

Abstract
In the current study, the design, synthetic feasibility and biochemical characterization of biodegradable peptidic PEG-based nanocarriers is described. The components were selected to influence the body elimination pathway upon nanocarrier biodegradation. Two prototypical nanocarriers were prepared using non-PEGylated and PEGylated peptidic cores [CH3CO-(Lys-βAla-βAla)X-Cys-CONH2 (X= 2, 4)]. A homodimeric nanocarrier with 4 copies of fluorescein-PEG5kDa was synthesized by linking two PEGylated peptidic cores (X=2) using a disulfide bond. A dual-labeled heterodimeric nanocarrier with 2 copies of fluorescein-PEG5kDa and 4 copies of Texas Red was also synthesized. Optimum conditions for linking imaging agents, PEG, or a peptidic core to a peptidic core were determined. Significantly higher yields (69% versus 30%) of the PEGylated peptidic core were obtained by using 2 copies of β-alanine as a spacer along with increasing DMSO concentrations, which resulted in reduced steric hindrance. Stoichiometric addition of the components was also demonstrated and found to be important for reducing polydispersity. Nanocarrier biodegradation was evaluated in simulated intracellular and extracellular/blood environments using 3mM and 10μM glutathione in buffer, respectively. The nanocarrier was 9-fold more stable in the extracellular environment. The results suggest selective intracellular degradation of the nanocarrier into components with known body elimination pathways.
Keywords: Peptide, PEG, nanocarrier, disulfide, GSH-dependent biodegradation, intra- cellular elimination
1. Introduction
PEGylation, the process of conjugating proteins and other drugs to poly(ethylene glycol) (PEG), has improved the clinical performance of drugs such as interferon α-2a (PEGASYS®) and interferon α-2b (PegIntron®) for treating diseases such as Hepatitis C infection and chronic myelogenous leukemia [1, 2]. The observed improvements in clinical efficacy are primarily due to reduced proteolytic degradation, enhanced physical stability, higher solubility, and reduced systemic clearance due to a longer circulating half-life [3–5]. PEGylation results in reduced immunogenicity and antigenicity as well as reduced toxicity by, among other mechanisms, reducing interactions with cells of the immune system such as dendritic cells and macrophages [6, 7]. These are the very same cells that are infected by the Human Immunodeficiency Virus (HIV). Therefore, while PEGylation has been enormously successful in maintaining plasma concentrations of various drugs, its utility as a targeting agent or drug delivery scaffold for eradicating HIV infection is low. Since many anti-HIV drugs have poor physicochemical and biopharmaceutical properties, our goal has been to design nanocarriers capable of delivering drugs specifically to HIV infected cells while improving the solubility, stability and pharmacokinetics of these potent drugs.
Of the many advantages of PEGylation, the reduction in systemic clearance and prolonged body exposure to drugs is potentially one of the most important for AIDS patients since the “pill” burden (i.e., the number of dose units and frequency of administration) is high and patient compliance is far from ideal. PEGylation reduces renal clearance since the kidneys filter substances according to their size although charge, deformability and shape are also considered significant factors [8, 9]. Since PEGylation results in a hydrophobic to hydrophilic shift in physical properties, the rate of phagocytic clearance is also reduced [7, 10–12]. For example, 12 copies of a 5 kDa PEG conjugated to bovine hemoglobin increased blood half-life by a factor of 14 and systemic availability by a factor of 8 [13, 14]. In addition to reduced renal filtration, prolonged blood residence time was attributed to lower immunogenicity and reduced phagocytosis [7, 10]. A comparative study in rats using rIL-2 modified to varying extents with PEGs of molecular weights ranging from 0.35 to 20 kDa showed that binding of several small PEGs (e.g., 350 Da) or one 4kDa PEG did not alter their pharmacokinetics [15]. They also observed a rapid decrease in systemic clearance as the effective molecular size increased from 21 to approximately 70 kDa. This suggests progressive exclusion from glomerular filtration consistent with the general belief that molecules less than 20kDa in size are well cleared by the kidneys. The clearance above 70kDa was very low corresponding to the molecular weight of human serum albumin (~67 kDa), a protein that is predominantly excluded from filtration by the kidney glomerular basement membrane [15]. Due to its flexibility and deformability, it has been suggested that the optimal PEG mass for reduced renal clearance is ~40–60 kDa [16, 17]. In the first phase of our studies, we developed a series of multi-arm and branched PEG nanocarriers containing multiple copies of the chemo-attractant peptide N-formyl-Met-Leu-Phe (fMLF) for the specific purpose of promoting macrophage uptake [18–20]. Maximal uptake in macrophage-like differentiated human U937 cells occurred at a scaffold size of 20kDa whereas further increases in molecular weight up to 40kDa resulted in lower uptake [20]. We observed similar results in vivo [19]. However, virtually no uptake was observed when fMLF was not present on the PEG nanocarrier consistent with reports of reduced interactions with macrophages due to PEG [6, 7]. These results also support previous observations demonstrating that receptor-mediated endocytosis is strongly size dependent [21–24].
Polymer shape and branching are also known to alter the properties of PEG carriers [5, 25–29]. For example, a branched PEG constructed of two linear molecules of succinimidyl carbonate PEG attached to the α and ε-amino groups of lysine demonstrated higher proteolytic stability and a longer half-life in the blood as compared to their native and linear polymer conjugate counterparts [26, 27]. The binding of a branched 10 kDa PEG to asparaginase reduced antigenicity by 10-fold as compared to the counterpart with a 5 kDa linear PEG [26, 27]. It was also found to reduce uricase immunogenicity and antigenicity more efficiently than the linear 5 kDa polymer [30, 31]. While the 10 kDa PEG2 is considerably smaller than the glomerular filtration threshold size, it was found to accumulate to a significant extent in the liver of Balb/c mice [30, 31] suggesting that molecular shape and volume may be important factors in determining the biodistribution and clearance pathways of PEG nanocarriers.
There is considerable interest in determining the biological fate of soluble nanocarriers as well as other nano-sized carriers such as nanoparticles. In the present study, we shift our focus from the initial body distribution of nanocarriers (i.e., actively targeting HIV-infected cells) to designing nanocarriers with preprogrammed body elimination properties (i.e., nanocarriers that selectively release their cargo inside cells, degrade and follow a predetermined systemic elimination pathway).
2. Materials and methods
2.1. Materials
2-arm peptidic core [Acetylated-Lys-βAla-βAla-Lys-βAla-βAla-Cys(TP)-Amidated MW 813 Da], 4-arm peptidic core [Acetylated-Lys-βAla-βAla-Lys-βAla-βAla-Lys-βAla-βAla-Lys-βAla-βAla-Cys(TP)-amidated MW 1317 Da] and 6-arm peptidic core [Fmoc-Cys(t-Butylthio)- βAla-Lys-βAla-Lys-βAla-Lys-βAla-Lys-βAla-Lys-βAla-Lys-Amidated MW 1627 Da] were purchased from W.M Keck Foundation Biotechnology Resource Laboratory (New Haven, CT). Fluorescein-PEG5kDa-NHS and m-PEG3.4kDa-NHS were purchased from Nektar Therapeutics Corp. (Huntsville, AL). Texas Red-NHS was obtained from Invitrogen (Eugene, OR). Sodium phosphate monobasic, sodium phosphate dibasic, tris buffer, dithiothreitol (DTT), 2,2’-dithiodipyridine (Aldrithiol-2; TP-TP), glutathione (GSH), ninhydrin, phenol, phenylalanine, potassium cyanide, triflouroacetic acid (TFA) and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich Corp. (St. Louis, MO). Dimethylformamide (DMF) was purchased from Acros Organics (Morris Plains, NJ). Centrifugal filters (Amicon Ultra 30kDa and Microcon 10kDa) were obtained from Millipore Corp. (Billerica, MA). DIEA (N,N’-Diisopropylethylamine) was purchased from Acros Organics (Geel, Belgium).
2.2. Spectral Analyses
UV spectra were recorded on a Beckman Coulter DU 640 spectrophotometer. Mass spectrometry using matrix-assisted-laser-desorption-ionization time-of-flight (MALDI- TOF) was performed on Voyager 4800. MALDI/MS data fully confirmed the structure of compounds.
2.3. Chromatography
Gel permeation chromatography was performed on a Sephadex G-75 (Amersham Bioscience; Uppsala, Sweden) using phosphate buffers (PB) pH 5.5±0.2 and 7.4±0.2 as eluents. The fluorescence readings of each fraction obtained from gel permeation chromatography were detected at Ex= 485 nm and Em= 535 nm (for fluorescein) using a Tecan GENios microplate reader (Durham, NC). Fluorescent compounds were also subjected to HPLC (Waters 2475 Multi λ Fluorescence Detector) using a size exclusion chromatographic column, TSKgel G4000PWxl, 7.8 mm × 30 cm, 10 μm (Tosoh Bioscience; Montgomery Ville, PA).
2.4. Quantification of peptidic cores
Nanocarriers were prepared using PEGylated and non-PEGylated peptidic cores (Figs. 1, 2 and 3). The concentrations of free ε-amine groups of lysines on the peptidic cores were calculated using a quantitative Kaiser chromogenic assay established by Sarin et.al with modification [32]. The standard curve was first generated with phenylalanine. Ninhydrin solution (6% w/v) in ethanol was added to various concentrations of phenylalanine solution (14.5 -60 nmoles) dissolved in double distilled water, followed by the addition of phenol (4g/ml) and potassium cyanide (0.65 mg/ml). The mixtures were heated at 110 °C for two minutes, followed by the addition of 2ml of 60% ethanol. Optical density was read at 570 nm and a standard curve was prepared (Fig. 4). DMSO was used to dissolve the 2-arm peptidic core (2 free amino groups on lysine) since it was sparingly soluble in water. The 4-arm peptidic core (4 free amino groups on lysine) and 6-arm peptidic core (6 free amino groups on lysine) were freely soluble in water. The concentrations of each of the peptidic cores were determined using the phenylalanine standard curve.
Fig. 1.

Schematic representation of a monodisperse biodegradable dimeric nanocarrier composed of peptidic backbone irreversibly or reversibly conjugated with one or more targeting ligand/drug either directly or through the distal ends of PEG.
Fig. 2.

Synthesis of homodimeric peptide-based PEG nanocarriera.
Fig. 3.

Synthesis of heterodimeric peptide-based PEG nanocarriera.
Fig. 4.

Calibration curve of primary amine group of phenylalanine as a function of optical density (570nm). This curve was obtained from quantitative Kaiser chromogenic assay. The concentration of primary amine groups on the 2-, 4- and 6-arm peptidic cores was determined using this chromogenic assay. All measurements were done in triplicate. (R2 =0.993)
2.5. PEGylated 6-arm peptidic core
The PEGylation reaction of the peptidic core with 6 possible attachment sites (6-arm) at lysines was performed with 3 molar excess of m-PEG3.4kDa-NHS. This reaction was carried out overnight using DMF:DIEA (99:1). The crude PEGylated 6-arm peptidic core reaction was analyzed using MALDI-TOF (Fig. 5).
Fig. 5.
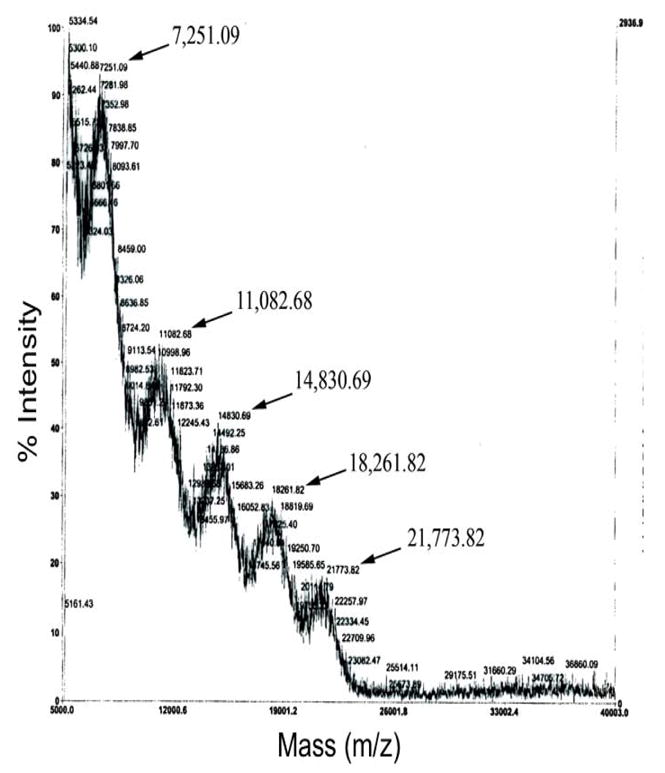
MALDI-TOF (m/z) spectrum of crude 6-arm PEGylated peptidic core reaction, showing the heterogeneity of products. The products contain a mixture of conjugation of 2 (7251 Da), 3 (11,082 Da), 4 (14,830 Da), 5 (18,261 Da) and 6 (21,773 Da) copies of m-PEG3.4kDa.
2.6. Fluorescein-labeled Cys-protected PEGylated 2-arm peptidic core
The PEGylation reaction of the Cys-protected peptidic core with 2 possible attachment sites (2-arm) at lysine was performed with 3 molar excess of fluorescein-PEG5kDa-NHS dissolved in 30% or 70% (v:v) of DMSO in 100 mM PB pH 7.4±0.2 (Fig. 2). The addition of activated PEG was performed over a period of one hour using four equal aliquots. The reaction was kept at room temperature for 16 hours. Sephadex G-75 (medium) was soaked in PB pH 7.41±0.2 overnight and loaded onto a 50 cm Sephadex column. The PEGylated product was purified using gel permeation chromatography at a rate of approximately 0.8 ml/min. The fluorescence intensities of each fraction were detected using a microplate reader at Ex = 485nm and Em = 535nm (Fig. 6). The first peak fractions for each chromatographic run were combined and concentrated using ultrafiltration (Amicon Ultra 30 kDa). The structure of the purified fluorescein-labeled PEGylated 2-arm peptidic core was confirmed using MALDI-TOF (Fig. 7).
Fig. 6.

Gel permeation chromatogram showing separation of labeled ‘Cys-protected fully PEGylated 2-arm peptidic core’ from labeled ‘Cys-protected partially PEGylated 2-arm peptidic core’ and unreacted fluorescein-PEG5kDa using Sephadex G-75 column in 100 mM phosphate buffer pH 7.4±0.2. The fluorescence measurements of each elution volume was detected at Ex = 485 nm; Em = 535 nm corresponding to the fluorescein dye. PEGylation reaction on the 2-arm peptidic core was carried out in two different reaction conditions (a) DMSO: 100 mM phosphate buffer pH 7.4±0.2 (3:7) (▴) (b) DMSO: 100 mM phosphate buffer pH 7.4±0.2 (7:3) (●).
Fig. 7.
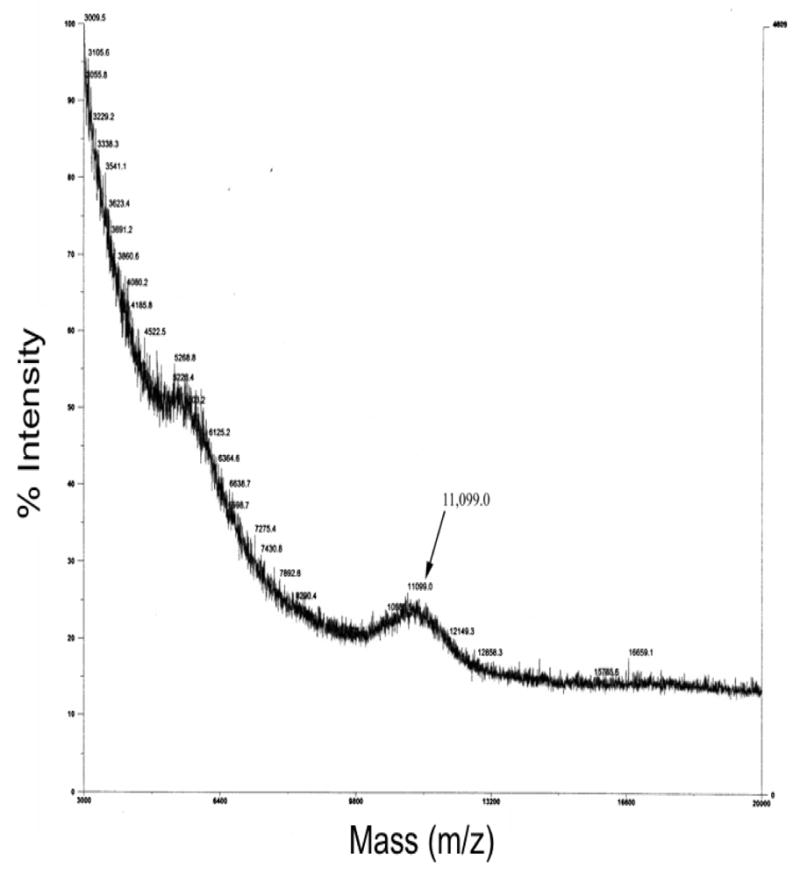
MALDI-TOF (m/z) spectrum of purified labeled Cys-protected fully PEGylated 2-arm peptidic core. The peak showing molecular weight of 11,099.0 Da confirms attachments of two fluorescein-PEG5kDa to the 2-arm peptidic core.
2.7. Deprotection of fluorescein-labeled Cys-protected PEGylated 2-arm peptidic core
In order to remove thiopyridine from the Cys-protected PEGylated 2-arm peptidic core, it was dissolved in 20 molar equivalents of DTT in 100mM PB at pH 8.0±0.2 and left at room temperature for 2 hours (Fig. 2). Unreacted DTT was removed using gel permeation chromatography on a Sephadex G-75 column in PB at pH 5.5±0.2. The effluent was concentrated and washed with PB pH 7.4±0.2 using a 10kDa MWCO Microcon filter.
2.8. Fluorescein-labeled homodimeric peptide-based PEG nanocarrier
A fluorescein-PEG5kDa-NHS standard curve was used to correlate fluorescence to molar concentration of the synthesized fluorescein-labeled Cys-protected PEGylated 2-arm peptidic core and its unprotected counterpart. The PEGylated peptidic cores dissolved in 100mM PB pH 7.4±0.2 were mixed and reacted for 16 hours at room temperature. The volume was reduced using ultrafiltration (Microcon 10kDa MWCO) and dimerization (Fig. 2) was confirmed using size exclusion HPLC (Fig. 8).
Fig. 8.

HPLC chromatogram of purified Cys-protected PEGylated 2- arm peptidic core (A, C), crude homodimeric peptide-based PEG nanocarrrier (E) and crude homodimeric nanocarrier spiked with purified Cys-protected PEGylated 2- arm peptidic core (B, D, F). Different flow rates have been used to obtain better resolution. Spiking was performed to confirm the formation of the homodimeric nanocarrier and for better visualization.
2.9. Texas Red-labeled Cys-protected 4-arm peptidic core
The Texas Red addition on the Cys-protected peptidic core with 4 possible attachment sites (4-arm) at lysine was performed with 3 molar excess of Texas Red-NHS dissolved in 30% DMSO in 100 mM PB pH 7.4±0.2 (Fig. 3). The reaction was performed overnight at room temperature. After the completion of labeling, a final concentration of 10 mM Tris buffer pH 7.0±0.2 was added in order to quench unreacted Texas Red.
2.10. Dual labeled heterodimeric peptide-based PEG nanocarrier
The dual labeled heterodimeric nanocarrier was prepared by reacting fluorescein-labeled Cys-unprotected PEGylated 2-arm peptidic core with equimolar amounts of Texas Red-labeled Cys-protected 4-arm peptidic core in 100mM PB pH 7.4±0.2 (Fig. 3). The reaction was performed at room temperature overnight. Using a 10kDa MWCO Microcon, the heterodimer was washed several times until the Texas Red fluorescence reading (Ex = 530nm, Em = 613nm) in the filtrate was constant and insignificant. MALDI-TOF mass spectrometry confirmed the formation of heterodimer (Fig. 9).
Fig. 9.

MALDI-TOF (m/z) spectrum of purified heterodimeric peptide based PEG nanocarrier doubly labeled with fluorescein and Texas Red, showing a peak at molecular weight of 13,375.6 Da confirming the product.
2.11. Biodegradation of heterodimeric nanocarrier
The dual labeled heterodimeric nanocarrier was evaluated for its potential to biodegrade under simulated intracellular and extracellular/blood environments using 3 mM or 10 μM glutathione, respectively. To carry out the degradation studies, the nanocarrier was dissolved in 100 mM PB pH =7.4±0.2 containing of either 3 mM or 10 μM reduced GSH and incubated at 37 °C. At each time points (0, 1, 3, 5, 7, 10 and 60 minutes; in case of 3 mM GSH) and (0, 15, 30, 60 and 90 minutes; in case of 10 μM GSH) solutions were aliquotted and treated with 1% TFA in order to stop the reaction. The TFA treated solution for each time-point was ultrafiltered (Microcon 10 kDa MWCO) for 20 minutes at 12,000 xg to remove the released smaller sized Texas Red-labeled 4-arm peptidic core monomeric component. The retentate was washed 2 times with 1% TFA followed by ultrafiltration. The biodegradation was assessed using a microplate reader at Ex = 530nm, Em = 613nm (for monitoring the reduction of Texas Red due to release of Texas Red-peptidic core monomer into the filtrate after degradation of the heterodimer) and at Ex = 485nm, Em = 535nm (for monitoring the constant signal of free fluorescein-PEGylated peptidic core monomer in the retentate after degradation of the heterodimer) (Figs. 10, 11). All experiments were performed in triplicate.
Fig. 10.
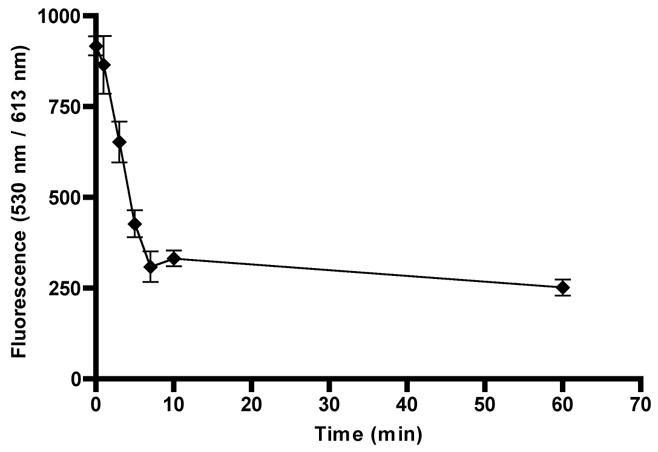
Time course release of the dual labeled biodegradable nanocarrier in 3 mM GSH at 37 °C: The heterodimeric nanocarrier was dissolved in 100 mM PB pH 7.4±0.2 in presence of 3 mM reduced GSH. At each time point (0,1,3,5,7,10 and 60 minutes), 1% TFA was added to the sample to stop the reduction reaction. The zero time point is identical to other sample with the exception that it did not contain any GSH. All experiments were performed in triplicate.
Fig. 11.
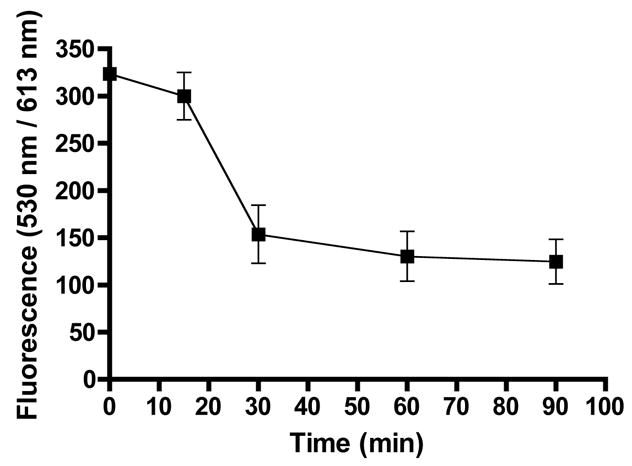
Time course release of the dual labeled biodegradable nanocarrier in 10 μM GSH at 37 °C: The heterodimeric nanocarrier was dissolved in 100 mM PB pH 7.4±0.2 in presence of 10 μM reduced GSH. At each time point (0,15,30,60 and 90 minutes), 1% TFA was added to the sample to stop the reduction reaction by GSH. The zero time point is identical to other sample with the exception that it did not contain any GSH. All experiments were performed in triplicate.
Since the oxidation (and loss) of GSH could confound the stability results in prolonged studies, especially at low concentrations such as 10 μM, a chromogenic assay using 2,2’-dithiodipyridine (TP-TP) was used to validate if the reduced GSH concentration remained constant for the duration of the biodegradation study. In this assay, the reduced form of GSH reacts with TP-TP and releases the chromogenic component 2-thiopyridine (2’-TP) that was monitored at 343nm [33, 34] using a microplate reader. The concentration of reduced GSH employed in this study was determined from the standard curve obtained by reacting 2–50 μM reduced GSH in 100 mM PB pH = 7.4±0.2 with a 10 molar excess of TP-TP (dissolved in DMSO). A linear relationship between the concentration of reduced GSH and 2’-TP was observed (R2 = 0.993). Absorbance at 343 nm remained constant during the 2 hours of incubation at 37 °C suggesting that GSH was not oxidized during the biodegradation experiment and the concentration of reduced GSH remained constant (~10 μM).
3. Results and Discussion
It is widely believed that by treating only disease affected cells; drug dosages and side effects can be reduced, thus improving therapeutic outcomes. As such, drug targeting is an important goal in the treatment of AIDS or cancer since specific cell populations are involved in those diseases. To date, most targeting strategies have focused on controlling the initial distribution of delivery vehicles to the site of the disease. The most commonly used approach involves the selective delivery of drugs to specific cell types using a particulate carrier (e.g., liposomes or nanoparticles) or soluble nanocarriers (e.g., drug-polymer conjugates) with attached cell surface targeting ligands. The specificity of delivery is related to many factors including the type and number of targeting ligands required for optimal cellular uptake. Intracellular disposition and fate are highly dependent on the type of cell surface receptor and may require an additional strategy to promote endosomal escape. Previously, we developed first generation nanocarriers using the chemo-attractant peptide, N-formyl-Met-Leu-Phe (fMLF), which were capable of actively targeting macrophages in vitro and in vivo [18–20]. fMLF was selected as the first targeting ligand for the nanocarriers since the goal was to target macrophages, a phagocytic cell that plays a significant role in the persistence of HIV infection. We also recently showed that intracellular distribution could be controlled by using a Tat peptide to facilitate endosomal escape [35]. This is particularly important for drugs that are hydrophilic and act in the cytosol or must gain access to the nuclear compartment. We now shift our focus to the post-initial body distribution phase. In the current studies, second-generation multimeric peptide-backbone PEG nanocarriers (Figs. 1, 2 and 3) were designed, synthesized, and characterized with the goal of building in specific body intracellular drug release and elimination properties. A biodegradable nanocarrier that is relatively stable in the blood circulation, while being selectively degraded inside target cells, was designed in order to exploit the natural extracellular-intracellular gradient of reducing conditions. The result is that the glutathione-sensitive disulfide bond between the monomeric peptidic units of the nanocarrier is cleaved releasing components with known body elimination pathways.
3.1. PEGylation reaction of nanocarrier monomer
In the current design, the free ε-amine groups of lysines on the 2-arm central peptidic core [acetylated-Lys-βAla-βAla-Lys-βAla-βAla-Cys(TP)-amidated] and the 4-arm central peptidic core [acetylated-Lys-βAla-βAla-Lys-βAla-βAla-Lys-βAla-βAla-Lys-βAla-βAla-Cys(TP)-amidated] are used for the attachment of fluorescein-PEG5kDa and Texas Red, respectively, (Figs. 2 and 3). Thus, it is necessary to quantify the amount of free amines present on the peptidic cores prior to PEGylation or labeling. The most common method used for quantification of peptides having primary amines is based on fluorescamine, a heterocyclic dione reagent that reacts with primary amines to form a fluorescent product [36]. However, fluorescamine also reacts with water at lower rates and peptides/polypeptides tend to absorb moisture from the air. This side reaction often leads to inaccurate measurements. Therefore, a Kaiser chromogenic assay was selected over the conventional fluorescamine assay in order to quantify the peptides used in the current study. Since this assay is typically used to quantify primary amines of peptides during solid phase synthesis [32], it was modified and standardized for quantification of peptides in the liquid phase. The assay was successfully adapted and a typical standard curve (Fig. 4) demonstrated a strong correlation (R2 =0.993) between the amount of primary amines in the 2- and 4-arm peptidic cores and the released chromogenic product.
An inherent problem associated with PEGs is polydispersity. This is particularly true at higher molecular weights [5, 37]. A goal of the current study was to design a nanocarrier with low polydispersity and high yield. As size is a critical determinant of the biodistribution and body persistence of nanocarrriers, high polydispersity is expected to lead to high bioavailability variability and possibly to negative therapeutic outcomes. Lower molecular weight PEGs (~3–5 kDa) have polydispersity values as low as 1.01 whereas they can be as high as 1.2 for larger molecular weight PEGs (~20kDa) [5, 37]. Another complicating factor is the presence of the impurity diol, which ranges from 1–15% depending on the molecular weight of PEG. The diol content in low-mass PEGs (~1%) is much lower than that for higher molecular weight PEGs (~15%) [5, 37]. High diol concentrations lead to unwanted aggregates or cross-linked products resulting in a low yield of the desired product. Therefore, it was hypothesized that attaching multiple low mass PEGs to the peptidic core would result in a higher yield of less polydisperse PEG nanocarriers as compared to attaching a single large PEG. Each PEG unit is attached in close proximity to each other on the peptide backbone resembling a branched or comb structure (Figs. 1, 2 and 3). The PEGylated portion of the nanocarrier resembles a branched PEG similar to PEG2, a second generation PEG [1, 30, 31]. Since branched PEGs have a relatively higher rate of hydration as compared to their linear counterparts, the viscosity radius of a protein that was PEGylated with four copies of a 5 kDa PEG was equivalent to PEGylation with a single 20 kDa PEG [38]. This also appears to hold true for the pharmacokinetics of PEGylated proteins. For example, Knauf et al. [15] demonstrated that the systemic clearance and elimination half-life of recombinant interleukin-2 that was PEGylated with multiple smaller PEGs or one larger PEG was essentially the same in rats. Taken together, these results suggest that the final topology and effective size of the nanocarrier is what determines biological functionality.
Achieving complete PEGylation of all of the ε-amine moieties on the peptidic core was challenging. As demonstrated by MALDI-TOF analysis, the initial PEGylation of a 6 lysine peptidic core where each lysine was separated by only one copy of β-alanine resulted in heterogeneous products containing 2–6 copies of PEG3.4kDa (Fig. 5). It was hypothesized that due to the close proximity of lysine, the PEGylation reaction was hindered. This was addressed by designing a peptidic core consisting of 4 internal lysines (i.e., a 4-arm central peptidic core) with two β-alanine residues repeated after each lysine moiety in order to provide adequate spacing. This spacing was found to be favorable for entry and conjugation of a large diameter (5 kDa) hydrated activated PEG. However, the analytical Kaiser assay indicated that only 50% of the total ε-amines reacted. This represented a significant improvement over the 30% PEGylation observed with the single β-alanine spacer in the peptidic core. However, total PEG content was still low considering that the target molecular size of the nanocarrier should ideally be between 20kDa and 40kDa. This issue was addressed by designing a peptidic core with 2 lysine PEG attachment sites and increasing PEG content by dimerizing the purified 2-arm PEGylated product (Fig. 2). The purification of the PEGylated product using a G-75 Sephadex column is shown in Fig. 6. When the fluorescein-PEG5kDa-NHS polymer was loaded by itself onto the column a single peak was obtained. The higher mobility peak corresponds to the ‘fully’ PEGylated product and the lower mobility peak represents a combination of excess unreacted fluorescein-PEG5kDa-NHS and ‘partially’ PEGylated peptidic core (Fig. 6). The mass of the higher mobility PEGylated product was determined by MALDI-TOF (Fig. 7). The expected molecular weight very closely matched the theoretical value confirming the formation of the PEGylated product (Fig. 2). Each ethylene glycol subunit is associated with two or three water molecules that impart a high hydrodynamic volume to the PEG resulting in a 5–10 fold increase in effective size. Higher DMSO concentrations (70%) resulted in a high PEGylated product yield (69%) whereas the low DMSO conditions resulted in a much lower yield of 32%. It appears that high aqueous conditions (i.e., low DMSO concentrations) lead to higher hydrodynamic volume and high steric hindrance resulting in dramatically reduced PEGylation product yield. Conversely, increasing the DMSO concentration resulted in an environment where PEG is not fully hydrated, steric hindrance is reduced and a higher PEGylated product yield is obtained. Therefore, higher concentrations of DMSO along with 2 copies of β-alanine spacer are crucial for successful PEGylation of peptidic core.
3.2. Homodimerization of peptide-based PEG nanocarrier
The presence of cysteine in the peptidic core enables the production of a dimeric second-generation nanocarrier. By using the multimer approach, the body elimination pathway can be precisely programmed into the nanocarrier design. This is achieved by controlling the degree of branching and the size of the monomers. The current design did not specifically include the cleavage of the monomer to its peptide and PEG components since the peptide did not contribute significantly to the overall size and the monomer size is below the renal filtration threshold. The homodimeric nanocarrier consisted of two monomers of a PEGylated 2-arm peptidic core monomer carrying 2 copies of 5kDa PEG-fluorescein) linked via a disulfide bond (Fig. 2). This design resulted in a nanocarrier with a MW of 21,950 Da. By reacting an equimolar amount of purified thiopyridine protected PEGylated 2-arm peptidic core with its unprotected counterpart, homodimerization was achieved (Fig. 2). The purified Cys-protected PEGylated 2-arm peptidic core generated single peaks at retention times of ~ 7 and ~ 11 minutes at flow rates of 0.7 and 0.5 ml/min, respectively, using size exclusion-HPLC (Fig. 8; Panels A and C). At flow rate of 0.3 ml/min, the crude homodimerization product exhibited an obvious shoulder with slower mobility (Fig. 8, Panel E). In order to further resolve the product, the crude product was spiked with purified Cys-protected PEGylated 2-arm peptidic core (Fig. 8; Panel F) at the same flow rate. This addition proved to be crucial for visualization of the dimerized product. At flow rates of 0.5 and 0.7 ml/min the homodimerized product was observed as a distinct shoulder with slower mobility (Fig. 8; Panels B and D) when spiked with free PEGylated 2-arm peptidic core. The flow rate of 0.3 ml/min gave the best resolution with minimal absorptive loss.
3.3. Heterodimeric peptide-based PEG nanocarrier and its biodegradation characteristics
A dual labeled heterodimeric nanocarrier consisting of a PEGylated 2-arm peptidic core monomer carrying two copies of Fluorescein-PEG5kDa and a non-PEGylated 4-arm peptidic core monomer carrying ~four copies of Texas Red linked via a disulfide bond was prepared for the purposes of assessing its potential for biodegradation (Fig. 3). The purified heterodimeric nanocarrier was subjected to MALDI-TOF mass spectrometry (Fig. 9). The obtained mass exhibited an increase to 13,375 Da consistent with the expected increase in molecular weight confirming the presence of the product. The biodegradation of the nanocarrier was investigated in environments that mimic the reducing concentration in blood (10 μM GSH) or inside cells (3mM GSH) [39, 40]. The reduction of the disulfide linkage and subsequent release of the nanocarrier monomers was monitored at each time-point by stopping the reaction using an acidifying solution. The small 4-arm peptidic core monomer carrying Texas Red passed through a 10 kDa MWCO filter while the free fluorescein-PEGylated 2-arm peptidic core is retained on the filter. Thus, simulated biodegradation was monitored by the reduction in the Texas Red fluorescence signal. The Texas Red fluorescence signal in the retentate is attributed to the intact heterodimeric nanocarrier. Biodegradation in the presence of 3 mM GSH at 37°C resulted in complete release of the Texas Red labeled 4-arm monomer from the intact heterodimeric nanocarrier in 7 minutes (Fig. 10). When the solution containing the heterodimer was modified to a final concentration of 1% TFA prior to the addition of GSH, release was not observed and the values were nearly identical to the zero time-point even after 2 hours of incubation at 37°C. This demonstrates that acidifying the solution stopped the reaction thus allowing for proper temporal quantification of biodegradation. In addition, the fluorescein tag remained attached to terminal end of PEG during the study as evidenced by a lack of signal at Ex = 485nm; Em = 535 nm from the filtrate. Therefore, at conditions that mimic the reducing environment inside the cell, the heterodimeric nanocarrier showed complete degradation to its monomeric components. Upon incubation of the doubly labeled nanocarrier in 10 μM GSH, biodegradation to its component monomers was complete after 60 minutes (Fig. 11). Prolonged incubation or addition of excess GSH (3 mM for one hour) did not further reduce the fluorescence reading. Since the oxidation and loss of GSH during the time course of the study was not observed, the stabilization method was considered valid. Although the nanocarrier was stable for ~1 hour in a reducing environment similar to the blood, the target blood stability is probably in the range of 24 hours or so in order to provide adequate exposure of the nanocarrier to affected cells. Our group and others have shown that the rate of biodegradation of the disulfide linkage can be manipulated by altering steric hindrance [34, 41, 42]. For example, a disulfide bond prepared from a sterically hindered cysteamine analogue linker, 1-amino-2-methyl-2-propanethiol, showed about 100-times slower degradation rate than that of the corresponding less hindered cysteamine linker (t1/2 of GSH-dependent disulfide degradation = 3min) [34]. The introduction of sterically hindered methyl groups to the peptidic core adjacent to the cysteine residue may result in a longer persistence for intact nanocarrier. Since the disulfide cleavage rate is proportional to glutathione concentration [42], the rate of biodegradation can be readily controlled by selection of such sterically hindered cysteines in the peptidic backbone of the nanocarrier prior to disulfide bond formation. In addition to using disulfide bonds, more stable carbamate or ester linkages can also be used to prolong the biodegradation rate.
4. Conclusions
In the current studies, a second-generation peptidic core monomer was identified that allowed for optimal attachment of multiple PEGs in stoichiometric amounts with low polydispersity. High product yields were obtained by selecting the optimal spacing requirements in the peptide core and by using high concentrations of DMSO to reduce the hydrodynamic volume of solvated PEG. Homodimeric and heterodimeric biodegradable nanocarriers were synthesized and characterized from PEGylated and non-PEGylated peptide cores. Selective intracellular biodegradation was observed in vitro. The modular synthesis of these nanocarriers has the advantage of minimizing polydispersity, a challenge that is always present with polymeric nanocarriers. The design is sufficiently flexible so that the component peptidic monomers could be used to link PEGs, imaging agents, drugs, targeting ligands or other peptidic cores. Two important design components, the biodegradable bonds between monomeric peptidic core units and using PEGs of the appropriate size in order to promote renal or hepatic elimination, allows for the pre-programming of body elimination properties into the nanocarrier.
Acknowledgments
Partial financial support from NIH AI51214 and the Parke-Davis Endowed Chair in Pharmaceutics and Drug Delivery is gratefully acknowledged. A fellowship from the American Foundation for Pharmaceutical Education to Matthew Palombo is also acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Perry CM, Jarvis B. Peginterferon-alpha-2a (40 kD): a review of its use in the management of chronic hepatitis C. Drugs. 2001;61(15):2263–88. doi: 10.2165/00003495-200161150-00013. [DOI] [PubMed] [Google Scholar]
- 2.Gupta S, Jen J, Kolz K, Cutler D. Dose selection and population pharmacokinetics of PEG-Intron in patients with chronic myelogenous leukaemia. Br J Clin Pharmacol. 2007;63(3):292–9. doi: 10.1111/j.1365-2125.2006.02757.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nucci ML, Shorr R, Abuchowski A. The therapeutic value of poly (ethylene glycol)-modified proteins. Advanced Drug Delivery Reviews. 1991;6(2):133–151. [Google Scholar]
- 4.Stigsnaes P, Frokjaer S, Bjerregaard S, van de Weert M, Kingshott P, Moeller EH. Characterisation and physical stability of PEGylated glucagon. International Journal of Pharmaceutics. 2007;330(1–2):89–98. doi: 10.1016/j.ijpharm.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 5.Kozlowski A, Harris JM. Improvements in protein PEGylation: pegylated interferons for treatment of hepatitis C. J Control Release. 2001;72(1–3):217–24. doi: 10.1016/s0168-3659(01)00277-2. [DOI] [PubMed] [Google Scholar]
- 6.Bailon P, Palleroni A, Schaffer CA, Spence CL, Fung WJ, Porter JE, et al. Rational design of a potent, long-lasting form of interferon: a 40 kDa branched polyethylene glycol-conjugated interferon alpha-2a for the treatment of hepatitis C. Bioconjug Chem. 2001;12(2):195–202. doi: 10.1021/bc000082g. [DOI] [PubMed] [Google Scholar]
- 7.Wattendorf U, Merkle HP. PEGylation as a tool for the biomedical engineering of surface modified microparticles. J Pharm Sci. 2008;97(11):4655–69. doi: 10.1002/jps.21350. [DOI] [PubMed] [Google Scholar]
- 8.Nakaoka R, Tabata Y, Yamaoka T, Ikada Y. Prolongation of the serum half-life period of superoxide dismutase by poly (ethylene glycol) modification. Journal of Controlled Release. 1997;46(3):253–261. [Google Scholar]
- 9.Venturoli D, Rippe B. Ficoll and dextran vs. globular proteins as probes for testing glomerular permselectivity: effects of molecular size, shape, charge, and deformability. Am J Physiol Renal Physiol. 2005;288(4):F605–13. doi: 10.1152/ajprenal.00171.2004. [DOI] [PubMed] [Google Scholar]
- 10.Ahsan F, Rivas IP, Khan MA, Torres Suarez AI. Targeting to macrophages: role of physicochemical properties of particulate carriers--liposomes and microspheres--on the phagocytosis by macrophages. J Control Release. 2002;79(1–3):29–40. doi: 10.1016/s0168-3659(01)00549-1. [DOI] [PubMed] [Google Scholar]
- 11.Foged C, Brodin B, Frokjaer S, Sundblad A. Particle size and surface charge affect particle uptake by human dendritic cells in an in vitro model. Int J Pharm. 2005;298(2):315–22. doi: 10.1016/j.ijpharm.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 12.Thiele L, Rothen-Rutishauser B, Jilek S, Wunderli-Allenspach H, Merkle HP, Walter E. Evaluation of particle uptake in human blood monocyte-derived cells in vitro. Does phagocytosis activity of dendritic cells measure up with macrophages? J Control Release. 2001;76(1–2):59–71. doi: 10.1016/s0168-3659(01)00412-6. [DOI] [PubMed] [Google Scholar]
- 13.Salazar VBY, Wettstein R, Cabrales P, Tsai AG, Intaglietta M. Microvascular experimental evidence on the relative significance of restoring oxygen carrying capacity vs. blood viscosity in shock resuscitation. Biochim Biophys Acta. 2008 doi: 10.1016/j.bbapap.2008.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Conover CD, Linberg R, Gilbert CW, Shum KL, Shorr RGL. Effect of polyethylene glycol conjugated bovine hemoglobin in both top-load and exchange transfusion rat models. Artificial organs. 1997;21(10):1066–1075. doi: 10.1111/j.1525-1594.1997.tb00444.x. [DOI] [PubMed] [Google Scholar]
- 15.Knauf MJ, Bell DP, Hirtzer P, Luo ZP, Young JD, Katre NV. Relationship of effective molecular size to systemic clearance in rats of recombinant interleukin-2 chemically modified with water-soluble polymers. Journal of Biological Chemistry. 1988;263(29):15064–15070. [PubMed] [Google Scholar]
- 16.Bailon P, Berthold W. Polyethylene glycol-conjugated pharmaceutical proteins. Pharmaceutical Science & Technology Today. 1998;1(8):352–356. [Google Scholar]
- 17.Yamaoka T, Tabata Y, Ikada Y. Distribution and tissue uptake of poly(ethylene glycol) with different molecular weights after intravenous administration to mice. J Pharm Sci. 1994;83(4):601–6. doi: 10.1002/jps.2600830432. [DOI] [PubMed] [Google Scholar]
- 18.Pooyan S, Qiu B, Chan MM, Fong D, Sinko PJ, Leibowitz MJ, et al. Conjugates bearing multiple formyl-methionyl peptides display enhanced binding to but not activation of phagocytic cells. Bioconjug Chem. 2002;13(2):216–23. doi: 10.1021/bc0100657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wan L, Pooyan S, Hu P, Leibowitz MJ, Stein S, Sinko PJ. Peritoneal macrophage uptake, pharmacokinetics and biodistribution of macrophage-targeted PEG-fMLF (N-formyl-methionyl-leucyl-phenylalanine) nanocarriers for improving HIV drug delivery. Pharm Res. 2007;24(11):2110–9. doi: 10.1007/s11095-007-9402-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wan L, Zhang X, Pooyan S, Palombo MS, Leibowitz MJ, Stein S, et al. Optimizing size and copy number for PEG-fMLF (N-formyl-methionyl-leucyl-phenylalanine) nanocarrier uptake by macrophages. Bioconjug Chem. 2008;19(1):28–38. doi: 10.1021/bc070066k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakai T, Kanamori T, Sando S, Aoyama Y. Remarkably size-regulated cell invasion by artificial viruses. Saccharide-dependent self-aggregation of glycoviruses and its consequences in glycoviral gene delivery. J Am Chem Soc. 2003;125(28):8465–75. doi: 10.1021/ja035636f. [DOI] [PubMed] [Google Scholar]
- 22.Osaki F, Kanamori T, Sando S, Sera T, Aoyama Y. A quantum dot conjugated sugar ball and its cellular uptake. On the size effects of endocytosis in the subviral region. J Am Chem Soc. 2004;126(21):6520–1. doi: 10.1021/ja048792a. [DOI] [PubMed] [Google Scholar]
- 23.Desai MP, Labhasetwar V, Walter E, Levy RJ, Amidon GL. The mechanism of uptake of biodegradable microparticles in Caco-2 cells is size dependent. Pharm Res. 1997;14(11):1568–73. doi: 10.1023/a:1012126301290. [DOI] [PubMed] [Google Scholar]
- 24.Prabha S, Zhou WZ, Panyam J, Labhasetwar V. Size-dependency of nanoparticle-mediated gene transfection: studies with fractionated nanoparticles. Int J Pharm. 2002;244(1–2):105–15. doi: 10.1016/s0378-5173(02)00315-0. [DOI] [PubMed] [Google Scholar]
- 25.Roberts MJ, Bentley MD, Harris JM. Chemistry for peptide and protein PEGylation. Adv Drug Deliv Rev. 2002;54(4):459–76. doi: 10.1016/s0169-409x(02)00022-4. [DOI] [PubMed] [Google Scholar]
- 26.Monfardini C, Schiavon O, Caliceti P, Morpurgo M, Harris JM, Veronese FM. A branched monomethoxypoly (ethylene glycol) for protein modification. Bioconjugate Chem. 1995;6(1):62–69. doi: 10.1021/bc00031a006. [DOI] [PubMed] [Google Scholar]
- 27.Veronese FM, Monfardini C, Caliceti P, Schiavon O, Scrawen MD, Beer D. Improvement of pharmacokinetic, immunological and stability properties of asparaginase by conjugation to linear and branched monomethoxy poly (ethylene glycol) Journal of Controlled Release. 1996;40(3):199–209. [Google Scholar]
- 28.Yamasaki N, Matsuo A, Isobe H. Novel polyethylene glycol derivatives for modification of proteins. Agricultural and biological chemistry. 1988;52(8):2125–2127. [Google Scholar]
- 29.Veronese FM, Caliceti P, Schiavon O. Branched and linear poly (ethylene glycol): influence of the polymer structure on enzymological, pharmacokinetic, and immunological properties of protein conjugates. Journal of Bioactive and Compatible Polymers. 1997;12(3):196. [Google Scholar]
- 30.Caliceti P, Schiavon O, Veronese FM. Biopharmaceutical properties of uricase conjugated to neutral and amphiphilic polymers. Bioconjug Chem. 1999;10(4):638–46. doi: 10.1021/bc980155k. [DOI] [PubMed] [Google Scholar]
- 31.Caliceti P, Schiavon O, Veronese FM. Immunological properties of uricase conjugated to neutral soluble polymers. Bioconjug Chem. 2001;12(4):515–22. doi: 10.1021/bc000119x. [DOI] [PubMed] [Google Scholar]
- 32.Sarin VK, Kent SB, Tam JP, Merrifield RB. Quantitative monitoring of solid-phase peptide synthesis by the ninhydrin reaction. Anal Biochem. 1981;117(1):147–57. doi: 10.1016/0003-2697(81)90704-1. [DOI] [PubMed] [Google Scholar]
- 33.Grassetti DR, Murray JF., Jr Determination of sulfhydryl groups with 2,2’- or 4,4’-dithiodipyridine. Arch Biochem Biophys. 1967;119(1):41–9. doi: 10.1016/0003-9861(67)90426-2. [DOI] [PubMed] [Google Scholar]
- 34.Huang SY, Pooyan S, Wang J, Choudhury I, Leibowitz MJ, Stein S. A polyethylene glycol copolymer for carrying and releasing multiple copies of cysteine-containing peptides. Bioconjug Chem. 1998;9(5):612–7. doi: 10.1021/bc980038p. [DOI] [PubMed] [Google Scholar]
- 35.Zhang X, Jin Y, Plummer M, Pooyan S, Gunaseelan S, Sinko P. Endocytosis and membrane potential are required for HeLa cell uptake of R.I.-CKTat9, a retro inverso Tat cell penetrating peptide. Mol Pharm. 2009 doi: 10.1021/mp800121f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Udenfriend S, Stein S, Bohlen P, Dairman W, Leimgruber W, Weigele M. Fluorescamine: a reagent for assay of amino acids, peptides, proteins, and primary amines in the picomole range. Science. 1972;178(63):871–2. doi: 10.1126/science.178.4063.871. [DOI] [PubMed] [Google Scholar]
- 37.Veronese FM. Peptide and protein PEGylation a review of problems and solutions. Biomaterials. 2001;22(5):405–417. doi: 10.1016/s0142-9612(00)00193-9. [DOI] [PubMed] [Google Scholar]
- 38.Fee CJ. Size comparison between proteins PEGylated with branched and linear poly(ethylene glycol) molecules. Biotechnol Bioeng. 2007;98(4):725–31. doi: 10.1002/bit.21482. [DOI] [PubMed] [Google Scholar]
- 39.Meister A. Glutathione deficiency produced by inhibition of its synthesis, and its reversal; applications in research and therapy. Pharmacol Ther. 1991;51(2):155–94. doi: 10.1016/0163-7258(91)90076-x. [DOI] [PubMed] [Google Scholar]
- 40.Meister A, Griffith OW, Novogrodsky A, Tate SS. New aspects of glutathione metabolism and translocation in mammals. Ciba Found Symp. 1979;(72):135–61. doi: 10.1002/9780470720554.ch9. [DOI] [PubMed] [Google Scholar]
- 41.Goff DA, Carroll SF. Substituted 2-iminothiolanes: reagents for the preparation of disulfide cross-linked conjugates with increased stability. Bioconjug Chem. 1990;1(6):381–6. doi: 10.1021/bc00006a003. [DOI] [PubMed] [Google Scholar]
- 42.Trimble SP, Marquardt D, Anderson DC. Use of designed peptide linkers and recombinant hemoglobin mutants for drug delivery: in vitro release of an angiotensin II analog and kinetic modeling of delivery. Bioconjug Chem. 1997;8(3):416–23. doi: 10.1021/bc970037h. [DOI] [PubMed] [Google Scholar]