cell culture wells containing Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 200 units/mL penicillin, 200 mg/mL streptomycin, and 100 mg/mL gentamycin and cultured for 4-6 hr. CGRP (250 nM) was added to the culture medium 5, 20, 40, 90 min prior to the collection of all slices (CGRP-treated or untreated) for assay. All cultures were maintained in a 10 % CO2 environment at 37 °C.
Statistical Analysis
The results from each study were presented as mean ± SE for n animals. Comparison between control and experimental groups was made by using one-way ANOVA followed by Dunnett’s test. Differences between means at a level of p≤0.05 were considered to be significant.
RESULTS
Colitis-induced changes in CGRP expression in lumbosacral DRG
CGRP immunoreactivity showed differential changes in lumbosacral DRG at 7 days of colitis (n=5) when compared to that from vehicle-treated control animals (n=7). In L1 DRG, there were significant increases in the number of cells immunoreactive for CGRP (Fig. 1A: 1.9-fold increase, p<0.05) and the percentage of DRG neurons expressing CGRP (control: 10.4 % vs TNBS: 21.8 %) when compared to control; colitis did not cause changes in CGRP profile in L6 DRG (Fig. 1B); however, significant decreases in the number of DRG neurons immunoreactive for CGRP (Fig. 1C: 50 % decrease, p<0.05) and the percentage of DRG neurons expressing CGRP (control: 15.3 % vs TNBS: 8.4 %) were detected in S1 DRG during colitis. Fluorescent densitometry analysis showed that there were no significant changes in the average CGRP immunoreactive density of individual neurons in L1 and S1 DRG from control and TNBS-treated animals.
Figure 1. Alterations of CGRP mRNA and protein in lumbosacral dorsal root ganglia during colitis.

TNBS colitis (n=5) causes increases in the number of CGRP immunoreactive cells in L1 DRG (A); no changes in L6 DRG (B) and decreases in S1 DRG (C) when compared to non-colitic control animals (n=7). TNBS colitis causes a time-dependent change in CGRP mRNA in L1 DRG (increases at 3 days, no changes at 7 days of colitis), L6 DRG (no changes at either 3 or 7 days of colitis), and S1 DRG (increases at both 3 and 7 days of colitis). *, p<0.05 vs control; #, p<0.05 vs TNBS 3 days.
To examine if the changes in CGRP protein in DRG was due to parallel changes in CGRP transcript during colitis, we examined the CGRP mRNA level in L1, L6 and S1 DRG with quantitative real-time PCR (n=4-5 animals for each treatment). Results showed no change in CGRP mRNA level in L1 DRG at 7 days of colitis, a time point when CGRP protein was increased (Fig. 1A). We then examined if CGRP mRNA was produced at an earlier time point in L1 DRG. Results showed that the level of CGRP mRNA in L1 DRG was significantly increased (2.5-fold, p<0.05) at 3 days of colitis when compared to that from vehicle-treated control animals (Fig. 1D), suggesting that increased CGRP protein level was due to increased CGRP transcription in L1 DRG. In L6 DRG (Fig. 1E), there were no significant differences in the level of CGRP mRNA at any time points examined. In S1 DRG (Fig. 1F), CGRP mRNA level was increased at both 3 (2.4-fold, p<0.05) and 7 days (1.9-fold, p<0.05) of colitis when compared to that from control animal, but showed a time-dependent declination (7 days vs 3 days, p<0.05).
CGRP immunoreactivity in colonic afferent neurons before and during colitis
CGRP immunoreactivity was examined in DiI-labeled colonic afferent neurons (Fig. 2) from animals with 7 days of colitis (n=7) or without colitis (n=5). In the DRG examined, a subpopulation of colonic afferent neurons (Fig. 2A, red cells labeled with DiI) expressed CGRP immunoreactivity (Fig. 2B and C, cells indicated with white arrows). Not all colonic afferent neurons expressed CGRP (Fig. 2A-C, cells indicated with yellow arrow) or all CGRP-immunoreactive neurons were colonic afferent neurons (Fig. 3A-C, cell indicated with purple arrow). In control animals, ~ 30-50 % of DiI-labeled cells expressed CGRP (specifically in L1 DRG: 32.6 ± 4.5 %, L6 DRG: 35.1 ± 2.4 % and S1 DRG: 54.2 ± 3.0 %). After TNBS-induced colitis, there were no significant changes in the percentage of DiI-labeled cells expressing CGRP in L1 or L6 DRG when compared to the matched segments from control animals (Fig. 2D), however, colitis caused a significant decrease in the percentage of DiI-labeled cells expressing CGRP in S1 DRG (50 % decrease, p<0.05) when compared to control (Fig. 2D). For this set of study, intracolonic instillation of 50 % EtOH (vehicle control) had no effects on CGRP expression in colonic afferent neurons when compared to saline, thus, data from these two groups were combined and served as control to TNBS treatment.
Figure 2. Distribution of CGRP immunoreactivity in colonic afferent neurons during colitis.

The distribution of CGRP (B, green cells, white and purple arrows) in colonic afferent neurons (A, DiI-labeled red cells, white and yellow arrows) was characterized by examining the percentage of DiI-labeled neurons expressing CGRP. In the merged image (C), colonic afferent neurons that contain CGRP are indicated with white arrows. Histogram (D) shows that the percentage of colonic afferent neurons expressing CGRP is decreased in S1 DRG following colitis (n = 7) compared to control (n = 5). There are no changes in the percentage of colonic afferent neurons expressing CGRP in L1 and L6 DRG. Sample photomicrographs show S1 DRG from control animal. Calibration bar = 80 μm. *, p<0.05.
Figure 3. CGRP mRNA expression in lumbosacral spinal cord before and during colitis.

Quantitative real-time PCR showed no changes in the level of CGRP mRNA in L1 and S1 spinal cord at both 3 and 7 days of colitis when compared to control (n= 3 to 5).
CGRP expression in lumbosacral spinal cord before and during colitis
Colitis may also affect CGRP expression in the lumbosacral spinal cord since sensory afferent axon terminates at the region of spinal superficial dorsal horn. CGRP immunoreactivity showed either increase or decrease in L1 and S1 spinal superficial dorsal horn, deeper laminae, and the intermediolateral area at 7 days of colitis (see Fig. 4), however, these changes were not due to changes in CGRP mRNA level (Fig. 3) because the level of CGRP mRNA did not alter in L1 and S1 spinal cord at either 7 days of colitis or 3 days of colitis when 3 to 5 animals in each treatment were analyzed.
Figure 4. CGRP immunoreactivity in L1 and S1 spinal cord before and during colitis.

CGRP immunoreactivity (IR) was mainly present in the superficial laminae (I-II) of the dorsal horn in both control and colitic animals (* in a, b, g, and i). In L1 spinal cord, colitis was associated with the increases in the density of CGRP-IR at the dorsal horn region (compare b to a, *), increases in the density of CGRP-IR fibers extending toward the central commissure (compare d to c, arrows), and increases in the number of CGRP neurons in the area of intermediolateral cell column (compare f to e, arrow). In S1 spinal cord, colitis causes decreases in the density of CGRP-IR in dorsal horn (compare i to h, *), but increases in CGRP-IR fibers in a location resembling the Lissauer’s tract. Dotted lines in a and b indicate the borders between gray and white matters in the spinal cord. Calibration bar = 200 μm in a, b; 80 μm in c, e, d, and f; 160 μm in h and i.
In L1 spinal cord, CGRP immunoreactivity was mainly expressed at the superficial dorsal horn laminae I and II (Fig. 4a,b). With colitis, significant increases in CGRP immunoreactivity were detected at dorsal horn region when compared to that from control animals (Fig. 4a,b and 5). In control animals, weak CGRP immunoreactive fibers were present in the deeper laminae region (Fig. 4c), and no apparent CGRP staining was present in the neurons of the intermediolateral cell column (Fig. 4e) in L1 spinal cord. With colitis, strong CGRP nerve fibers were present in the deep laminae region extending to the gray commissure (Fig. 4d, arrows). There were also increases in CGRP immunoreactivity in the area of intermediolateral cell column during colitis (compare f to e, arrow).
Figure 5. Changes in the density of the CGRP immunoreactivity in L1 and S1 spinal dorsal horn during colitis.

Histogram shows increases in L1 and decreases in S1 spinal CGRP immunoreactivity during 7 days of colitis when compared to control. n= 4-5 animals for each treatment. *, p<0.05.
In S1 spinal cord, CGRP immunoreactivity was mainly expressed at the superficial dorsal horn laminae I and II (Fig. 4g,h,i). With colitis, no changes in CGRP immunoreactivity were observed in lamina I but a significant decrease in lamina II when compared to that from control animals (Fig. 4h,i and 5). In addition, strong CGRP fiber bundles (Fig. 4i, arrows) were present along the lateral edge of the superficial dorsal horn extending to the region of sacral parasympathetic nucleus (SPN). These CGRP fibers were not present in the control animals (Fig. 4h).
Effects of CGRP on ERK1/2 and Akt phosphorylation in cultured spinal cord slices
We previously reported that the level of phospho-ERK1/2 was increased in L1 and S1 spinal cord during colitis, and that exogenous BDNF increased ERK1/2 activation in the cultured spinal cord slices within 30 min of incubation (Qiao et al., 2008). In the present study, we further examined whether CGRP was also able to activate ERK1/2 in the cultured slices. Results from three independent experiments showed that exogenous CGRP (250 nM) was unable to activate ERK1/2 within 90 min of incubation (Fig. 6A, B), however, CGRP activated Akt in the same preparation at 20 min after stimulation and lasted till 90 min as examined (Fig. 6C, D), suggesting differential intracellular signaling pathways activated by BDNF and CGRP in the spinal cord.
Figure 6. Effects of CGRP on spinal ERK1/2 and Akt phosphorylation.
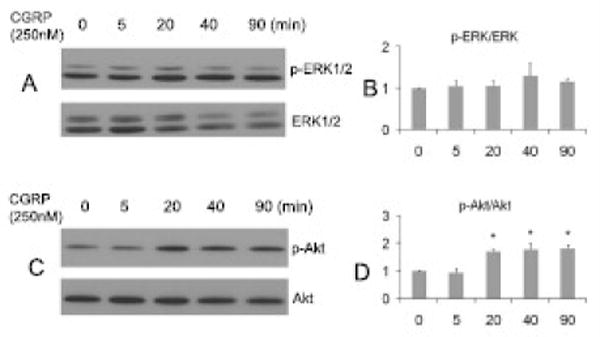
In A-D, S1 spinal cord was transversely sectioned at a thickness of 250 μm and incubated with CGRP (250 nM) for a designated time period indicated in A and C. Results showed that exogenous CGRP was able to increase Akt activation (D) but not ERK1/2 activation (B) in the spinal cord slices at 20 min of incubation (n=3 independent experiments).
Increases in the level of phospho-Akt in lumbosacral spinal cord in vivo during colitis
To examine if Akt was activated in the lumbosacral spinal cord during colitis in vivo, the L1, L6 and S1 spinal cord from control and colitic animals was homogenized for western blot analysis (Fig. 7A). Results showed that the phosphorylation level of Akt was significantly increased in L1, L6 and S1 spinal cord at 7 days of colitis (p<0.05) when compared to segment-matched controls (Fig. 7B). This is very different from the changes in the phosphorylation level of ERK1/2 in lumbosacral spinal cord during colitis, where phospho-ERK1/2 was increased in L1 and S1 spinal cord but not in L6 spinal cord (Qiao et al., 2008), suggesting region-specific changes in Akt and ERK1/2 during colitis.
Figure 7. Increases in Akt phosphorylation level in the spinal cord during colitis.

Lumbosacral spinal cord L1, L6 and S1 from control and animals treated with TNBS were homogenized and phospho-Akt was measured by western blot (A). An increased expression of phospho-Akt was observed in L1, L6 and S1 spinal cord during colitis (n=3 for each points). Histogram (B) shows relative levels of phospho-Akt expression in each segmental level examined. *p<0.05.
Further analysis with immunohistochemical staining showed that colitis increased phospho-Akt immunoreactivity in both L1 and S1 spinal cord (Fig. 8). In L1 spinal cord, colitis induced increases in phospho-Akt immunoreactivity at the spinal superficial dorsal horn mainly lamina II (Fig. 8B, *) and increases in the number of phospho-Akt immunoreactive neurons at the region near the central commissure (Fig. 8B, arrows) where CGRP nerve fibers extended to (compare to Fig. 4B). In S1 spinal cord, phospho-Akt immunoreactivity was increased at the dorsal horn laminae I and II (Fig. 8D, *).
Figure 8. Phospho-Akt immunoreactivity in L1 and S1 spinal cord before and during colitis.

Immunohistochemistry results showed increases in the intensity of phospho-Akt immunoreactivity at the superficial dorsal horn in both L1 and S1 spinal cord (compare B to A; D to C). The number of phospho-Akt immunoreactive neurons was increased in L1 spinal cord at the region of gray commissure during colitis (B, arrows). Bar = 300 μm.
DISCUSSION
The present studies demonstrate that colitis induces differential changes in CGRP mRNA and protein in lumbosacral dorsal root ganglia and spinal cord, with increases in CGRP mRNA and protein in L1 DRG; no changes in CGRP mRNA and protein in L6 DRG; and increases in CGRP mRNA and decreases in CGRP protein in S1 DRG. These results suggest that there may be more CGRP released from S1 DRG after increased production (transcription and translation) when compared to other DRG such as L1 DRG because we found that CGRP immunoreactivity was decreased in S1 colonic afferent neurons but not in L1 and L6 colonic afferent neurons during colitis. The differential changes in CGRP expression in different segmental levels of DRG may be due to the differential projections from colon to DRG as demonstrated in our previous study showing that retrograde dye only intensely labeled colonic afferent neurons in S1 DRG but with mixed intensity in other DRG, suggesting that the neurons in S1 DRG may have relatively higher responses to colonic irritation than the neurons in other DRG. These results may also reflect differential functional responses of lumbar and sacral sensory pathways to colonic inflammation. Differential mechanical sensory and chemosensory responses of these two afferent pathways have been reported in mice (Brierley SM et al., 2004; 2005). In addition, the current study shows differential changes in CGRP in lumbosacral spinal cord, with increased intensity of CGRP immunoreactivity in L1 spinal dorsal horn but decreased intensity of CGRP immunoreactivity in S1 spinal dorsal horn; however, no change in the level of CGRP mRNA was noted in these spinal segments. There were also marked increases in the intensity of CGRP nerve fibers in L1 and S1 spinal cord. The changes in CGRP protein but not mRNA in the spinal cord during colitis may be due to the following biological processes: 1) an anterograde transport of CGRP from DRG to the spinal cord (Kashihara et al., 1989; Schäfers et al., 2002); 2) release of CGRP from the nerve terminals (Wick et al., 2006; Eberhardt et al., 2008; Merighi et al., 2008); and 3) degradation of CGRP once it binds to receptor (Padilla et al., 2007). The later may cause intracellular signaling kinase activation such as increases in Akt phosphorylation in these spinal segments.
Our previous study has demonstrated the segmental distribution pattern of colonic primary afferents in rat lumbosacral DRG which is that the colonic afferent neurons are located in L1, L2, L6 and S1 DRG (Qiao and Grider, 2007a). The two peaks of labeling are likely to be due to innervation from the hypogastric/ splanchnic and pelvic afferents (Berthould et al., 2004; Bielefeldt et al., 2005) respectively. Colitis had no effects on the number of cells in DRG labeled by the retrograde tracing dye (Qiao and Grider, 2007a). Recent studies demonstrated that TNBS-induced colitis increased the expression of TTX-R Na+ current density (Nav1.8) and transient receptor potential ankyrin-1 (TRPA1) in colonic afferent neurons (Beyak MJ et al., 2004; Yang J, et al., 2008). These results suggest that colitis induced minimum number of DRG cell death if any, and colitis-induced decrease in CGRP expression in S1 colonic afferent neurons is not due to cell loss.
The involvement of CGRP in inflammatory conditions has been extensively studied. Previous studies utilizing CGRP knockout mice (Zhang et al., 2001, Thompson et al., 2008) or a CGRP receptor antagonist (Plourde, et al., 1997; Delafoy et al., 2006) demonstrated a protective role of CGRP in experimentally-induced colitis and a role in mediating inflammation-induced hyperalgesia, visceromoter reflex and colonic hypersensitivity in rats with colonic inflammation. However, the central role of CGRP in inflammatory bowel disease is unknown. There are also no studies on CGRP mRNA and protein expression, and distribution patterns in the colonic extrinsic afferent pathways with colitis. Our present study showed that CGRP mRNA and/or protein were synthesized in lumbosacral DRG and suggested that they may anterogradely transport to the spinal cord during colitis. In the dorsal horn of the spinal cord where primary afferent axons terminate, the primary afferent terminals contain and secrete numerous excitatory neurotransmitters including glutamate, substance P, somatostatin, VIP, CGRP and neurotrophins (Donnerer et al., 1993; Honoré et al., 2002; Malcangio et al., 2002; Szekely et al., 2002; Schmidtko et al., 2005). The level of these neuropeptides can be significantly affected by peripheral conditions such as inflammation or injury, and their release from the nerve terminals can further contribute to phenotypic and physiologic plasticity in the central nervous system (Krenz et al., 1999; Thompson et al., 1999; Sung et al., 2003).
The changes in CGRP level in response to peripheral inflammation or injury have been controversial but appear to vary in a time-dependent fashion: short-term (up to 72 h) induction of colitis decreased CGRP content in colonic afferent neurons (Traub et al., 1999) but chronic inflammation of bladder increased CGRP immunoreactivity in bladder afferent neurons (Vizzard, 2001); acute somatic inflammation (6 h) decreased CGRP content in dorsal spinal cord (Galeazza et al., 1995) while chronic somatic injury (up to several months) increased the level of CGRP protein in DRG and spinal cord (Smith et al., 1992; Hanesch et al., 1993; Galeazza et al., 1995). To determine if the time-dependent changes in CGRP is due to an initial release of CGRP centrally and then is balanced with increased CGRP transcription after chronic inflammation or injury, we examined CGRP mRNA levels and measured changes in CGRP in different segments of DRG/spinal cord in a time-dependent manner after colonic inflammation. To further assess the changes in CGRP in primary afferent pathways during colitis, we examined the levels of CGRP mRNA and protein in both lumbar (innervation from the hypogastric/splanchnic afferents) and sacral (innervation from the pelvic afferents) segments at 3 and 7 days of colitis. Results showed that colitis caused increases in CGRP transcripts primarily in the lumbosacral DRG, followed by differential changes in CGRP content in the lumbar and sacral levels. More specifically, in L1 DRG, CGRP mRNA was increased at 3 days but declined at 7 days of colitis, and CGRP protein was increased at 7 days of colitis; in S1 DRG, CGRP mRNA was increased at both 3 and 7 days of colitis, and CGRP protein was decreased at 7 days of colitis. One explanation may be that colitis induces CGRP expression, which results in CGRP release from colonic afferent neurons in a possible autocrine mechanism (Segond et al., 1992).
The diameter of the DRG neurons that express CGRP ranges from 10-25 μm in all DRG examined except in S1 DRG with colitis almost all of the DRG neurons having a diameter of 10-15 μm (data not shown). No further characterization was made to identify if these neurons were colonic afferents or bladder afferents. It is possible that while CGRP is released from the colonic afferent neurons to the spinal cord during colonic irritation, a paracrine action of other factors in the DRG increases CGRP expression in the surrounding neurons innervating other organ such as the urinary bladder (Qiao and Grider, 2007a). We found that colitis increased CGRP expression in bladder afferent neurons from L1 and S1 DRG (Qiao and Grider, 2007a). It is noted that we did not detect changes in CGRP immunoreactivity in either colonic afferent neurons or bladder afferent neurons in L6 DRG during colitis. Our previous studies also showed no changes in BDNF expression and ERK1/2 activation in L6 spinal level (Qiao et al., 2008). These results suggest that L6 spinal level is not primarily affected by colonic inflammation.
Previous studies showed that increases in BDNF expression level in DRG may affect CGRP expression locally during colitis (Qiao and Grider, 2007a). Increases in NGF in the inflamed colon (di Mola et al., 2000) may also contribute to CGRP expression in colonic afferent neurons via retrograde transport (Campenot and MacInnis, 2004). Retrograde NGF signaling can activate ERK5 in the DRG, induce the phosphorylation of cAMP-responsive element-binding protein (CREB) and increases CGRP transcripts (Lanigan and Russo, 1997). Recent studies showed that injection of NGF (alphaNGF) antiserum decreased the levels of CGRP protein in sensory neurons in DRG (Shadiack et al., 2001). Also in support of this notion, CGRP mRNA was absent in DRG from TrkA-/- mice as well as in NGF-deprived DRG explants (Patel et al., 2000).
CGRP is present within C and A-delta fibers that terminate in the dorsal horn (Chung et al., 1988). CGRP-positive fibers formed a dense plexus in lamina I and II, the reticulated region of lamina V, and the tract of Lissauer at the cervical, thoracic, lumbar, and sacral levels of the human spinal cord (Harmann et al., 1988). The present study shows a similar regional distribution of CGRP in the superficial dorsal horn, the region of dorso-lateral tract, and the lateral collateral path in rat lumbosacral spinal cord. These areas are known to contain the main targets for termination of visceral fibers and contribute to visceral sensation and/or autonomic control of pelvic organs (Holzer, 1988; Hwang et al., 2005; Zinck et al., 2007). The results showing the presence of an intense CGRP fiber bundle extending from Lissauer’s tract to the region of the sacral parasympathetic nucleus in S1 spinal cord suggest a potential CGRP release pathway during colitis. It is reported that CGRP expressed in the lateral collateral of Lissauer’s tract may play roles in bladder micturition control (Hwang et al., 2005; Zvara and Vizzard, 2007), and thus may also play roles in colitis-induced bladder cross-sensitization (Pezzone et al., 2005).
The cellular physiological function of CGRP is dependent on CGRP binding to its specific receptor and inducing intracellular signaling activation. Recent studies demonstrated that CGRP is capable of activating PI-3 kinase/Akt and MAPK pathways in cultured cells and tissues (Parameswaran et al., 2000). Our previous study showed that the phosphorylation level of ERK1/2 was increased in L1 and S1 spinal cord during colitis (Qiao et al., 2008). To examine if CGRP contributes to ERK1/2 activation, we incubated spinal cord slices in vitro with CGRP and found that CGRP could not activate ERK1/2 after up to 90 min of stimulation; however, parallel studies showed that ERK1/2 was activated by BDNF within 30 min of incubation in these spinal slices (Qiao et al., 2008). In contrast, after CGRP incubation, the level of Akt phosphorylation was significantly increased at 20 min and sustained till 90 min as demonstrated in S1 spinal cord slices. The differential responses of ERK1/2 and Akt were also exhibited in vivo during colitis showing that ERK1/2 was activated in L1 and S1 but not L6 spinal cord; however, Akt was activated in L1, S1 and L6 spinal cord, suggesting that colitis causes a differential signaling activation in the spinal cord.
In summary, the present study demonstrated that colitis induced segment and region- specific changes in the expression of neurotransmitters, neuropeptides and the activation of intracellular signaling pathways in the primary afferents during colitis, and increased expression and release of CGRP in DRG may not have a role in the activation of ERK1/2 in the spinal cord during colitis. Although CGRP is able to activate Akt in culture, the likelihood of CGRP in activating Akt in the spinal cord in vivo during colitis needs to be further investigated.
Acknowledgments
Supporting Grants: The AGAF Bridging Grant (LYQ); NIH DK 077917 (LYQ); NIH DK 034153 (JRG)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abercrombie M. Estimation of nuclear population from microtome section. Anat Rec. 1946;94:239–247. doi: 10.1002/ar.1090940210. [DOI] [PubMed] [Google Scholar]
- Bernstein CN, Niazi N, Robert M, Mertz H, Kodner A, Munakata J, Naliboff B, Mayer EA. Rectal afferent function in patients with inflammatory and functional intestinal disorders. Pain. 1996;66:151–161. doi: 10.1016/0304-3959(96)03062-x. [DOI] [PubMed] [Google Scholar]
- Berthoud HR, Blackshaw LA, Brookes SJ, Grundy D. Neuroanatomy of extrinsic afferents supplying the gastrointestinal tract. Neurogastroenterol Motil. 2004;16:28–33. doi: 10.1111/j.1743-3150.2004.00471.x. [DOI] [PubMed] [Google Scholar]
- Beyak MJ, Ramji N, Krol KM, Kawaja MD, Vanner SJ. Two TTX-resistant Na+ currents in mouse colonic dorsal root ganglia neurons and their role in colitis-induced hyperexcitability. Am J Physiol Gastrointest Liver Physiol. 2004;287:G845–855. doi: 10.1152/ajpgi.00154.2004. [DOI] [PubMed] [Google Scholar]
- Bielefeldt K, Christianson JA, Davis BM. Basic and clinical aspects of visceral sensation: transmission in the CNS. Neurogastroenterol Motil. 2005;17:488–499. doi: 10.1111/j.1365-2982.2005.00671.x. [DOI] [PubMed] [Google Scholar]
- Brierley SM, Carter R, Jones W, 3rd, Xu L, Robinson DR, Hicks GA, Gebhart GF, Blackshaw LA. Differential chemosensory function and receptor expression of splanchnic and pelvic colonic afferents in mice. J Physiol. 2005;567(Pt 1):267–281. doi: 10.1113/jphysiol.2005.089714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brierley SM, Jones RC, 3rd, Gebhart GF, Blackshaw LA. Splanchnic and pelvic mechanosensory afferents signal different qualities of colonic stimuli in mice. Gastroenterology. 2004;127:166–178. doi: 10.1053/j.gastro.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Burton MB, Gebhart GF. Effects of intracolonic acetic acid on responses to colorectal distension in the rat. Brain Res. 1995;672:77–82. doi: 10.1016/0006-8993(94)01382-r. [DOI] [PubMed] [Google Scholar]
- Clifton MS, Hoy JJ, Chang J, Idumalla PS, Fakhruddin H, Grady EF, Dada S, Corvera CU, Bhargava A. Role of calcitonin receptor-like receptor in colonic motility and inflammation. Am J Physiol Gastrointest Liver Physiol. 2007;293:G36–44. doi: 10.1152/ajpgi.00464.2006. [DOI] [PubMed] [Google Scholar]
- Carrasquillo Y, Gereau RW., 4th Activation of the extracellular signal-regulated kinase in the amygdala modulates pain perception. J Neurosci. 2007;27:1543–1551. doi: 10.1523/JNEUROSCI.3536-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campenot RB, MacInnis BL. Retrograde transport of neurotrophins: fact and function. J Neurobiol. 2004;58:217–229. doi: 10.1002/neu.10322. [DOI] [PubMed] [Google Scholar]
- Chung K, Lee WT, Carlton SM. The effects of dorsal rhizotomy and spinal cord isolation on calcitonin gene-related peptide-labeled terminals in the rat lumbar dorsal horn. Neurosci Lett. 1988;90:27–32. doi: 10.1016/0304-3940(88)90781-1. [DOI] [PubMed] [Google Scholar]
- Coutinho SV, Meller ST, Gebhart GF. Intracolonic zymosan produces visceral hyperalgesia in the rat that is mediated by spinal NMDA and non-NMDA receptors. Brain Res. 1996;736:7–15. doi: 10.1016/0006-8993(96)00661-0. [DOI] [PubMed] [Google Scholar]
- Delafoy L, Gelot A, Ardid D, Eschalier A, Bertrand C, Doherty AM, Diop L. Interactive involvement of BDNF, NGF and CGRP in colonic hypersensitivity in the rat. Gut. 2006;55:940–945. doi: 10.1136/gut.2005.064063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- di Mola FF, Friess H, Zhu ZW, Koliopanos A, Bley T, Di Sebastiano P, Innocenti P, Zimmermann A, Buchler MW. Nerve growth factor and Trk high affinity receptor (TrkA) gene expression in inflammatory bowel disease. Gut. 2000;46:670–679. doi: 10.1136/gut.46.5.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dömötör A, Peidl Z, Vincze A, Hunyady B, Szolcsányi J, Kereskay L, Szekeres G, Mózsik G. Immunohistochemical distribution of vanilloid receptor, calcitonin-gene related peptide and substance P in gastrointestinal mucosa of patients with different gastrointestinal disorders. Inflammopharmacology. 2005;13:161–177. doi: 10.1163/156856005774423737. [DOI] [PubMed] [Google Scholar]
- Donnerer J, Schuligoi R, Stein C, Amann R. Upregulation, release and axonal transport of substance P and calcitonin gene-related peptide in adjuvant inflammation and regulatory function of nerve growth factor. Regul Pept. 1993;46:150–154. [PubMed] [Google Scholar]
- Drewes AM, Frøkjaer JB, Larsen E, Reddy H, Arendt-Nielsen L, Gregersen H. Pain and mechanical properties of the rectum in patients with active ulcerative colitis. Inflamm Bowel Dis. 2006;12:294–303. doi: 10.1097/01.MIB.0000209365.09189.04. [DOI] [PubMed] [Google Scholar]
- Eberhardt M, Hoffmann T, Sauer SK, Messlinger K, Reeh PW, Fischer MJ. Calcitonin gene-related peptide release from intact isolated dorsal root and trigeminal ganglia. Neuropeptides. 2008 Mar 5; doi: 10.1016/j.npep.2008.01.002. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Evangelista S, Tramontana M. Involvement of calcitonin gene-related peptide in rat experimental colitis. J Physiol Paris. 1993;87:277–280. doi: 10.1016/0928-4257(93)90017-n. [DOI] [PubMed] [Google Scholar]
- Galeazza MT, Garry MG, Yost HJ, Strait KA, Hargreaves KM, Seybold VS. Plasticity in the synthesis and storage of substance P and calcitonin gene-related peptide in primary afferent neurons during peripheral inflammation. Neuroscience. 1995;66:443–458. doi: 10.1016/0306-4522(94)00545-g. [DOI] [PubMed] [Google Scholar]
- Gebhart GF, Bielefeldt K, Ozaki N. Gastric hyperalgesia and changes in voltage gated sodium channel function in the rat. Gut. 2002;51(Suppl 1):i15–18. doi: 10.1136/gut.51.suppl_1.i15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grider JR, Piland BE. The peristaltic reflex induced by short-chain fatty acids is mediated by sequential release of 5-HT and neuronal CGRP but not BDNF. Am J Physiol Gastrointest Liver Physiol. 2007;292:G429–437. doi: 10.1152/ajpgi.00376.2006. [DOI] [PubMed] [Google Scholar]
- Guillery RW. On counting and counting errors. J Comp Neurol. 2002;447:1–7. doi: 10.1002/cne.10221. [DOI] [PubMed] [Google Scholar]
- Harmann PA, Chung K, Briner RP, Westlund KN, Carlton SM. Calcitonin gene-related peptide (CGRP) in the human spinal cord: a light and electron microscopic analysis. J Comp Neurol. 1988;269:371–380. doi: 10.1002/cne.902690305. [DOI] [PubMed] [Google Scholar]
- Hanesch U, Pfrommer U, Grubb BD, Schaible HG. Acute and chronic phases of unilateral inflammation in rat’s ankle are associated with an increase in the proportion of calcitonin gene-realted peptide-immunoreactive dorsal root ganglion cells. Eur J Neurosci. 1993;5:154–161. doi: 10.1111/j.1460-9568.1993.tb00481.x. [DOI] [PubMed] [Google Scholar]
- Holzer P. Local effector functions of capsaicin-sensitive sensory nerve endings: Involvement of tachykinins, calcitonin gene-related peptide and other neuropeptides. Neuroscience. 1988;24:739–768. doi: 10.1016/0306-4522(88)90064-4. [DOI] [PubMed] [Google Scholar]
- Honoré P, Kamp EH, Rogers SD, Gebhart GF, Mantyh PW. Activation of lamina I spinal cord neurons that express the substance P receptor in visceral nociception and hyperalgesia. J Pain. 2002;3:3–11. doi: 10.1054/jpai.2002.27001. [DOI] [PubMed] [Google Scholar]
- Hwang SJ, Oh JM, Valtschanoff JG. The majority of bladder sensory afferents to the rat lumbosacral spinal cord are both IB4- and CGRP-positive. Brain Res. 2005;1062:86–91. doi: 10.1016/j.brainres.2005.09.026. [DOI] [PubMed] [Google Scholar]
- Kashihara Y, Sakaguchi M, Kuno M. Axonal transport and distribution of endogenous calcitonin gene-related peptide in rat peripheral nerve. J Neurosci. 1989;9:3796–3802. doi: 10.1523/JNEUROSCI.09-11-03796.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenz NR, Meakin SO, Krassioukov AV, Weaver LC. Neutralizing intraspinal nerve growth factor blocks autonomic dysreflexia caused by spinal cord injury. J Neurosci. 1999;19:7405–7414. doi: 10.1523/JNEUROSCI.19-17-07405.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau AM, Yashpal K, Cahill CM, St Louis M, Ribeiro-da-Silva A, Henry JL. Sensory neuron and substance P involvement in symptoms of a zymosan-induced rat model of acute bowel inflammation. Neuroscience. 2007;145:699–707. doi: 10.1016/j.neuroscience.2006.11.066. [DOI] [PubMed] [Google Scholar]
- Lanigan T, Russo A. Binding of upstream stimulatory factor and a cell-specific activator to the calcitonin/calcitonin gene-related peptide enhancer. J Biol Chem. 1997;272:18316–18324. doi: 10.1074/jbc.272.29.18316. [DOI] [PubMed] [Google Scholar]
- Larsson MH, Rapp L, Lindström E. Effect of DSS-induced colitis on visceral sensitivity to colorectal distension in mice. Neurogastroenterol Motil. 2006;18:144–152. doi: 10.1111/j.1365-2982.2005.00736.x. [DOI] [PubMed] [Google Scholar]
- Malcangio M, Getting SJ, Grist J, Cunningham JR, Bradbury EJ, Charbel Issa P, Lever IJ, Pezet S, Perretti M. A novel control mechanism based on GDNF modulation of somatostatin release from sensory neurones. FASEB J. 2002;16:730–732. doi: 10.1096/fj.01-0971fje. [DOI] [PubMed] [Google Scholar]
- Merighi A, Bardoni R, Salio C, Lossi L, Ferrini F, Prandini M, Zonta M, Gustincich S, Carmignoto G. Presynaptic functional trkB receptors mediate the release of excitatory neurotransmitters from primary afferent terminals in lamina II (substantia gelatinosa) of postnatal rat spinal cord. Dev Neurobiol. 2008;68:457–475. doi: 10.1002/dneu.20605. [DOI] [PubMed] [Google Scholar]
- Miampamba M, Sharkey KA. Distribution of calcitonin gene-related peptide, somatostatin, substance P and vasoactive intestinal polypeptide in experimental colitis in rats. Neurogastroenterol Motil. 1998;10:315–329. doi: 10.1046/j.1365-2982.1998.00111.x. [DOI] [PubMed] [Google Scholar]
- Morris GP, Beck PL, Herridge MS, Depew WT, Szewczuk MR, Wallace JL. Hapten-induced model of chronic inflammation and ulceration in the rat colon. Gastroenterology. 1989;96:795–803. [PubMed] [Google Scholar]
- Nahin RL, Byers MR. Adjuvant-induced inflammation of rat paw is associated with altered calcitonin gene-related peptide immunoreactivity within cell bodies and peripheral endings of primary afferent neurons. J Comp Neurol. 1994;349:475–485. doi: 10.1002/cne.903490311. [DOI] [PubMed] [Google Scholar]
- Ohno T, Hattori Y, Komine R, Ae T, Mizuguchi S, Arai K, Saeki T, Suzuki T, Hosono K, Hayashi I, Oh-Hashi Y, Kurihara Y, Kurihara H, Amagase K, Okabe S, Saigenji K, Majima M. Roles of calcitonin gene-related peptide in maintenance of gastric mucosal integrity and in enhancement of ulcer healing and angiogenesis. Gastroenterology. 2008;134:215–225. doi: 10.1053/j.gastro.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Okajima K, Harada N. Regulation of inflammatory responses by sensory neurons: molecular mechanism(s) and possible therapeutic applications. Curr Med Chem. 2006;13:2241–2251. doi: 10.2174/092986706777935131. [DOI] [PubMed] [Google Scholar]
- Padilla BE, Cottrell GS, Roosterman D, Pikios S, Muller L, Steinhoff M, Bunnett NW. Endothelin-converting enzyme-1 regulates endosomal sorting of calcitonin receptor-like receptor and beta-arrestins. J Cell Biol. 2007;179:981–997. doi: 10.1083/jcb.200704053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parameswaran N, Disa J, Spielman WS, Brooks DP, Nambi P, Aiyar N. Activation of multiple mitogen-activated protein kinases by recombinant calcitonin gene-related peptide receptor. Eur J Pharmacol. 2000;389:125–130. doi: 10.1016/s0014-2999(99)00874-2. [DOI] [PubMed] [Google Scholar]
- Patel TD, Jackman A, Rice FL, Kucera J, Snider WD. Development of sensory neurons in the absence of NGF/TrkA signaling in vivo. Neuron. 2000;25:345–357. doi: 10.1016/s0896-6273(00)80899-5. [DOI] [PubMed] [Google Scholar]
- Pezzone MA, Liang R, Fraser MO. A model of neural cross-talk and irritation in the pelvis: implications for the overlap of chronic pelvic pain disorders. Gastroenterology. 2005;128:1953–1964. doi: 10.1053/j.gastro.2005.03.008. [DOI] [PubMed] [Google Scholar]
- Plourde V, St-Pierre S, Quirion R. Calcitonin gene-related peptide in viscerosensitive response to colorectal distension in rats. Am J Physiol. 1997;273:G191–196. doi: 10.1152/ajpgi.1997.273.1.G191. [DOI] [PubMed] [Google Scholar]
- Qiao LY, Grider JR. TNBS colitis elicits differential changes in CGRP expression in sympathetic and parasympathetic spinal cord and dorsal root ganglia (DRG) Gastroenterology. 2005;128:A375. [Google Scholar]
- Qiao LY, Grider JR. Up-regulation of calcitonin gene-related peptide and receptor tyrosine kinase TrkB in rat bladder afferent neurons following TNBS colitis. Exp Neurol. 2007a;204:667–679. doi: 10.1016/j.expneurol.2006.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao LY, Grider JR. Alterations in CGRP mRNA expression in lumbosacral DRG and spinal cord following trinitrobenzene sulfonic acid (TNBS) colitis. Gastroenterology. 2007b;132:A719. [Google Scholar]
- Qiao LY, Gulick MA, Bowers J, Kuemmerle JF, Grider JR. Differential changes in brain-derived neurotrophic factor and extracellular signal-regulated kinase in rat primary afferent pathways with colitis. Neurogastroenterol Motil. 2008 Mar 26; doi: 10.1111/j.1365-2982.2008.01119.x. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Qiao LY, Vizzard MA. Cystitis-induced upregulation of tyrosine kinase (TrkA, TrkB) receptor expression and phosphorylation in rat micturition pathways. J Comp Neurol. 2002;454:200–211. doi: 10.1002/cne.10447. [DOI] [PubMed] [Google Scholar]
- Schäfers M, Geis C, Brors D, Yaksh TL, Sommer C. Anterograde transport of tumor necrosis factor-alpha in the intact and injured rat sciatic nerve. J Neurosci. 2002;22:536–545. doi: 10.1523/JNEUROSCI.22-02-00536.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidtko A, Del Turco D, Coste O, Ehnert C, Niederberger E, Ruth P, Deller T, Geisslinger G, Tegeder I. Essential role of the synaptic vesicle protein synapsin II in formalin-induced hyperalgesia and glutamate release in the spinal cord. Pain. 2005;115:171–181. doi: 10.1016/j.pain.2005.02.027. [DOI] [PubMed] [Google Scholar]
- Segond N, Gerbaud P, Cressent M, Lasmoles F, Taboulet J, Jullienne A, Raynaud F, Moukhtar MS, Evain-Brion D. Calcitonin gene-related peptide: an autocrine growth factor with regulatory activity in vitro. Biochem Biophys Res Commun. 1992;187:381–388. doi: 10.1016/s0006-291x(05)81504-9. [DOI] [PubMed] [Google Scholar]
- Shadiack AM, Sun Y, Zigmond RE. Nerve growth factor antiserum induces axotomy-like changes in neuropeptide expression in intact sympathetic and sensory neurons. J Neurosci. 2001;21:363–371. doi: 10.1523/JNEUROSCI.21-02-00363.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GD, Harmar AJ, McQueen DS, Seckl JR. Increase in substance P and CGRP, but not somatostatin content of innervating dorsal root ganglia in adjuvant monoarthritis in the rat. Neurosci Lett. 1992;137:257–260. doi: 10.1016/0304-3940(92)90417-6. [DOI] [PubMed] [Google Scholar]
- Sung B, Lim G, Mao J. Altered expression and uptake activity of spinal glutamate transporters after nerve injury contribute to the pathogenesis of neuropathic pain in rats. J Neurosci. 2003;23:2899–2910. doi: 10.1523/JNEUROSCI.23-07-02899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szekely JI, Torok K, Mate G. The role of ionotropic glutamate receptors in nociception with special regard to the AMPA binding sites. Curr Pharm Des. 2002;8:887–912. doi: 10.2174/1381612024607126. [DOI] [PubMed] [Google Scholar]
- Thompson SW, Bennett DL, Kerr BJ, Bradbury EJ, McMahon SB. Brain-derived neurotrophic factor is an endogenous modulator of nociceptive responses in the spinal cord. Proc Natl Acad Sci U S A. 1999;96:7714–7718. doi: 10.1073/pnas.96.14.7714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson BJ, Washington MK, Kurre U, Singh M, Rula EY, Emeson RB. Protective roles of alpha-calcitonin and beta-calcitonin gene-related peptide in spontaneous and experimentally induced colitis. Dig Dis Sci. 2008;53:229–241. doi: 10.1007/s10620-007-9848-7. [DOI] [PubMed] [Google Scholar]
- Traub RJ, Hutchcroft K, Gebhart GF. The peptide content of colonic afferents decreases following colonic inflammation. Peptides. 1999;20:267–273. doi: 10.1016/s0196-9781(98)00157-0. [DOI] [PubMed] [Google Scholar]
- Vizzard MA. Alterations in neuropeptide expression in lumbosacral bladder pathways following chronic cystitis. J Chem Neuroanat. 2001;21:125–138. doi: 10.1016/s0891-0618(00)00115-0. [DOI] [PubMed] [Google Scholar]
- Wick EC, Pikios S, Grady EF, Kirkwood KS. Calcitonin gene-related peptide partially mediates nociception in acute experimental pancreatitis. Surgery. 2006;139:197–201. doi: 10.1016/j.surg.2005.08.024. [DOI] [PubMed] [Google Scholar]
- Winston JH, Toma H, Shenoy M, He ZJ, Zou L, Xiao SY, Micci MA, Pasricha PJ. Acute pancreatitis results in referred mechanical hypersensitivity and neuropeptide up-regulation that can be suppressed by the protein kinase inhibitor k252a. J Pain. 2003;4:329–337. doi: 10.1016/s1526-5900(03)00636-9. [DOI] [PubMed] [Google Scholar]
- Yang J, Li Y, Zuo X, Zhen Y, Yu Y, Gao L. Transient receptor potential ankyrin-1 participates in visceral hyperalgesia following experimental colitis. Neurosci Lett. 2008;440:237–241. doi: 10.1016/j.neulet.2008.05.093. [DOI] [PubMed] [Google Scholar]
- Zhang L, Hoff AO, Wimalawansa SJ, Cote GJ, Gagel RF, Westlund KN. Arthritic calcitonin/alpha calcitonin gene-related peptide knockout mice have reduced nociceptive hypersensitivity. Pain. 2001;89:265–273. doi: 10.1016/s0304-3959(00)00378-x. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Xu H, Clapham DE, Ji RR. Phosphatidylinositol 3-kinase activates ERK in primary sensory neurons and mediates inflammatory heat hyperalgesia through TRPV1 sensitization. J Neurosci. 2004;24:8300–8309. doi: 10.1523/JNEUROSCI.2893-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinck ND, Rafuse VF, Downie JW. Sprouting of CGRP primary afferents in lumbosacral spinal cord precedes emergence of bladder activity after spinal injury. Exp Neurol. 2007;204:777–790. doi: 10.1016/j.expneurol.2007.01.011. [DOI] [PubMed] [Google Scholar]
- Zvara P, Vizzard MA. Exogenous overexpression of nerve growth factor in the urinary bladder produces bladder overactivity and altered micturition circuitry in the lumbosacral spinal cord. BMC Physiol. 2007;7:9–20. doi: 10.1186/1472-6793-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]