

Abstract
Alzheimer’s disease (AD) neuropathology is characterized by loss of synapses and neurons, neuritic plaques consisting of β-amyloid (Aβ) peptides, and neurofibrillary tangles consisting of intracellular aggregates of hyperphosphorylated tau protein in susceptible brain regions. Aβ oligomers trigger a cascade of pathogenic events including tau hyperphosphorylation and aggregation, inflammatory reactions, and excitotoxicity that contribute to the progression of AD. The molecular chaperone Hsp90 facilitates the folding of newly synthesized and denatured proteins and is believed to play a role in neurodegenerative disorders in which the defining pathology results in misfolded proteins and the accumulation of protein aggregates. Some agents that inhibit Hsp90 protect neurons against Aβ toxicity and tau aggregation, and assays for rapidly screening potential Hsp90 inhibitors are of interest. We used the release of the soluble cytosolic enzyme lactate dehydrogenase (LDH) as an indicator of the loss of cell membrane integrity and cytotoxicity resulting from exposure to Aβ peptides to evaluate the neuroprotective properties of novel novobiocin analogues and established Hsp90 inhibitors. Compounds were assessed for potency in protecting proliferating and differentiated SH-SY5Y neuronal cells against Aβ-induced cell death; the potential of each agent alone was also determined. The data indicated that several of the compounds decreased Aβ toxicity even at low nanomolar concentrations and, unexpectedly, were more potent in protecting the undifferentiated cells against Aβ. The novobiocin analogues alone were not toxic even up to 10 μM concentrations whereas GDA and the parent compound, novobiocin, were toxic at 1 and 10 μM, respectively. The results suggest that novobiocin analogues may provide novel leads for the development of neuroprotective drugs.
1. Introduction
Considerable evidence suggests that the accumulation of β-amyloid (Aβ) oligomers or fibrils lead to the neurodegeneration that occurs in Alzheimer’s disease (AD).1, 2 Deposits of insoluble Aβ are found in the brains of patients with AD and are one of the pathological hallmarks of this disease. These Aβ aggregates exhibit toxic properties and are likely linked to the induction of inflammatory processes that result in neuronal cell death.3 In addition to the Aβ aggregates, the microtubule-associated protein Tau is hyperphosphorylated and misfolded, leading to neurofibrillary tangles (NFTs) that are also hallmarks of AD pathology. Tau is normally expressed in the cytoplasm of neurons where it serves to stabilize the microtubule network in axons. In AD, tau becomes hyperphosphorylated and dissociates from microtubules, forming filamentous aggregates of misfolded proteins that polymerize into NFTs.4 The presence of misfolded proteins suggests that enhancement of the protein-folding machinery may exhibit therapeutic potential.
Hsp90 is a pivotal ATP-dependent molecular chaperone that interacts with many co-chaperones to fold proteins or target misfolded proteins for degradation. Hsp90 contains two nucleotide binding sites. The N-terminal domain binds the natural products geldanamycin (GDA), radicicol and their derivatives, which modulate at least two different conformational states. Recently, novobiocin, a coumarin-containing DNA gyrase inhibitor that binds to the C-terminal nucleotide binding site and inhibits Hsp90 function was elucidated.5,6,7 The C-terminal region modulates the N-terminal ATPase activity of Hsp90.8,9,10 Binding of ATP to the N-terminal domain is required in order for the C-terminal ATP site to become available for nucleotide binding. Based on previous studies, there is only a small therapeutic window for N-terminal inhibitors because of toxicity that is produced upon client protein degradation.11,12 Consequently, the development of such compounds to treat neurodegenerative diseases is limited. Novobiocin analogues have proven to be the most promising class of C-terminal inhibitors yet identified. Although other DNA gyrase inhibitors may possess similar activities, they remain untested for Hsp90 inhibition. The natural product itself induces degradation of Hsp90 clients at high concentration (~700 μM in SKBr3 cells),13 and has therefore required subsequent development to produce more efficacious compounds.
In these studies we used the release of the cytosolic enzyme lactate dehydrogenase (LDH) from immortalized neuronal SH-SY5Y cells as a measure of cell viability in testing the protective effects of several Hsp90 inhibitors. LDH catalyzes the conversion of pyruvate to lactate with concomitant conversion of NADH to NAD+. The protein is released into the medium following disruption of the cell membrane, which leads to cell death. Therefore the LDH activity released is not only used as an indicator of cell membrane integrity, but also as a useful method to determine cytotoxicity. Although similar methods have been previously reported, the goal herein was to utilize these conditions and apply them to a high-throughput screening format that has now been optimized for 96-well plates.
A series of novobiocin analogues, including A4, A4-dimer and an additional analogue (KU32)14–15 from our laboratory were evaluated along with several previously identified Hsp90 natural product inhibitors such as celastrol,16 gedunin,17 EGCG,18 GDA19 and gamendazole.20
To evaluate these compounds for their ability to protect neuronal cells against Aβ-induced toxicity, an assay was developed employing the SH-SY5Y cell line that resulted in a reproducible Z-factor for this system. A Z-factor of 0.76, which was obtained via this protocol, indicates a highly reproducible and accurate measure of robustness of the assay. Furthermore, it greatly reduces the probability that a “hit” has occurred by random coincidence. Utilizing this assay in an HTS format will allow rapid detection of chemical modulators that protect such cells from Aβ-induced toxicity.21
2. Experimental
2.1 A library of Novobiocin Analogues
A library of novobiocin analogues was designed to probe the essential nature of several residues found on the natural product and to expeditiously reveal modifications that could enhance Hsp90 inhibition.14, 15 All compounds were prepared as a 1 M stock solution (DMSO).
2.2 Chemicals
Epigallocatechin Gallate (EGCG) and retinoic acid (RA) were purchased from Sigma (St. Louis, MO, USA). Celastrol and gedunin were from Cayman Chemical (Michigan, USA). All compounds were solubilized in DMSO. All drugs were prepared as 1M stock and stored as single use aliquots at −20°C. β-Amyloid (Aβ25–35) was purchased from AnaSpec Inc. (San Jose, CA, USA).
2.3 Cell culture of SH-SY5Y cells
The SH-SY5Y neuroblastoma cells were initially grown in DMEM medium supplemented with 10% fetal bovine serum (FBS), 1% penicillin, and 100 μg/ml streptomycin in 75 cm2 vented culture flasks. The cells were grown at 37°C in 5% CO2, 95% humidified air. Medium was changed every 2–3 days. When cells had reached 80–90% confluency in the flask, they were trypsinized and seeded onto 96 wells plates.
2.4 LDH assays
The activity of LDH released into the medium was used to determine cell viability under the various experimental conditions. Following treatment with the indicated concentrations of drugs or DMSO alone, cells were exposed to 10 μM Aβ25–35 in serum-free DMEM medium for 48 h. The medium collected from the control and drug-treated wells was centrifuged at 10,000 × g for 10 min to remove floating cells, and aliquots (5 μL) of the supernatants were transferred to news 96-well plates for measurement of LDH activity. The reaction mixture consisted of 50 mM phosphate buffer, pH 7.4, 1 mM pyruvate and 0.2 mM NADH in a total volume of 0.2 mL/well. The oxidation of NADH was followed kinetically by measuring the change in absorbance at 490 nm over 5 min at 25°C. The LDH activity released in the presence of 10 μM Aβ for 48 h was considered to be the maximal level of cell death, i.e., 100%. Effects of the Hsp90 inhibitors in the presence of Aβ were evaluated as the percent of cell death that occurred when the indicated concentrations of the drugs were added to the cultures 2 h before the Aβ. When the potential toxicity of the agents added without Aβ was determined, the actual LDH activity in the medium was compared with that in DMSO-treated cells. Cell viability was determined using the trypan blue (0.04%) exclusion assay.
2.5 Sensitivity of proliferating vs differentiated cells to drugs
In order determine if SH-SY5Y cells differentiated to a more neuronal phenotype by exposure to retinoic acid (RA) differed in their sensitivity to the drug treatments, cells were plated at densities of 3×104 in 48 well plates and allowed to grow for 7 days in the presence of 10 μM of RA, during which time they stopped proliferating and began developing neurites similar to those in primary neurons in culture. Following the differentiation step, the cells were exposed to various concentrations of the drugs in the presence of Aβ as was done for proliferating cells. The cells were examined daily under a phase contrast microscope for assessment of morphologic changes.
2.6 Statistical analysis
All values are expressed as mean ± S.E., and statistical analysis was carried out using Student’s t-test for comparisons between two groups. P values less than 0.05 were considered significant.
2.7 Suitability for high throughput screening
Once optimal conditions were found, the Z-factor was calculated as described by Zhang and colleagues with a value > 0.5 indicating an excellent assay with high statistical reliability. The Z-factor was determined in 10 positive controls (with 100 nM drugs) and 10 negative controls (with vehicle DMSO). Plates were shaken for approximately 30 s to ensure homogeneity. After incubation and shaking at 23 ºC for 30 min, the absorbance was monitored at 490 nm. The Z-factor was determined by solution of the following equation. 16
3. Results
It has been shown that treatment of embryonic primary neurons and neuronal cell cultures with Aβ25–35 produces distinct morphological changes and eventual cell death, similar to the conditions observed in human patients with AD. Pretreatment of neuronal cells with neuroprotective agents can reduce or abolish these toxic effects.22 Neuroprotective effects of drugs are typically determined in primary neurons derived from embryonic rat brain exposed to 10 μM Aβ in the presence or absence of drug for 48 h. This requires the sacrifice of a large number of embryonic pups and labor-intensive preparations that lead to a small number of primary neurons. Such preparations are not amenable to high throughput screening, suggesting the use of immortalized cell lines such as SH-SY5Y neuroblastoma cells that proliferate rapidly, but can also be stimulated to differentiate can serve as an appropriate surrogate. Aβ25–35 rather than the more physiological Aβ1–42 peptide was used for all of the initial screening. In a limited series of assays we tested effects of the agents against Aβ1–42 – induced toxicity and confirmed our findings with the shorter peptide as the toxic stimulus. Pretreatment of non-differentiated SH-SY5Y cells with a series of Hsp90 inhibitors markedly decreased Aβ-induced toxicity in a dose-dependent manner as illustrated in the left column figure 3. Minor neuroprotective effects were associated with drug concentrations as low as 0.01 nM for KU32, A4 and A4 dimer, but >50 % protection was not observed below ~1 nM for these agents and below 100 nM for other drugs. Treatment of SH-SY5Y neuronal cells with each drug alone at 100 nM did lead to some cell death, suggesting that higher concentrations of the agents may have some adverse effects when cells are not under the stress induced by the Aβ peptide. Comparison of the neuroprotective activity of these agents, determined that KU32, A4 and A4-dimer exhibit greater efficacy than the other compounds. (Table 1)
Figure 3.

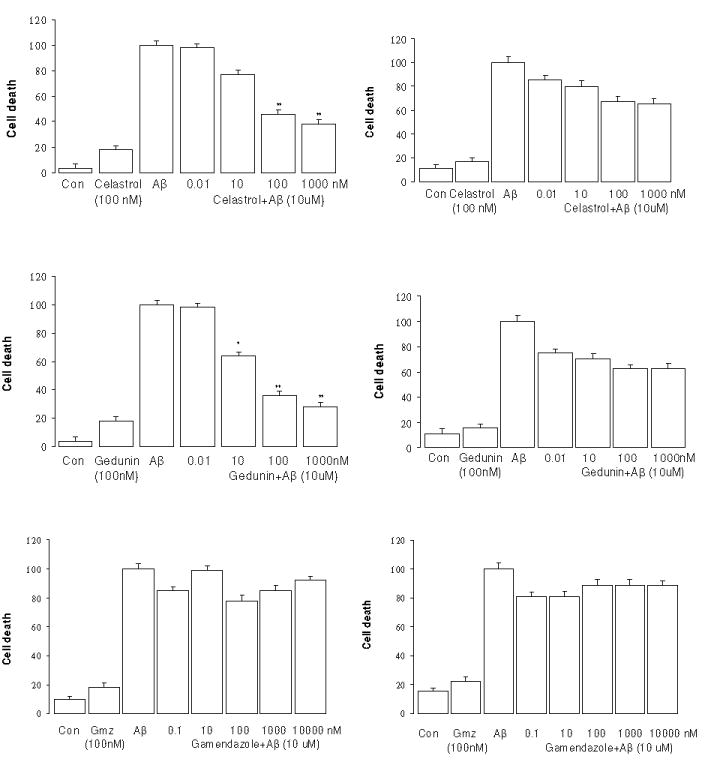
Dose dependence of protection against Aβ25–35 toxicity in the SH-SY5Y by various drugs. (Left, non-differentiated; right, differentiated by retinoic acid). * p<0.05 and ** p<0.01 vs Aβ Control
Table 1.
EC50 values for neuroprotection
| Groups | EC50 (nM) |
|---|---|
| KU32 | 1.490 ± 0.421 |
| A4 | 4.850 ± 0.336 |
| A4 dimer | 4.482 ± 0.365 |
| GDA | 42.69 ± 3.412 |
| Novobiocin | 48.39 ± 3.432 |
| EGCG | 39.87 ± 3.336 |
| Celastrol | 37.64 ± 3.621 |
| Gedunin | 38.95 ± 3.225 |
When the neuroprotective effects of the agents were tested in the SH-SY5Y cells differentiated by the addition of RA, efficacy of the drugs was significantly lower and the dose-dependence shifted to require higher concentrations to achieve even 40% protection (Fig. 3, right panel, p>0.05). Only KU-32 provided nearly 70% protection at 1 μM. This was somewhat unexpected given our previous report that A4 (at 100 nM) provided nearly complete protection against Aβ in primary embryonic neurons.23 Whether genotypic differences in the responses of primary neurons and RA-differentiated SH-SY5Y cells to Aβ or to the drugs are responsible for the differential efficacy remains undetermined. Gamendazole did not affect either type of cell.
We have previously reported that GDA exhibited marked anti-proliferative activity against SkBr3 and MCF-7 cell lines at (1 μM) and induced significant cell death in primary neurons at 10 μM.23 The novobiocin analogue A4, on the other hand, showed no anti-proliferative activity and no neuronal toxicity at 10 μM.23 We tested the effects of the parent compound, novobiocin, and GDA on the SH-SY5Y cells to determine if they also protect against Aβ toxicity. These 2 agents did protect the cells at concentrations of 10 nM or higher, and they also did this much more effectively in the non-differentiated cells (Fig. 4). The data indicate that GDA and novobiocin were less potent than the novobiocin analogues in protecting against Aβ (Table 1). Even though most of the Hsp90 inhibitors did afford some protection against Aβ, visual examination of the cultures suggested that higher concentrations of both GDA and novobiocin alone led to some cell death. Upon increasing the concentration of drugs, ranging from 0.001 to 10 μM, only GDA and novobiocin led to an ~3-fold increase in cell death above controls in both non-differentiated and differentiated cells. In the assay, novobiocin and GDA produced nearly equivalent EC50 values for neuroprotection, however, Hsp90 exhibits ~ 100–1000 fold lower binding affinity for novobiocin compared to GDA, which suggests that inhibitors of the N- and C-termini manifest alternative mechanisms for Hsp90 modulation. No toxicity was observed with KU-32, A4, or the A4-dimer even at 10 μM (data not shown).
Figure 4.
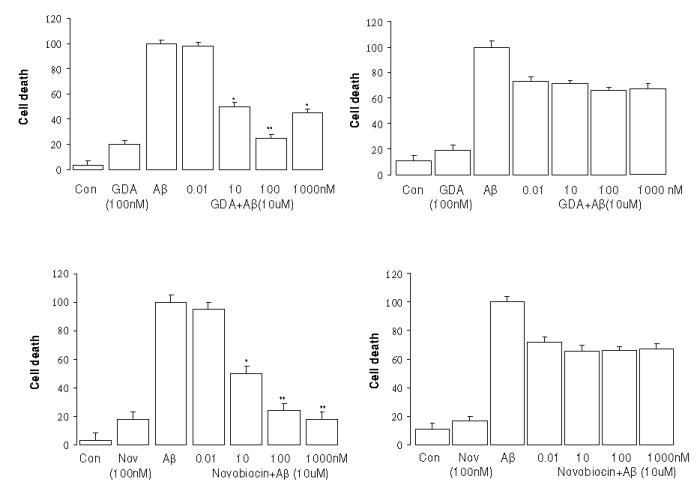
Dose dependent GDA and novobiocin protection against Aβ25–35 toxicity. (Left, without retinoic acid; Right, with retinoic acid). * p<0.05 and ** p<0.01 vs Aβ Control
The differential toxicity of GDA and novobiocin compared to the analogues was visually most apparent in cultures of the differentiated cells, as shown in the representative images provided in figure 5, the cells elaborate neurites that lead to the formation of cell-to-cell networks. Progressive network formation with long neuritic projections was clearly visible in the control cells as well as in those exposed to 10 μM KU-32, A4, and A4-dimer (Fig. 5A,B,C, and D) for 24 h. However the cells exposed to 10 μM novobiocin or GDA consistently had very short neurites in the case of novobiocin and many enlarged and beaded projections with GDA. These latter morphological changes are indicative of damage to the differentiated cells. These observations suggest that KU32, A4 and A4-dimer are likely to be more suitable than GDA and novobiocin as leads for the development of neuroprotective agents.
Figure 5.


The effects of Hsp90 inhibitors on the morphological properties of SH-SY5Y cells differentiated as described in the Methods. Phase contrast images were taken 24 h after addition of drugs. A. Control cells were exposed only to DMSO. B, C, and D represent cells exposed to 10 μM KU32, A4 and A4 dimer respectively. E and F are representative of cells exposed to 10 μM novobiocin or GDA respectively. G. Cell viablility was quantified by the trypan blue exclusion assay. The images are representative of numerous wells in which the high concentrations of the drugs were tested.
4. Discussion
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder, which ultimately results in brain cell death.24 The etiology of the sporadic form of AD is complex, with an interaction of both genetic and environmental risk factors.25 A key event in AD pathogenesis appears to be the conversion of Aβ from its soluble monomeric form into various oligomers or aggregates within the brain, followed by the hyperphosphorylation and misfolding of Tau. Aggregates of Aβ1–42 and tau have been proposed to cause death by activation of components of the apoptotic pathway, induction of oxidative stress, and formation of free radicals,26–30 all of which contribute to cell death.
Current treatments for AD are relatively ineffective and do little besides delay the onset of certain symptoms. Consequently, there is an increasing need for therapeutics that can effectively treat the disease as demographics are leading to increased incidence of AD. 31
Hsp90 has a profound effect on critical cellular processes as a consequence of its diverse biological roles. 32, 33 Hsp90 exerts its effects through other co-chaperones and partner proteins, forming a major chaperone complex that can protect cells against toxicity resulting from the accumulation of protein aggregates. It has been proposed that increased chaperone activity may refold aggregated proteins and thus have potential therapeutic implications for the numerous neurodegenerative diseases characterized by misfolded proteins. Recently, Greengard and coworkers demonstrated that increased expression of Hsp90 and Hsp70 in response to inhibition of Hsp90 by GDA resulted in decreased tau aggregates and provided neuroprotection in cells that expressed mutant tau that becomes hyperphosphorylated as in AD brain.34 Similarly we reported that A4 protects primary neurons against Aβ toxicity and led to up-regulation of Hsp70, a chaperone linked to the regulation of abnormal tau in neurons23. Therefore, regulation of Hsp90 expression levels can be pursued as a novel approach toward the treatment of diseases caused by the accumulation of protein aggregates.
The limited therapeutic potential of previously identified Hsp90 inhibitors for neurodegenerative diseases, including GDA and novobiocin, suggests that improved analogues are necessary to provide clinically useful alternatives. Development of novobiocin analogues to improve the poor Hsp90 inhibitory activity of the agent has been attempted. 8 We have generated a library of novobiocin analogues,14,15 several of which demonstrated little or no cell toxicity, and this includes the KU32, A4 and the A4-dimer which were used in these studies.
Similarly, celastrol was reported to regulate the Hsp90 pathway and induce heat shock response similar to other Hsp90 inhibitors. 16 By conducting molecular modeling studies, researchers proposed that celastrol blocked Cdc37 binding to Hsp90 instead of binding to the Hsp90 ATP binding pockets. Immunoprecipitation studies confirmed that celastrol did indeed disrupt Hsp90-Cdc37 interactions.35 Gedunin was found to be an Hsp90 inhibitor as determined by high-throughput gene expression studies.17 Further biochemical studies demonstrated that the mechanism of modulating Hsp90 function was distinct from that of GDA and its analogues, suggesting an additional class of Hsp90 inhibitors. 36
Epigallocatechin-3-gallate (EGCG), a widely used anti-oxidant abundant in green tea, was reported to modulate the cytotoxicity of rotenone in human neuroblastoma SH-SY5Y cells. 18 It appears to manifest these activities through inhibition and release of the aryl hydrocarbon receptor (Ah receptor or AhR) from Hsp90. In addition, ECGC is capable of antagonizing AhR-mediated toxic effects against numerous environmental insults as a result of its redox-active functionalities. 37
Gamendazole was recently identified as an orally active antispermatogenic compound with antifertility effects. Recent reports suggest that gamendazole binds to Hsp90, inhibits luciferase rematuration, and is not displaced by the addition of other Hsp90 inhibitors such as GDA or the novobiocin analogue, A4. Consequently, gamendazole may represent yet a different class of Hsp90 inhibitors.38
Previous studies indicated that A4 induced the degradation of Hsp90-dependent client proteins at ~70-fold lower concentration than novobiocin. 14 The structure of A4 includes a shortened N-acetyl amide side chain, removal of the 4-hydroxy substituent and an unmodified diol on the noviose appendage as compared to novobiocin. 15, 39 Compound A4 is unique in its ability to induce Hsp90 levels at concentrations 1000–10000-fold lower than that required for client protein degradation and this observation provided the basis for subsequent studies aimed at its development as a neuroprotective agent. A4 is also unique as compared to N-terminal inhibitors in that it completely lacks toxicity.23 In contrast to the monomeric species, dimers of A4 based on the structure of coumermycin A1 were found to manifest anti-proliferative activity against some cancer cells.40 Subsequent studies demonstrated that conversion of nontoxic molecules into those that exhibit anti-proliferative activity occurs through modification of the amide side chain. Confirmation was based on preparation of a series of monomeric species representing the A4 scaffold, which resulted in compounds that manifest submicromolar EC50‘s against several cancer cell lines.40
In our present studies, three novobiocin analogues provided excellent neuroprotective activity and little if any toxicity. Thus these compounds may represent novel leads for influencing neurodegenerative diseases as they appear to offer a wide therapeutic window for actual in vivo testing.
5. Conclusion
In this study, we have optimized the LDH assay to screen Hsp90 inhibitors for neuroprotective and cytotoxic activity in SH-SY5Y neuronal cells exposed to lethal concentrations of Aβ. The assay described herein has been optimized for each component in an effort to provide a highly reproducible method of evaluating an array of Hsp90 inhibitors for neuroprotective properties. In our studies, the novobiocin analogues, A4, A4-dimer and KU32 exhibited significant protection against the Aβ-induced toxicity at low concentrations, but with a potentially large therapeutic window. The results suggest these novobiocin analogues may represent an effective class of novel compounds to pursue for the treatment of neurodegenerative diseases. In addition, the LDH activity assay was optimized to be a highly reproducible assay with a Z-factor of 0.76 for moderate and high-throughput screening.
Figure 1.

Structures of Hsp90 inhibitors.
Figure 2.

Structure of novobiocin analogues A4, A4-dimer, KU32 and other Hsp90 inhibitor
Acknowledgments
The authors gratefully acknowledge support of this project by the NIH (U54 HD055763 and U01 AG031106).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hölscher C. Rev Neurosci. 2005;16:181–212. doi: 10.1515/revneuro.2005.16.3.181. [DOI] [PubMed] [Google Scholar]
- 2.Small Cappai. J Neurochem. 2006;99:708–710. doi: 10.1111/j.1471-4159.2006.04212.x. [DOI] [PubMed] [Google Scholar]
- 3.Yan Q, Zhang J, Liu H, Babu KS, Vassar R, Biere AL, Citron M, Landreth G. J Neurosci. 2003;23:7504–9. doi: 10.1523/JNEUROSCI.23-20-07504.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative Tauopathies. Annu Rev Neurosci. 2001;24:1121. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 5.Garnier C, Lafitte D, Tsvetkov PO, Barbier P, Leclerc-Devin J, Millot JM, Briand C, Makarov AA, Catelli MG, Peyrot V. J Biol Chem. 2002;277:12208–12214. doi: 10.1074/jbc.M111874200. [DOI] [PubMed] [Google Scholar]
- 6.Marcu MG, Schulte TW, Neckers L. J Natl Cancer Inst. 2000;92:242–248. doi: 10.1093/jnci/92.3.242. [DOI] [PubMed] [Google Scholar]
- 7.Hartson SD, Thulasiraman V, Huang W, Whitesell L, Matts RL. Biochemistry. 1999;38:3837–3849. doi: 10.1021/bi983027s. [DOI] [PubMed] [Google Scholar]
- 8.Scheibel T, Weikl T, Buchner J. Proc Natl Acad Sci USA. 1998;95:1495–1499. doi: 10.1073/pnas.95.4.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson BD, Chadli A, Felts SJ, Bouhouche I, Catelli MG, Toft DO. J Biol Chem. 2000;275:32499–32507. doi: 10.1074/jbc.M005195200. [DOI] [PubMed] [Google Scholar]
- 10.Owen BA, Sullivan WP, Felts SJ, Toft DO. J Biol Chem. 2002;277:7086–92. doi: 10.1074/jbc.M111450200. [DOI] [PubMed] [Google Scholar]
- 11.Le Brazidec JY, Kamal A, Busch D, Thao L, Zhang L, Timony G, Grecko R, Trent K, Lough R, Salazar T, Khan S, Burrows F, Boehm MF. J Med Chem. 2004;47:3865–73. doi: 10.1021/jm0306125. [DOI] [PubMed] [Google Scholar]
- 12.Andrus MB, Hicken EJ, Meredith EL, Simmons BL, Cannon JF. Org Lett. 2003;5:3859–62. doi: 10.1021/ol035400g. [DOI] [PubMed] [Google Scholar]
- 13.Marcu MG, Chadli A, Bouhouche I, Catelli M, Neckers LM. J Biol Chem. 2002;275:37181–37186. doi: 10.1074/jbc.M003701200. [DOI] [PubMed] [Google Scholar]
- 14.Yu XM, Neckers L, Blake H, Holzbeierlein J, Cronk B, Brian SJ, Blagg J Am Chem Soc. 2005;127:12778–12779. doi: 10.1021/ja0535864. [DOI] [PubMed] [Google Scholar]
- 15.Burlison JA, Neckers L, Smith AB, Maxwell A, Brian SJ, Blagg J Am Chem Soc. 2006;128:15529–36. doi: 10.1021/ja065793p. [DOI] [PubMed] [Google Scholar]
- 16.Westerheide SD, Bosman JD, Mbadugha BN. J Biol Chem. 2004;279:56053–60. doi: 10.1074/jbc.M409267200. [DOI] [PubMed] [Google Scholar]
- 17.Hieronymus H, Lamb J, Ross KN, Peng XP, Clement C, Rodina A, Nieto M, Du J, Stegmaier K, Raj SM, Maloney KN, Clardy J, Hahn WC, Chiosis G, Golub TR. Cancer Cell. 2006;10:321–330. doi: 10.1016/j.ccr.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 18.Chung WG, Miranda CL, Maier CS. Brain Res. 2007;1176:133–42. doi: 10.1016/j.brainres.2007.07.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bedin M, Gaben AM, Saucier C, Mester J. Int J Cancer 2004. 1;109:643–52. doi: 10.1002/ijc.20010. [DOI] [PubMed] [Google Scholar]
- 20.Joseph ST, Ramappa C, Sudhakar RJ, Brian SJ, Blagg, Gunda IG. Biology of Reproduction. 2008;78:1139–1152. [Google Scholar]
- 21.Avila C, Kornilayev BA, Brian SJ, Blagg Bioor Med Chem. 2006;14:1134–1142. doi: 10.1016/j.bmc.2005.09.027. [DOI] [PubMed] [Google Scholar]
- 22.Pike CJ, Walencewicz-Wasserman AJ, Kosmoski J, Cribbs DH, Glabe CG, Cotman CW. J Neurochem. 1995;64:253–65. doi: 10.1046/j.1471-4159.1995.64010253.x. [DOI] [PubMed] [Google Scholar]
- 23.Ansar S, Burlison JA, Hadden MK, Yu XM, Kelly ED, Bean J, Neckers L, Audus KL, Michaelis ML, Blagg BSJ. Bioorg Med Chem Let. 2007;17:1984–1990. doi: 10.1016/j.bmcl.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 24.Jellinger KA. J Neural Transm. 2006;113:1603–1623. doi: 10.1007/s00702-006-0578-3. [DOI] [PubMed] [Google Scholar]
- 25.Blennow K, De Leon MJ, Zetterberg H. Lancet. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- 26.Estus S, Tucker HM, van Rooyen C, Wright S, Brigham EF, Wogulis M, Rydel RE. J Neurosci. 1997;17:7736–7745. doi: 10.1523/JNEUROSCI.17-20-07736.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mattson MP, Partin J, Begley JG. Brain Res. 1998;807:167–176. doi: 10.1016/s0006-8993(98)00763-x. [DOI] [PubMed] [Google Scholar]
- 28.Pike CJ, Ramezan-Arab N, Cotman CW. J Neurochem. 1997;69:1601–1611. doi: 10.1046/j.1471-4159.1997.69041601.x. [DOI] [PubMed] [Google Scholar]
- 29.Keller JN, Kindy MS, Holtsberg FW, St Clair DK, Yen HC, Germeyer A, Steiner SM, Bruce-Keller AJ, Hutchins JB, Mattson MP. J Neurosci. 1998;18:687–697. doi: 10.1523/JNEUROSCI.18-02-00687.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo Q, Sebastian L, Sopher BL, Miller MW, Ware CB, Martin GM, Mattson MP. J Neurochem. 1999;72:1019–1029. doi: 10.1046/j.1471-4159.1999.0721019.x. [DOI] [PubMed] [Google Scholar]
- 31.Goedert M. Nat Rev Neurosci. 2001;2:492–501. doi: 10.1038/35081564. [DOI] [PubMed] [Google Scholar]
- 32.Burrows F, Zhang H, Kamal A. Cell Cycle. 2004;3:1530–6. doi: 10.4161/cc.3.12.1277. [DOI] [PubMed] [Google Scholar]
- 33.Sreedhar AS, Kalmar E, Csermely P, Shen YF. FEBS Lett. 2004;562:11–15. doi: 10.1016/s0014-5793(04)00229-7. [DOI] [PubMed] [Google Scholar]
- 34.Dou F, Netzer WJ, Tanemura K, Li F, Hartl FU, Takashima A, Gouras GK, Greengard P, Xu H. Proc Natl Acad Sci USA. 2002;100:721–6. doi: 10.1073/pnas.242720499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang T, Hamza A, Cao XH, Wang B, Yu S, Zhan CG, Sun D. Mol Cancer Ther. 2008;7:163–70. doi: 10.1158/1535-7163.MCT-07-0484. [DOI] [PubMed] [Google Scholar]
- 36.Justin L, Emily DC, Todd RG. Science. 2006;313:1929–35. [Google Scholar]
- 37.Christine M, Palermo CM, Westlake CA, Gasiewicz TA. Biochemistry. 2005;44:5041–52. doi: 10.1021/bi047433p. [DOI] [PubMed] [Google Scholar]
- 38.Burlison JA, Avila C, Vielhauer G, Lubbers DJ, Holzbeierlein J, Blagg BS. J Org Chem. 2008;73:2130–2137. doi: 10.1021/jo702191a. [DOI] [PubMed] [Google Scholar]
- 39.Burlison JA, Blagg BS. Org Lett. 2006;8:4855–4858. doi: 10.1021/ol061918j. [DOI] [PubMed] [Google Scholar]
- 40.Zhang J, Chung TD, Oldenburg KR. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]