

Abstract
The crucial role played by HLA antigens and natural killer (NK) cell activating ligands in the interactions of malignant cells with components of the host's immune system has stimulated interest in the characterization of their expression by malignant cells. Convincing evidence generated by the immunohistochemical staining of surgically removed malignant lesions with monoclonal antibodies (mAb) recognizing HLA antigens and NK cell activating ligands indicates that the surface expression of these molecules is frequently altered on malignant cells. These changes appear to have clinical significance, since in some types of malignant disease they are associated with the histopathological characteristics of the lesions as well as with disease free interval and survival. These associations have been suggested to reflect the effect of HLA antigen and NK cell activating ligand abnormalities on the interactions of tumor cells with antigen-specific cytotoxic T lymphocytes (CTL) and with NK cells. Nevertheless, there are examples in which disease progresses in the face of appropriate HLA antigen and/or NK cell activating ligand as well as tumor antigen expression by malignant cells and of functional antigen-specific CTL in the investigated patient. In such scenarios, it is likely that the tumor microenvironment is unfavorable for CTL and NK cell activity and contributes to tumor immune escape. Many distinct escape mechanisms have been shown to protect malignant cells from immune recognition and destruction in the tumor microenvironment. In this paper, following the description of the structural and functional characteristics of soluble HLA antigens and NK cell activating ligands, we will review changes in their serum level in malignant disease and discuss their potential role in the escape mechanisms utilized by tumor cells to avoid recognition and destruction.
Keywords: antigen processing machinery, cancer, classical HLA class I antigen, HLA class II antigen, HLA-G, immune escape, immunotherapy, MIC, NK cells, NK cell activating ligand, non-classical HLA class I antigen
I. Introduction
The revival of the cancer immunosurveillance theory [1] along with the disappointing clinical results obtained in T cell-based immunotherapy trials conducted in patients with cancer [2-5] has re-emphasized the role of immune escape mechanisms in their pathogenesis and clinical course of malignant diseases. As a result, tumor immunologists have been focusing their investigations on the identification and molecular characterization of the multiple mechanisms by which tumor cells evade immune recognition and destruction [6-11]. Among them, changes in classical [6] and non-classical [6,8,11-15] HLA class I as well as HLA class II [16] antigen expression by tumor cells are of particular interest to tumor immunologists and clinical oncologists because of the critical role they play in the generation of tumor antigen (TA)-specific immune responses (Figure 1A-C), as well as their ability to modulate the interactions of natural killer (NK) cells and T cell subpopulations with target cells (Figure 1A-C). More recently, these studies have been expanded to include the analysis of the expression and functional characteristics of the phylogenetically distant MHC class I related chain (MIC) as well as the UL16-binding proteins (ULBP) in malignant lesions given their ability to act as NK cell activating ligands (Figure 1A-C) [8,12,17-19]. The potential clinical relevance of these escape mechanisms is suggested by the statistically significant association of changes in classical [6] and non-classical [6,12] HLA class I as well as HLA class II [20,21] antigen expression with the clinical course of the disease at least in certain malignancies.
Figure 1. Molecular mechanisms underlying the functional properties of HLA and NK cell activating ligand expression by malignant cells.

(A) Once transported to the plasma membrane, the classical HLA class I-β2m-peptide complex plays a major role in the interactions between target cells and (a) activation of peptide-specific CTL through TCR; and (b) inhibition of T cell subpopulations through inhibitory receptors KIR. (B) In contrast to classical HLA class I, the non-classical HLA class I, HLA-G, inhibits CTL, CD4(+) T cells and NK cells through its interaction with the NK cells receptor CD94/NKG2. (C) MICA/B as well as ULBP ligand expression by tumor cells may be potentially beneficial to TA-specific immune responses through their interaction with the NK cells receptor CD94/NKG2 on NK cells and subsets of T cells, resulting in the activation of NK and T cell-mediated killing.
The above findings have been suggested to reflect the effect of HLA antigen and NK cell activating ligand abnormalities on the interactions of tumor cells with TA-specific CTL and NK cells. Nevertheless, there are examples in which disease progresses in the face of appropriate HLA antigen and/or NK cell activating ligand as well as TA expression by malignant cells and of functional TA-specific CTL [6]. In such scenarios, the tumor microenvironment is thought to be unsuitable for the host's TA-specific immune response and contributes to tumor cell immune escape (reviewed in 20-25). In this regard, to date the potential effects of soluble HLA antigens (sHLA) and NK-cell activating ligands (sNKAL), either released by tumor cells and/or derived from tumor cell lysis, on immune cells present in the tumor microenvironment has been investigated to a limited extent [26-79]. The low interest in sHLA and in sNKAL is surprising given the increase in their serum level in several pathological conditions, including but not limited to malignant diseases [33,34,37,48,58,61,62,70-72,75,77]. Moreover, both sHLA and sNKAL have been shown to induce apoptosis of CTL and NK cells through its interaction with CD8 or inhibitory receptor superfamily (IRS) [33,34,43,47,52,54,58,61,62,69,76,80-82]. The potential clinical relevance of these findings is suggested by the statistically significant association of classical [41,53,55,58,67,68,74] and non-classical [42,47-49,53,55,60,71] sHLA, as well as sNKAL [61-64,72,73,76,78,79] level with stage of disease as well as the clinical course of the disease in certain malignancies.
In this paper, we will summarize the available information about the structural characteristics of sHLA and sNKAL as well as the level of their expression in malignant diseases. Furthermore, we will review the potential role sHLA and sNKAL may play in tumor immune escape and in the clinical course of malignant disease. In organizing this paper we have not attempted to review each subject in its entirety, but rather have focused on what we believe are the critical issues in this field.
II. Classical and non-classical sHLA and sNKAL expression in malignant disease
II.A. Classical HLA class I antigen
Like their counterparts in other animal species, classical HLA class I antigens are detectable not only on the membrane of most nucleated cells, but also in plasma and urine [33,34,37,58,74]. The existence of classical sHLA in the serum of healthy individuals was first reported in 1970, when sHLA-A2 and sHLA-A7 were identified in human serum and in the low-density β-lipoprotein fraction in serum from normal individuals, respectively [33,34,37,74]. The structure of classical sHLA molecules is similar to that of their cell-bound counterparts. They consist of a membrane spanning polymorphic heavy chain, β2-microglobulin (β2m) and an 8–10 amino acid peptide. However, at variance with their membrane anchored counterparts, classical sHLA heavy chains are heterogeneous in their structure and molecular size. In this regard, three isoforms of classical sHLA heavy chains, which are generated through distinct mechanisms, have been identified in plasma (Figure 2A). One with the approximate molecular weight (mw) of 45 kDa represents the full-length gene product. One with the approximate mw of 39 kDa is synthesized from alternatively spliced transcripts lacking exon 5 encoding the hydrophobic transmembrane anchor [27]. Lastly, one with the approximate mw of 35 kDa lacks the transmembrane and cytoplasmic domain. This heterogeneity is most likely the result of the multiple mechanisms underlying the generation of classical sHLA heavy chains. The larger 45 kDa isoforms are likely to be the result of shed membrane-bound molecules as they contain both a transmembrane domain and a cytoplasmic domain. The smaller 35 kDa isoforms are thought to represent products of proteolytic cleavage, since they contain neither the transmembrane nor the cytoplasmic domain [31,39,74]. Finally the intermediate 39 kDa isoforms appear to be the products of alternate mRNA splicing that generates truncated sHLA lacking the transmembrane domain [72]. It is noteworthy that classical sHLA have been found in plasma, not only as sHLA class I heavy chain-β2m-peptide complexes, but also as β2m-free heavy chains [28,31,58,68,74].
Figure 2. Structural properties of soluble HLA antigens.
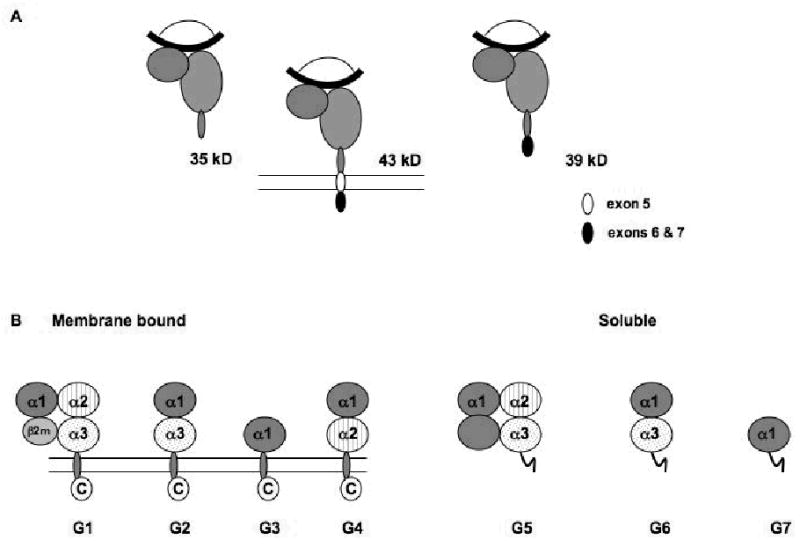
(A) HLA class I heavy chains ( ) are detectable in plasma as 43kDa, 39kDa and 35kDa moieties which represent membrane associated, alternatively spliced and metalloprotease cleaved forms, respectively. Splicing of exon 5 results in the removal of amino acids depicted in white (●), but not residues encoded by exons 6 and 7 (●). Metalloprotease mediated cleavage likely results in removal of amino acids encoded by exons 5–7. All three forms are shown to be associated with β2m (
) are detectable in plasma as 43kDa, 39kDa and 35kDa moieties which represent membrane associated, alternatively spliced and metalloprotease cleaved forms, respectively. Splicing of exon 5 results in the removal of amino acids depicted in white (●), but not residues encoded by exons 6 and 7 (●). Metalloprotease mediated cleavage likely results in removal of amino acids encoded by exons 5–7. All three forms are shown to be associated with β2m ( ) and peptide (
) and peptide ( ); however, β2m-free heavy chains have also been detected. (B) HLA-G is detectable in seven forms. Four of them, HLA-G1, -G2, -G3 and -G4, are bound to the cell surface, while the remaining three, HLA-G5, -G6 and -G7 are soluble. HLA-G1 is the only isoform derived from the translation of the total HLA-G transcript. The other membrane bound isoforms lack one or two globular domains. The structure of the soluble isoforms resembles that of the corresponding membrane bound isoforms in the extracellular part, but differs at the C-terminus. The extracellular domain and the intracytoplasmic tail, which are present in the membrane bound isoforms, are replaced in the secreted isoforms by a short hydrophilic tail. These differences provide a marker to distinguish shed or proteolytically cleaved HLA-G isoforms from secreted HLA-G isoforms.
); however, β2m-free heavy chains have also been detected. (B) HLA-G is detectable in seven forms. Four of them, HLA-G1, -G2, -G3 and -G4, are bound to the cell surface, while the remaining three, HLA-G5, -G6 and -G7 are soluble. HLA-G1 is the only isoform derived from the translation of the total HLA-G transcript. The other membrane bound isoforms lack one or two globular domains. The structure of the soluble isoforms resembles that of the corresponding membrane bound isoforms in the extracellular part, but differs at the C-terminus. The extracellular domain and the intracytoplasmic tail, which are present in the membrane bound isoforms, are replaced in the secreted isoforms by a short hydrophilic tail. These differences provide a marker to distinguish shed or proteolytically cleaved HLA-G isoforms from secreted HLA-G isoforms.
Stable amounts of classical sHLA molecules circulate in all healthy individuals at a concentration of about 1 μg/ml [33,34,37,58,74], although some individuals with particular HLA haplotypes have substantially higher concentrations than others [33,34,37,58,74]. In this regard it has been reported that individuals with the HLA-A9 allotype have higher serum levels of sHLA molecules [74]. Race also appears to play a role in determining serum levels of sHLA molecules in normal individuals [74]. For example, HLA-A23 and HLA-A24 allotypes are associated with high serum sHLA level in both Caucasians and African Americans [74], while HLA-A29 allotype is associated with high sHLA levels in Caucasians but not in African Americans [74]. The reverse is true for the HLA-A33 allotype, which is associated with high sHLA levels in African Americans but not in Caucasians [74].
To date only a limited number of studies have investigated the level of classical sHLA in patients with malignant disease. The limited number of studies most likely reflects the limited interest in these molecules in spite of the experimental evidence, which is compatible with their role in immunological events. Nevertheless, the level of classical sHLA appears to change in some malignant diseases. Shimura et. al. have demonstrated that patients with Stage IV advanced gastric cancer had significantly lower levels of classical sHLA compared to normal healthy volunteers and also compared to patients with less advanced Stage I and Stage II gastric cancers [32]. Furthermore, this study also showed that classical sHLA levels were significantly lower in all gastric cancer patients with the HLA-A24 allotype, regardless of stage [32]. In a study by Westhoff et. al., patients with metastatic malignant melanoma showed no significant difference in classical sHLA levels compared to healthy controls; although patients with less advanced primary malignant melanoma demonstrated significantly lower levels of serum classical sHLA levels compared to healthy controls [36]. In contrast, Shimura et. al. have demonstrated significantly elevated levels of classical sHLA molecules in Japanese patients with pancreatic cancer [41]. The level of classical sHLA has also been investigated in hematologic malignancies. In this regard, classical sHLA and β2m levels have been reported to correlate with disease aggressiveness in multiple myeloma (MM) [53,55,], chronic myelogenous leukemia (CML) [74], acute myeloid leukemia (AML) [31,67], and myelodysplastic syndrome (MDS) [55,67]. In NHL [68,74] and HD [68,74], classical sHLA and β2m levels have been shown to be elevated compared to healthy controls and to normalize in NHL and HD patients in complete remission. Interestingly, in some cases, NHL patients who experienced a relapse demonstrated an increase in sHLA levels [74]. The upregulation of sHLA molecules in malignant disease as well as in other pathological processes appears to be caused by the increased production of cytokines, such as interferon- α (IFN-α) and interferon-γ (IFN-γ), since the level of classical sHLA is increased in the spent medium of cells as well as in the plasma of patients treated with either IFN-α or IFN-γ [29,30,33,57-59]. It is noteworthy that changes in the level of sHLA are not unique of malignant diseases since they have also been documented in patients with autoimmune disease, transplant rejection, and infections [58,74].
II.B. Non-classical sHLA class I antigens and sNKAL
Evidence accumulated during the last few years has convincingly shown that the non-classical HLA class I antigens HLA-E, -F and -G may serve as immunosuppressive molecules [6,8,11-14], while the phylogenetically distant MHC class I chain-related surface glycoproteins MICA and MICB and the UL16-binding proteins ULBP1, ULBP2, ULBP3 and ULBP4 may act as NK cell activating ligands [8,12,17-19]. These findings have stimulated interest in the characterization of soluble non-classical HLA class I antigen and sNKAL in patients with malignant disease, since the interaction of these molecules with host's immune system may be affected by these antigens. Here we review the characteristics of non-classical sHLA and sNKAL as well as the level of their expression in malignant diseases. It is noteworthy that the available information is still limited, since the field is in an early stage and progress in this area is hindered by the lack and/or limited availability of non-classical sHLA- and sNKAL-specific monoclonal antibodies (mAb).
II.B.1. Non-classical sHLA
To the best of our knowledge, no study has investigated sHLA-F expression in healthy individuals and/or in patients with malignant diseases, while sHLA-E expression has only been investigated in melanocytes and primary melanoma cell lines in vitro. In this regard, Derre et. al. demonstrated that melanocytes and melanoma cell lines can produce a 37 kDa sHLA-E chain [59]. This size corresponds to that of the extracellular portion of the membrane-bound HLA-E chain, suggesting that it is produced by the cleavage of membrane HLA-E by a membrane-bound and/or extracellular protease. The latter mechanism seems likely, since membrane bound and extracellular matrix metalloproteinases (MMP) have been shown to be involved in proteolytic cleavage and shedding of membrane bound classical HLA class I antigen, MICA/B and HLA-E in a number of cell lines [31,39,45,59,74].
Among the non-classical HLA class I antigens, HLA-G has been the most extensively studied. HLA-G exists in seven isoforms that are generated by alternative splicing of the primary HLA-G transcript [71]. Four of them, HLA-G1, -G2, -G3 and -G4, are bound to the cell surface, while the remaining three, HLA-G5, -G6 and -G7 are soluble (Figure 2B) [71]. The latter three are the counterparts of HLA-G1, -G2 and -G3, respectively [71]. The major isoforms are HLA-G1 and HLA-G5, which both share immunosuppressive activities mediated by their binding to the receptors CD85j (ILT-2), CD85d (ILT-4), CD158d (KIR2DL4) and CD160 (BY55) [45,72,73]. These receptors have a differential cellular distribution, since CD85j (ILT-2) is expressed by B, NK and T cells, CD160 (BY55) by endothelial, NK and T cells, CD85d (ILT-4) only by macrophages and CD158d (KIR2DL4) only by NK cells [8,9,12,13,15,71].
As indicated above, sHLA-G derives from the secretion of soluble isoforms, especially HLA-G5, as well as from the shedding of proteolytically cleaved surface isoforms, like HLA-G1. In physiological conditions, monocytes/macrophages together with myeloid and plasmacytoid dendritic cells are the major producers of sHLA-G [71]. sHLA-G has been detected in serum from healthy individuals utilizing immunoassays, which either detect sHLA-G1 in combination with sHLA-G5 or only sHLA-G5. In this regard, stable amounts of sHLA-G1+G5 have been found to range from 27.7 to 95.9 ng/ml, while those of sHLA-G5 from 14.6 to 85.8 ng/ml in plasma of healthy individuals [71]. It is noteworthy that the isoforms of sHLA-G appear to be differentially expressed in some body fluids, since amniotic fluid contains only sHLA-G1, while ascites contains only sHLA-G5 [71]. The biologic and functional significance of these findings remains to be determined.
sHLA-G plasma levels, which are markedly lower than those of classical sHLA, appear to be influenced by several variables. Among them is the gender of the donors, since the level of sHLA-G is higher in women than in men [71]. Furthermore, there is suggestive evidence that sHLA-G levels are increased in serum and amniotic fluid during pregnancy [71]. However, these results were obtained using different assays with different antibodies; therefore the conclusions must be interpreted with caution, since one cannot exclude that the reported differences reflect different sensitivity of the assays used by different investigators. Lastly, as previously observed for classical sHLA [71], another important variable is represented by the HLA-G polymorphism. Healthy individuals carrying the HLA-G*01013 allele or the “null” allele HLA-G*0105N have significantly lower sHLA-G levels than subjects carrying the more frequent HLA-G*01011 and HLA-G*01012 alleles [71]. In addition, subjects with the latter alleles have significantly lower sHLA-G levels than individuals with the HLA-G*01041 allele. Polymorphisms in the 3′UTR and the 5′UTR of the HLA-G gene may further influence the sHLA-G level [71].
As observed for classical sHLA, changes in the level of sHLA-G have been detected in serum from patients with solid tumors and lymphoproliferative disorders. Increased levels of sHLA-G have been detected in the plasma of patients with glioblastoma multiforme (GBM) [49], breast [49] and ovarian cancer [49], lymphoblastic and monocytic acute leukemia [48,60], malignant melanoma [42], MM [55] and neuroblastoma (NB) [71]. Whether the increased sHLA-G levels detected in patients with malignant diseases reflects increased shedding of sHLA-G by tumor cells and/or up-regulation of surface HLA-G or secretion of sHLA-G by peripheral blood monocytes through tumor derived cytokines such as IL-10, TGF-β1, remains to be determined [42,60,76]. The latter mechanism is suggested by the induction of in vitro sHLA-G production by monocytes following incubation with tumor-cell derived supernatants [71]. In GBM patients, sHLA-G serum levels did not differ from those found in healthy controls, but patients with high sHLA-G levels had a significantly shorter survival than those with low sHLA-G levels [49]. In acute myeloid leukemia (AML), seventy-four percent of patients, especially of the FABM4 and FABM5 subtypes, showed markedly increased sHLA-G serum levels [48,60]. This figure reached 89% in acute lymphoblastic leukemia (ALL) patients, with a higher frequency of upregulated sHLA-G serum levels in T than in B-ALL [48,60]; specifically, high serum levels of sHLA-G were found in 16 out of 17 patients with T-ALL versus 7 out of 11 patients with B-ALL. In melanoma [42], MM [55], as well as NB [71] serum sHLA-G levels have been found to be significantly higher in patients than in age-matched healthy controls. Malignant ascitis from patients with breast or ovarian cancer has significantly higher levels of sHLA-G than ascitis of non-neoplastic origin, suggesting that sHLA-G level may represent a useful adjunct to cytology in the differential diagnosis of malignant versus benign ascitis [49]. It is noteworthy that as for classical sHLA antigens, alterations in the level of sHLA-G does not appear to be restricted to patients with malignant disease since they have been found also in patients with autoimmune diseases, transplant rejection, and infections [71].
II.B.2. sNKAL
The stress-inducible MHC class I chain-related surface glycoproteins MICA and MICB and the UL16-binding proteins ULBP1, ULBP2, ULBP3 and ULBP4 are ligands of the C-type lectin-like receptor NKG2D [8,12,17-19]. The latter potently activates NK cells, even overcoming inhibitory signals by MHC class I molecules. MICA, and most likely MICB, has a restricted distribution in normal tissues; their expression is induced by inflammatory stress only in gastric and small and large intestinal epithelium [8,12,17-19]. However immunohistochemical staining of only a limited number of surgically removed malignant lesions and analysis of cell lines in long term culture of different embryological origin have shown that MICA and MICB have a much broader distribution in malignant tumors [17-19]. This expression pattern has been suggested to reflect the induction of MICA and MICB by the DNA repair pathway in response to genotoxic insults that have resulted in genetic mutations contributing to the malignant transformation of the cell.
Stable amounts of sMICA and sMICB have been found to circulate in all healthy individuals at a concentration of about <30 pg/ml and <50 pg/ml, respectively [61,62,64]. Analysis of sera from about 600 patients with various malignant diseases including carcinomas of the breast, esophagus, stomach, liver, pancreas, colon, ovaries, cervix, endometrium, kidney, prostate as well as oral squamous cell carcinoma, osteosarcoma, glioma, neuroblastoma, leukemia, MM and melanoma has shown that the levels of sMICA and sMICB (median 161 pg/ml and 216 pg/ml, respectively) are significantly higher in patients with malignant disease than in age-matched healthy controls [43-45,50,52-54,61-64,70,72,73,75-79]. This finding is not restricted to patients with malignant disease since increase in the level of sMICA and sMICB has been found also in patients with autoimmune diseases, transplant rejection, and infections [70,72,77].
To the best of our knowledge, except for a limited number cell lines in long-term culture [44,50,52,65-67] and surgically removed glioblastoma, neuroblastoma and leukemic samples [50,52], no study has investigated the level of sULBP ligand in serum from patients with malignant disease.
II.C. Clinical relevance of sHLA, sHLA-G, and sMICA/B in patients with malignant disease
In general, changes in the level of sHLA, sHLA-G and sMICA/B appear to be related with disease progression, since at least in some malignant diseases, they are associated with advanced tumor stage [32,34,36,41,42,47,49,52,55,58,60,61-64,67,68,71,73-75,78,79]. These findings suggest that, in at least some tumors, the level of sHLA, sHLA-G, and sMICA/B may be clinically relevant. Nevertheless, analysis of several different types of malignant diseases has generated conflicting results about the association of sHLA, sHLA-G and sMICA/B with the clinical course of the disease. Moreover, the clinical significance of the sHLA, sHLA-G and sMICA/B serum level in patients with malignant disease appears to vary among tumors of different histotypes. For example, sHLA-G level is inversely correlated with survival in GBM [49], but does not appear to be predictive of survival in NB in spite of the significant increase in patients [71]. In MM, serum sHLA-G levels are significantly higher in patients than in healthy controls and the level of sHLA-G is positively correlated with the advanced stage of the disease and with tumor load, but not with the clinical course of the disease [55]. As noted above, in breast and ovarian carcinoma, the level of sHLA-G has been found to be increased in ascitis of patients with these diseases, suggesting that, in addition to cytology, it may be a useful marker to differentiate malignant from benign ascites [49]. In the case of MIC ligands, the level of sMICA has only been found to be associated with overall as well as progression-free survival in oral squamous cell carcinoma [79], prostate carcinoma [63] and MM [73,78].
It is likely that both technical and biological mechanisms confound the data and underlie the different findings in tumors of different histiotype in regards to the prognostic significance of sHLA, sHLA-G, and sMICA/B in patients with malignant disease. Among the technical reasons are differences in the sensitivity of the assays employed, characteristics of the mAb utilized and/or number of lesions analyzed. The latter is likely to play a significant role, since those studies, which have found no association between sHLA or MICA/B and clinical course of the disease have analyzed a considerably lower number of lesions than those studies that have demonstrated an unfavorable association [32,34,36,41,42,47,49,52,55,58,60,61-64,67,68,71,73-75,78,79]. Among the biological variables, which generally have not been taken into account in the design of the studies as well as in the analysis of the results, are heterogeneity of the patient populations investigated in terms of clinical stage, histopathological characteristics of the lesions, previous therapy, and length of the follow-up periods. Furthermore, it should be stressed that the detection of sHLA, sHLA-G, and sMICA/B in serum does not exclude that they are non-functional or dysfunctional because of mutations and/or changes in their conformation. The complexity of malignant disease, which is not a single disease entity, can also play a role, since the level of cell membrane expressed classical and non-classical HLA class I antigen may vary among histologic subtypes of malignant cells [6]. Furthermore, the analysis of the clinical significance of sHLA, sHLA-G, and sMICA/B expression in patients with malignant disease has, in general, not taken into account the expression of other types of histocompatibility antigens, as well as other molecules that are likely to greatly affect the interactions of malignant cells with host's immune system. The limitation of this approach is underscored by the positive [83] or negative [84] association of HLA class II antigen expression in cutaneous metastatic melanoma lesions with the clinical course of the disease only when the level of co-stimulatory molecule B7.1 [83] and HLA class I antigen [84] expression, respectively, is included in the analyses.
IV. Potential role of sHLA and sNKAL in immune escape
The major unanswered question in human tumor immunology today is why the presence of TA-specific immune responses, which can be detected in a variable percentage of patients, is not paralleled by a clinical response in the majority of immunized patients despite the presence of all the necessary components, i.e. TA, APC, immune effector cells and cytokines, for the successful development of these responses [1,6-8,16]. In such scenarios, the tumor microenvironment is thought to be unfavorable for TA-specific immune responses and to contribute to ineffective host's immune responses. Many distinct escape mechanisms have been shown to protect malignant cells from immune recognition and destruction in the tumor microenvironment (Figure 3). Interested readers are referred to recent reviews [6,7,15,20-25,85] on this topic. In the following section we will discuss the potential role of sHLA and sNKAL in the protection of tumor cells from host's immune response.
Figure 3. Immune escape mechanisms utilized by tumor cells.

Escape mechanisms utilized by tumor cells include: i) HLA class I antigen-TA derived peptide complex loss which can result from loss of a) TA, b) APM antigen processing machinery components, or c) HLA class I antigens;ii) release of immune suppressive small molecules such as PGE2, INOS and/or H2O2; iii) release of immune suppressive cytokines resulting in altered immune cell function; iv) Fas ligand expression resulting in the killing of Fas+ lymphocytes; g) over-expression of anti-apoptotic proteins in melanoma cells resulting in apopotitic resistance; and v) expression of tumor associated gangliosides which can inhibit IL-2 dependent lymphocyte proliferation as well as induce apoptotic signals.
Although the exact functional role of sHLA and sNKAL is not known, it is likely that they play a role in immunological phenomena, since they are immunologically active, as indicated by their reactivity with antibodies and with T cell receptors and by their immunogenicity in allogeneic and xenogeneic combinations [37,38,40,46,51,54,58,61,62,71,74]. Furthermore, the potential clinical relevance of sHLA and sNKAL is suggested by the statistically significant association of increased levels of sHLA or sNKAL with the clinical course of the disease in certain malignancies. The potential role sHLA and sNKAL may play in the escape of tumor cells from the host's immune response is summarized in figure 4.
Figure 4. Potential role of sHLA and sNKAL in the escape of tumor cells from the host's immune response.
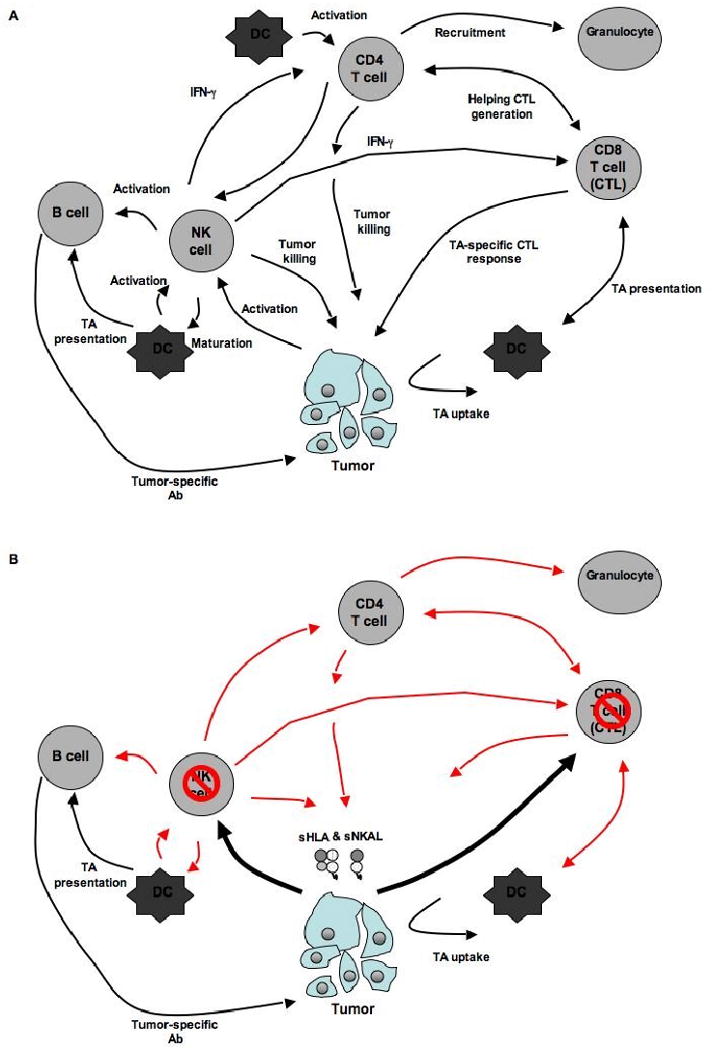
The immune system can target tumor cell growth through several mechanisms. (A) It is thought that the most effective way of mounting a TA-specific immune response is through the combined action of CD8(+) and IFN-γ-secreting CD4(+) T helper cells (Th1). TA-specific CD8(+) T cells can be activated by antigen presenting cells (APC) and kill tumors cells directly. The survival and persistence of CD8(+) T cells is dependent upon CD4(+) T helper cells. Naïve CD4(+) Th1 cells recognize HLA class II antigen-peptide complexes, through their T cell receptor (TCR), on the surface of APC. This interaction leads to the generation of i) Th1 helper cells which promote survival and proliferation of CD8(+) T cells and ii) cytotoxic CD4(+) T cells, which can directly kill HLA class II antigen expressing tumor cells. In addition, both CD8(+) and CD4(+) T cells secrete IFN- γ, which can further sensitize tumor cells to CD8(+) T cell-mediated killing by upregulating HLA class I antigens and APM components, promoting the recruitment of natural killer (NK) cells, granulocytes and macrophages, as well as inhibiting angiogenesis within tumor stroma. Tumor growth can also be controlled by IL-5-secreting CD4(+) T helper cells (Th2). APC activate IL-5 Th2 cells, which induce the accumulation of eosinophils and/or provide help for the generation of an antibody-based TA-specific immune response. NK cells might also play a role by recognizing ‘stress’ or ‘danger’ signals that are produced by tumors. Once activated NK cells may contribute to the tumor immune response through (i) direct lysis of tumor cells, (ii) indirectly providing TA to APC for presentation to CTL, (iii) activating CD4+ T and B-cells as well as CTL through the secretion of cytokines such as IFN- γ. (B) The release of soluble classical and non-classical HLA antigens as well as NK cell activating ligands may lead to downregulation of the host's TA-specific immune response through (i) induction of apoptosis of both CTL as well as NK cells, (ii) indirect downregulation of CD4+ T helper as well as B cell reponses via suppression of IFN- γ secondary to NK cell apoptosis, and (iii) altering the level of membrane bound HLA antigen and NKAL ligand expression thereby hindering their recognition by T and NK cells, respectively.
Convincing evidence has shown that classical β2m-free as well as β2m-associated sHLA can induce apoptosis of activated CD8+ T cells in vitro [86]. Whether dimeric or tetrameric forms of classical sHLA are required to induce apoptosis, as shown in other in vitro systems [87-90], remains to be determined. Conflicting mechanisms have been shown to underlie the induction of apoptosis in activated T cells. Zavazava et. al. have shown that apoptosis results from interaction of soluble classical sHLA with the T cell receptor [91]. In contrast, Puppo and colleagues have shown that apoptosis mediated by classical sHLA is dependent on CD8 in both T and NK cells suggesting that interaction with the TCR is not required for this effect [86,92,93]. However there is agreement on the requirement of Fas and FasL in the induction of apoptosis by classical sHLA [86,91,92,94]. This mechanism is supported by the observation that CD8 cross-linking by specific antibodies, like classical sHLA, generates signals to induce Fas mRNA and soluble Fas ligand [95]. An additional mechanism is suggested by the earlier report [58] that classical sHLA may prolong allograft survival by inducing unresponsiveness. In light of our current understanding of the requirements for naïve T cell activation, induction of anergy or tolerance by classical sHLA may reflect lack of associated costimulatory molecules. However, direct in vitro evidence for non-responsiveness of T cells induced by classical sHLA has not been documented possibly due to the use in most studies of activated T cells, activated NK cells, clones or hybridomas which do not typically require costimulation. Studies in mice demonstrate the potential of soluble MHC class I antigens to modulate the immune response. The injection of MHC class I dimers or tetramers, but not monomers, induced tolerance of alloreactive T cells [87,96]. Most relevant to the malignant setting, injection of appropriate MHC class I-peptide complexes into tumor-bearing mice suppressed T cell-mediated control of tumor growth suggesting a potential in vivo function for naturally occurring soluble MHC class I molecules [88,97]. Whether such approaches may have an impact on the control of tumor growth in patients with malignant diseases has yet to be tested.
The effects of classical sHLA do not appear to be limited to T cells, since interaction of classical sHLA with either IRS or CD8 on NK cells leads to NK cell apoptosis [80,81]. As for T cells, the mechanism underlying classical sHLA induced NK cell apoptosis appears to be related to Fas/FasL interaction, since classical sHLA induces de novo transcription of Fas and FasL mRNA in NK cells [98-100]. Furthermore, classical sHLA induced apoptosis of NK cells can be inhibited by Fas- and FasL-specific mAb [98]. The interaction of classical sHLA with CD8 or IRS on NK cells not only leads to their death, but also can induce the production and secretion of detectable amounts of IFN-γ and TGF-β [100]. The physiological significance of this effect is still to be defined. The release of TGF-β from NK cells upon interaction with classical sHLA may further down-regulate NK cell survival by inducing their apoptosis and impairment of NK cell-mediated cytotoxicity [101,102]. One can speculate that by activating antigen-presenting cells and thus by favoring optimal antigen presentation to T lymphocytes, IFN-γ can induce a Th1-type immune response, typical of the antiviral host response. In this context, IFN-γ production which accompanies NK cell death induced by classical sHLA may be useful in switching from the innate to the adaptive immune response. In addition, IFN-γ can also up-regulate the expression of classical HLA class I antigens on target cells, possibly leading to a stronger inhibitory effect on NK cell-mediated functional activities via interaction of these molecules with the inhibitory counter IRS. At the same time, IFN-γ can increase the shedding of classical sHLA from tumor cells and thus increase the degree of NK cell apoptosis upon interaction with CD8 and/or activating IRS both in the tumor microenvironment and in distant sites. Thus, classical sHLA can interact with CD8, thereby inducing NK cell death without the need of NK cell-target cell interaction. On the other hand, NK cells, during interaction with target cells, might receive an apoptotic signal through the direct binding of CD8 and/or activating IRS with classical HLA class I antigens expressed by the target cell. These phenomena may play a key role in regulating the response of innate immunity against tumors. In this regard, classical sHLA can induce apoptosis of T and NK cells at concentrations as low as 1pg/ml [81]. It is therefore possible that tumor cells can downregulate T and NK cell activity by releasing classical sHLA actively and/or passively through necrosis of tumor cells. This phenomenon could explain, at least in part, how tumors with a low expression of classical cell surface HLA class I antigens can escape both T cell- and NK cell-mediated killing.
For non-classical sHLA antigen much of the available information is limited to sHLA-G. In this regard, there is convincing evidence that, like classical sHLA [71], sHLA-G induces apoptosis of activated CD8+ T cells and CD8+ NK cells, utilizing the same pathway: their binding to CD8 leads to FasL upregulation, soluble FasL secretion and activated CD8+ cell apoptosis by Fas/sFasL interaction [71]. However, there is conflicting information in the literature about the potency of sHLA-G and classical sHLA in triggering activated CD8+ cell apoptosis [71]. Whether these discrepancies reflect the different source of antigens used, the different methods of purification and/or other technical issues remains to be determined. Furthermore, it is not known whether sHLA-G cooperates with classical sHLA in inducing apoptosis and whether this effect is additive or synergistic. In any case, it is likely that in physiological conditions sHLA-G molecules do not play a major role, since their level in serum is about one order of magnitude below that required to induce CD8+ T cell and CD8+ NK cell apoptosis in vitro [71]. Thus, the potential role of HLA-G in the regulation of the immune response would be restricted to pathological conditions associated with a marked increase in the level of sHLA-G in serum or in a given anatomic site. In this regard, the production of inflammatory molecules by tumor cells may play a role in modulating the host immune response by increasing sHLA-G expression by peripheral monocytes [103]. Over the years, a number of tumor-derived factors with immunosuppressive activity have been identified (Table 1) [85]. Notably many of these molecules have been demonstrated to be inducers of HLA-G production in monocytes [71,85].
Table 1. Tumor-Associated Suppressive Factorsa.
| 1. The TNF family ligands: Induce leukocyte apoptosis via the TNF family receptors [85] | |
| FasL | Fas |
| TRAIL | TRAIL-Rs |
| TNF | TNFR1 |
| 2. Small molecules | |
| ProstagladinE2 (PGE2) | Inhibits leukocyte functions through increased camp [85] |
| Histamine | Inhibits leukocyte functions through increased cAMP [85] |
| Epinephrine | Inhibits leukocyte functions through increased cAMP [85] |
| Inducible Nitric Oxide Synthase | Promotes or inhibits Fas-mediated apoptosis by regulation of nitric oxide levels [85] |
| H202 | Has pro-oxidant activity, increases cAMP levels, causes apoptosis in NK cells, inhibits tumor-specific CTL [85] |
| 3. Cytokines | |
| TGF-β | Inhibits perforin and granzyme mRNA expression; inhibits lymphocyte proliferation [85] |
| IL-10 | Inhibits production of IL-1β, IFN-γ, IL-12 and TNFα [85] |
| Granulocyte-macrophage colony-stimulating factor | Promotes expansion of immunosuppressive tumor-associated macrophages [85] |
| 4. Tumor-associated gangliosides | Inhibit IL-2 dependent lymphocyte proliferation or induce apoptotic signals [85] |
A partial list of immunosuppressive factors selected to demonstrate their diversity and a wide spectrum of effects on immune cells.
Like classical sHLA, sHLA-G1 inhibits the cytotoxic activity of HLA class I antigen restricted, antigen-specific CTL [71]. Moreover, sHLA-G5 can inhibit CD4 and CD8 T cell proliferation by blocking cell cycle progression through yet undefined mechanisms. Rajagopalan et al. [104] have recently showed that sHLA-G, following binding to the CD158d receptor on the surface of human resting NK cells, is endocytosed and triggers the expression of a set of chemokines and cytokines driving a pro-inflammatory/proangiogenic response. These findings suggest that NK cells can exert beneficial effects at sites of HLA-G expression, such as stimulation of vascularization in the maternal decidua during pregnancy. Furthermore, a novel effect of sHLA-G on angiogenesis has been recently described. Specifically, sHLA-G1 has been shown to inhibit in vitro and in vivo angiogenesis by inducing endothelial cell apoptosis upon binding to the CD160/BY55 receptor [105]. Since the latter receptor was detected also in the vasculature of a murine tumor, it is tempting to speculate that CD160 may represent an attractive therapeutic target to inhibit tumor-associated neoangiogenesis.
In addition to classical and non-classical sHLA, sNKAL may also play a role in immune escape of tumor cells. As previously mentioned, sMICA and sMICB are released by tumor cells and are present in the sera of patients with certain malignant disease. Thus it is conceivable that the NKG2D activating receptor on cytolytic effector T (either TCRαβ+or TCRγδ+) and NK cells can bind to sMICA or sMICB instead of MIC-A/B expressed by tumor cells. In addition, it has been reported that sMIC ligands impair NKG2D expression and consequently impair NKG2D functionality in TA-specific cytotoxic lymphocytes and NK cells [38,40,43-45,50,106-108]. These two mechanisms may allow tumor cells to evade immune system-mediated control. If this were true, it would be essential to analyze the mechanisms by which tumor cells can shed MICA/B in order to prevent or block this loss. Thus by preserving MICA/B at the tumor cell surface, effector cells could deliver the lethal hit and eliminate tumor cells. Like MICA/B, other NKG2D ligands, such as ULBPs, which are found in the sera of patients with neuroblastoma and leukemia [50,52], might be released by tumor cells and contribute to tumor escape (Fig. 4).
V. Conclusion
The data we have discussed suggest that changes which are likely to take place in the tumor microenvironment may have a negative impact on the recognition of tumor cells by host's immune system and on the ability of cytotoxic cells to control tumor growth. Accumulation in the microenvironment of sHLA antigens and sNKAL, which are produced by tumor cells an/or stromal cells as well as infiltrating leukocytes, may interfere with interactions of CTL or NK cells with tumor cells. Both sHLA and sNKAL may lead to reduced CTL and NK cell-mediated killing by directly inducing apoptosis of CTL and NK cells. Moreover, if HLA antigens are released from the surface of tumor cells, one might expect a reduced level of HLA antigens on the cell membrane and a consequent reduction in HLA class I antigen–TA-derived peptide complex recognition by CTLs. Furthermore, sHLA may inhibit the cytotoxic effects of NK cells even when tumor cells downregulate cell surface HLA class I antigens, which would otherwise enhance NK cell mediated killing. A similar mechanism of NK inactivation has been described for soluble non-classical sHLA antigens HLA-G as well as sNKAL MICA that can also be released from tumor cells. These phenotypic changes of tumor cells in the tumor microenvironment provide a potential mechanism for the lack of correlation between in vitro susceptibility of tumor cells to lysis mediated by HLA class I antigen-restricted, TA-specific CTL and control of tumor growth in vivo. Because of the lack of exposure to cytokines and MMP which are present in the tumor microenvironment, tumor cells cultured in vitro restore the expression HLA class I antigen-TA-derived peptide complexes on their membranes.
The potential changes which may be induced in tumor cells by the microenvironment have implications for the design of approaches to characterize their phenotype and of immunotherapeutic strategies for the treatment of malignant diseases. Together these findings emphasize the importance to characterize the interplay between tumor microenvironment and immune effector cells. This information may suggest strategies to overcome the barriers posed by the microenvironment to an effective destruction of tumor cells mediated by immunological mechanisms. Moreover, the data we have reviewed indicate that classical and non-classical HLA class I antigens as well as NK-cell activating ligands released by tumor cells in the microenvironment may interfere with the function of cytotoxic cells utilizing multiple mechanisms. As a result, cytotoxic cells are not likely to be able to control tumor growth in spite of the expression of HLA class I antigen-TA derived peptide complexes and/or of NK-cell activating ligands by tumor cells. In this case, however, the data published by Dranoff and his collaborators [109,110] and corroborated by Marten et al's [111] recent results, suggest that administration or induction of antibodies recognizing the molecules involved, if not responsible for tumor progression may represent an effective therapeutic approach. In conclusion, the changes which may take place in tumor cells in the microenvironment as well as the potential effects of histocompatibility antigens released by tumor cells on immune cells provide mechanisms for the lack of clinical responses in spite of a TA-specific immune response. In addition, the information we have discussed emphasizes the need to characterize tumor cells within the microenvironment in order to optimize immunotherapeutic strategies for the treatment of malignant disease.
References
- 1.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–998. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 2.Marchand M, Weynants P, Rankin E, et al. Tumor regression responses in melanoma patients treated with a peptide encoded by gene MAGE-3. Int J Cancer. 1995;63:883–885. doi: 10.1002/ijc.2910630622. [DOI] [PubMed] [Google Scholar]
- 3.Jaeger E, Bernahrd H, Romero P, Ringhoffer M, Arand M, Karbach J, Ilsemann C, Hagedorn M, Knuth A. Generation of cytotoxic T-cell responses with synthetic peptides in vivo: implications for tumor vaccines with melanoma-associated antigens. Int J Cancer. 1996;66:162–169. doi: 10.1002/(SICI)1097-0215(19960410)66:2<162::AID-IJC4>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 4.Parmiani G, Castelli C, Dalerba P, Mortarini R, Rivoltini L, Marincola FM, Anichini A. Cancer immunotherapy with peptide-based vaccines: what have we achieved? Where are we going? J Natl Cancer Inst. 2002;94:805–818. doi: 10.1093/jnci/94.11.805. [DOI] [PubMed] [Google Scholar]
- 5.Durrant LG, Spendlove I. Cancer vaccines entering Phase III clinical trials. Expert Opin Emerg Drugs. 2003;8:489–500. doi: 10.1517/14728214.8.2.489. [DOI] [PubMed] [Google Scholar]
- 6.Chang CC, Campoli M, Ferrone S. Classical and nonclassical HLA class I antigen and NK cell-activating ligand changes in malignant cells: current challenges and future directions. Adv Cancer Res. 2005;93:189–234. doi: 10.1016/S0065-230X(05)93006-6. [DOI] [PubMed] [Google Scholar]
- 7.Knutson KL, Disis ML. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol Immunother. 2005;54:721–728. doi: 10.1007/s00262-004-0653-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moretta L, Bottino C, Pende D, Vitale M, Mingari MC, Moretta A. Human natural killer cells: Molecular mechanisms controlling NK cell activation and tumor cell lysis. Immunol Lett. 2005;100:7–13. doi: 10.1016/j.imlet.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 9.Chang CC, Ferrone S. NK cell activating ligands on human malignant cells: molecular and functional defects and potential clinical relevance. Semin Cancer Biol. 2006;16:383–92. doi: 10.1016/j.semcancer.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 10.Chang CC, Ferrone S. Immune selective pressure and HLA class I antigen defects in malignant lesions. Cancer Immunol Immunother. 2007;56:227–36. doi: 10.1007/s00262-006-0183-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim R, Emi M, Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology. 2007;121:1–14. doi: 10.1111/j.1365-2567.2007.02587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Freiss N. HLA-G molecules: from maternal-fetal tolerance to tissue acceptance. Adv Immunol. 2003;81:199–252. doi: 10.1016/s0065-2776(03)81006-4. [DOI] [PubMed] [Google Scholar]
- 13.Seliger B, Abken H, Ferrone S. HLA-G and MIC expression in tumors and their role in anti-tumor immunity. Trends Immunol. 2003;24:82–87. doi: 10.1016/s1471-4906(02)00039-x. [DOI] [PubMed] [Google Scholar]
- 14.Algarra I, García-Lora A, Cabrera T, Ruiz-Cabello F, Garrido F. The selection of tumor variants with altered expression of classical and nonclassical MHC class I molecules: implications for tumor immune escape. Cancer Immunol Immunother. 2004;53:904–910. doi: 10.1007/s00262-004-0517-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wischhusen J, Waschbisch A, Wiendl H. Immune-refractory cancers and their little helpers--an extended role for immunetolerogenic MHC molecules HLA-G and HLA-E? Semin Cancer Biol. 2007;17:459–68. doi: 10.1016/j.semcancer.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 16.Marsman M, Jordens I, Griekspoor A, Neefjes J. Chaperoning antigen presentation by MHC class II molecules and their role in oncogenesis. Adv Cancer Res. 2005;93:129–158. doi: 10.1016/S0065-230X(05)93004-2. [DOI] [PubMed] [Google Scholar]
- 17.Bahram S. MIC genes: from genetics to biology. Adv Immunol. 2000;76:1–60. doi: 10.1016/s0065-2776(01)76018-x. [DOI] [PubMed] [Google Scholar]
- 18.Carosella ED, Moreau P, Le Maoult J, Le Discorde M, Dausset J, Rouas-Vivier E, Tomasello E, Paul P. Lymphocyte activation via NKG2D: towards a new paradigm in immune recognition? Curr Opin Immunol. 2002;14:306–311. doi: 10.1016/s0952-7915(02)00337-0. [DOI] [PubMed] [Google Scholar]
- 19.Burgess SJ, Maasho K, Masilamani M, Narayanan S, Borrego F, Coligan JE. The NKG2D receptor: immunobiology and clinical implications. Immunol Res. 2008;40:18–34. doi: 10.1007/s12026-007-0060-9. [DOI] [PubMed] [Google Scholar]
- 20.Campoli M, Chang CC, Oldford SA, Edgecombe AD, Drover S, Ferrone S. HLA antigen changes in malignant tumors of mammary epithelial origin: molecular mechanisms and clinical implications. Breast Dis. 2004;20:105–125. doi: 10.3233/bd-2004-20112. [DOI] [PubMed] [Google Scholar]
- 21.Campoli M, Ferrone S. A Fresh Look at an Old Story: Revisiting HLA class II antigen expression by melanoma cells. Expert Rev Dermatol. 2006;1:737–755. [Google Scholar]
- 22.Waldmann TA, Dubois S, Tagaya Y. Contrasting roles of IL-2 and IL-15 in the life and death of lymphocytes: implications for immunotherapy. Immunity. 2001;14:105–110. [PubMed] [Google Scholar]
- 23.Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet. 2001;29:117–129. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 24.Toutirais O, Chartier P, Dubois D, Bouet F, Leveque J, Catros-Quemener V, Genetet N. Constitutive expression of TGF-beta1, interleukin-6 andinter- leukin-8 by tumor cells as a major component of immune escape in human ovarian carcinoma. Eur Cytokine Netw. 2003;14:246–255. [PubMed] [Google Scholar]
- 25.Letterio JJ. TGF-beta signaling in T cells: roles in lymphoid and epithelial neoplasia. Oncogene. 2005;24:5701–5712. doi: 10.1038/sj.onc.1208922. [DOI] [PubMed] [Google Scholar]
- 26.Giacomini P, Aguzzi A, Pestka S, Fisher PB, Ferrone S. Modulation by recombinant DNA leukocyte (alpha) and fibroblast (beta) interferons of the expression and shedding of HLA- and tumor-associated antigens by human melanoma cells. J Immunol. 1984;133:1649–1655. [PubMed] [Google Scholar]
- 27.Krangel MS. Secretion of HLA-A and -B antigens via an alternative RNA splicing pathway. J Exp Med. 1986;163:1173–1190. doi: 10.1084/jem.163.5.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dobbe LM, Stam NJ, Neefjes JJ, Giphart MJ. Biochemical complexity of serum HLA class I molecules. Immunogenetics. 1988;27:203–210. doi: 10.1007/BF00346587. [DOI] [PubMed] [Google Scholar]
- 29.Aulitzky WE, Grosse-Wilde H, Westhoff U, Tilg H, Aulitzky W, Gastl G, Herold M, Huber C. Enhanced serum levels of soluble HLA class I molecules are induced by treatment with recombinant interferon-gamma (IFN- gamma) Clin Exp Immunol. 1991;86:236–239. doi: 10.1111/j.1365-2249.1991.tb05802.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giacomini P, Fraioli R, Calabro AM, Di Filippo F, Natali PG. Class I major histocompatibility complex enhancement by recombinant leukocyte interferon in the peripheral blood mononuclear cells and plasma of melanoma patients. Cancer Res. 1991;51:652–656. [PubMed] [Google Scholar]
- 31.Demaria S, Schwab R, Gottesman SR, Bushkin Y. Soluble beta 2-micro- globulin-free class I heavy chains are released from the surface of activated and leukemia cells by a metalloprotease. J Biol Chem. 1994;269:6689–6694. [PubMed] [Google Scholar]
- 32.Shimura T, Hagihara M, Yamamoto K, Takebe K, Munkhbat B, Ogoshi K, Mitomi T, Nagamachi Y, Tsuji K. Quantification of serum-soluble HLA class I antigens in patients with gastric cancer. Hum Immunol. 1994;40:183–6. doi: 10.1016/0198-8859(94)90067-1. [DOI] [PubMed] [Google Scholar]
- 33.Puppo F, Scudeletti M, Indiveri F, Ferrone S. Serum HLA class I antigens: markers and modulators of an immune response? Immunol Today. 1995;6:124–127. doi: 10.1016/0167-5699(95)80127-8. [DOI] [PubMed] [Google Scholar]
- 34.Puppo F, Indiveri F, Scudeletti M, Ferrone S. Soluble HLA antigens: new roles and uses. Immunol Today. 1997;18:154–155. doi: 10.1016/s0167-5699(97)84660-9. [DOI] [PubMed] [Google Scholar]
- 35.O'Callaghan CA, Bell JI. Structure and function of the human MHC class Ib molecules HLA-E, HLA-F and HLA-G. Immunol Rev. 1998;163:129–138. doi: 10.1111/j.1600-065x.1998.tb01192.x. [DOI] [PubMed] [Google Scholar]
- 36.Westhoff U, Fox C, Otto FJ. Soluble HLA class I antigens in plasma of patients with malignant melanoma. Anticancer Res. 1998;18:3789–92. [PubMed] [Google Scholar]
- 37.Aultman D, Adamashvili I, Yaturu K, Langford M, Gelder F, Gautreaux M, Ghali GE, McDonald J. Soluble HLA in human body fluids. Hum Immunol. 1999;60:239–44. doi: 10.1016/s0198-8859(98)00122-0. [DOI] [PubMed] [Google Scholar]
- 38.Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, Spies T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science. 1999;285:727–729. doi: 10.1126/science.285.5428.727. [DOI] [PubMed] [Google Scholar]
- 39.Demaria S, Bushkin Y. Soluble HLA proteins with bound peptides are released from the cell surface by the membrane metalloproteinase. Hum Immunol. 2000;61:1332–1338. doi: 10.1016/s0198-8859(00)00213-5. [DOI] [PubMed] [Google Scholar]
- 40.Cosman D, Mullberg J, Sutherland CL, Chin W, Armitage R, Fanslow W, Kubin M, Chalupny NJ. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity. 2001;14:123–133. doi: 10.1016/s1074-7613(01)00095-4. [DOI] [PubMed] [Google Scholar]
- 41.Shimura T, Tsutsumi S, Hosouchi Y, Kojima T, Kon Y, Yonezu M, Kuwano H. Clinical significance of soluble form of HLA class I molecule in Japanese patients with pancreatic cancer. Hum Immunol. 2001;62:615–619. doi: 10.1016/s0198-8859(01)00246-4. [DOI] [PubMed] [Google Scholar]
- 42.Ugurel S, Rebmann V, Ferrone S, Tilgen W, Grosse-Wilde H, Reinhold U. Soluble human leukocyte antigen-G serum level is elevated in melanoma patients and is further increased by interferon-alpha immunotherapy. Cancer. 2001;92:369–376. doi: 10.1002/1097-0142(20010715)92:2<369::aid-cncr1332>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 43.Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature. 2002;419:734–738. doi: 10.1038/nature01112. [DOI] [PubMed] [Google Scholar]
- 44.Pende D, Rivera P, Marcenaro S, Chang CC, Biassoni R, Conte R, Kubin M, Cosman D, Ferrone S, Moretta L, Moretta A. Major histocompatibility complex class I-related chain A and UL16-binding protein expression on tumor cell lines of different histotypes: analysis of tumor susceptibility to NKG2D-dependent natural killer cell cytotoxicity. Cancer Res. 2002;62:6178–6186. [PubMed] [Google Scholar]
- 45.Salih HR, Rammensee HG, Steinle A. Cutting edge: down-regulation of MIC-A on human tumors by proteolytic shedding. J Immunol. 2002;169:4098–5102. doi: 10.4049/jimmunol.169.8.4098. [DOI] [PubMed] [Google Scholar]
- 46.Vivier E, Tomasello E, Paul P. Lymphocyte activation via NKG2D: towards a new paradigm in immune recognition? Curr Opin Immunol. 2002;14:306–311. doi: 10.1016/s0952-7915(02)00337-0. [DOI] [PubMed] [Google Scholar]
- 47.Wiendl H, Mitsdoerffer M, Hofmeister V, Wischhusen J, Bornemann A, Meyermann R, et al. A functional role of HLA-G expression in human gliomas: an alternative strategy of immune escape. J Immunol. 2002;168:4772–4780. doi: 10.4049/jimmunol.168.9.4772. [DOI] [PubMed] [Google Scholar]
- 48.Amiot L, Le Friec G, Sebti Y, Drenou B, Pangault C, Guilloux V, et al. HLA- G and lymphoproliferative disorders. Semin Cancer Biol. 2003;13:379–385. doi: 10.1016/s1044-579x(03)00029-4. [DOI] [PubMed] [Google Scholar]
- 49.Rebmann V, Regel J, Stolke D, Grosse-Wilde H. Secretion of sHLA-G molecules in malignancies. Semin Cancer Biol. 2003;13:371–377. doi: 10.1016/s1044-579x(03)00028-2. [DOI] [PubMed] [Google Scholar]
- 50.Salih HR, Antropius H, Gieseke F, Lutz SZ, Kanz L, Rammensee HG, Steinle A. Functional expression and release of ligands for the activating immunoreceptor NKG2D in leukemia. Blood. 2003;102:1389–1396. doi: 10.1182/blood-2003-01-0019. [DOI] [PubMed] [Google Scholar]
- 51.Cao W, He W. UL16 binding proteins. Immunobiology. 2004;209:283–290. doi: 10.1016/j.imbio.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 52.Raffaghello L, Prigione I, Airoldi I, Camoriano M, Levreri I, Gambini C, Pende D, Steinle A, Ferrone S, Pistoia V. Downregulation and/or release of NKG2D ligands as immune evasion strategy of human neuroblastoma. Neoplasia. 2004;6:558–568. doi: 10.1593/neo.04316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carbone E, Neri P, Mesuraca M, Fulciniti MT, Otsuki T, Pende D, Groh V, Spies T, Pollio G, Cosman D, Catalano L, Tassone P, Rotoli B, Venuta S. HLA class I, NKG2D, and natural cytotoxicity receptors regulate multiple myeloma cell recognition by natural killer cells. Blood. 2005;105:251–258. doi: 10.1182/blood-2004-04-1422. [DOI] [PubMed] [Google Scholar]
- 54.Jinushi M, Takehara T, Tatsumi T, Hiramatsu N, Sakamori R, Yamaguchi S, Hayashi N. Impairment of natural killer cell and dendritic cell functions by the soluble form of MHC class I-related chain A in advanced human hepatocellular carcinomas. J Hepatol. 2005;43:1013–1020. doi: 10.1016/j.jhep.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 55.Leleu X, Le Friec G, Facon T, Amiot L, Fauchet R, Hennache B, Coiteux V, Yakoub-Agha I, Dubucquoi S, Avet-Loiseau H, Mathiot C, Bataille R, Mary JY. Intergroupe Francophone du Myélome. Total soluble HLA class I and soluble HLA-G in multiple myeloma and monoclonal gammopathy of undetermined significance. Clin Cancer Res. 2005;11:7297–7303. doi: 10.1158/1078-0432.CCR-05-0456. [DOI] [PubMed] [Google Scholar]
- 56.Raffaghello L, Prigione I, Bocca P, Morandi F, Camoriano M, Gambini C, et al. Multiple defects of the antigen-processing machinery components in human neuroblastoma: immunotherapeutic implications. Oncogene. 2005;24:4634–4644. doi: 10.1038/sj.onc.1208594. [DOI] [PubMed] [Google Scholar]
- 57.Rodgers JR, Cook RG. MHC class Ib molecules bridge innate and acquired immunity. Nat Rev Immunol. 2005;5:459–471. doi: 10.1038/nri1635. [DOI] [PubMed] [Google Scholar]
- 58.Bangia N, Ferrone S. Antigen presentation machinery (APM) modulation and soluble HLA molecules in the tumor microenvironment: do they provide tumor cells with escape mechanisms from recognition by cytotoxic T lymphocytes? Immunol Invest. 2006;35:485–503. doi: 10.1080/08820130600808246. [DOI] [PubMed] [Google Scholar]
- 59.Derré L, Corvaisier M, Charreau B, Moreau A, Godefroy E, Moreau-Aubry A, Jotereau F, Gervois N. Expression and release of HLA-E by melanoma cells and melanocytes: potential impact on the response of cytotoxic effector cells. J Immunol. 2006;177:3100–3107. doi: 10.4049/jimmunol.177.5.3100. [DOI] [PubMed] [Google Scholar]
- 60.Gros F, Sebti Y, de Guibert S, Branger B, Bernard M, Fauchet R, Amiot L. Soluble HLA-G molecules increase during acute leukemia, especially in subtypes affecting monocytic and lymphoid lineages. Neoplasia. 2006;8:223–230. doi: 10.1593/neo.05703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Holdenrieder S, Stieber P, Peterfi A, Nagel D, Steinle A, Salih HR. Soluble MICA in malignant diseases. Int J Cancer. 2006;118:684–687. doi: 10.1002/ijc.21382. [DOI] [PubMed] [Google Scholar]
- 62.Holdenrieder S, Stieber P, Peterfi A, Nagel D, Steinle A, Salih HR. Soluble MICB in malignant diseases: analysis of diagnostic significance and correlation with soluble MICA. Cancer Immunol Immunother. 2006;55:1584–1589. doi: 10.1007/s00262-006-0167-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Märten A, von Lilienfeld-Toal M, Büchler MW, Schmidt J. Soluble MIC is elevated in the serum of patients with pancreatic carcinoma diminishing gammadelta T cell cytotoxicity. Int J Cancer. 2006;119:2359–2365. doi: 10.1002/ijc.22186. [DOI] [PubMed] [Google Scholar]
- 64.Salih HR, Goehlsdorf D, Steinle A. Release of MICB molecules by tumor cells: mechanism and soluble MICB in sera of cancer patients. Hum Immunol. 2006;67:188–95. doi: 10.1016/j.humimm.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 65.Song H, Kim J, Cosman D, Choi I. Soluble ULBP suppresses natural killer cell activity via down-regulating NKG2D expression. Cell Immunol. 2006;239:22–30. doi: 10.1016/j.cellimm.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 66.Waldhauer I, Steinle A. Proteolytic release of soluble UL16-binding protein 2 from tumor cells. Cancer Res. 2006;66:2520–2526. doi: 10.1158/0008-5472.CAN-05-2520. [DOI] [PubMed] [Google Scholar]
- 67.Albitar M, Johnson M, Do KA, Day A, Jilani I, Pierce S, Estey E, Kantarjian H, Keating M, Verstovsek S, O'brien S, Giles FJ. Levels of soluble HLA-I and beta2M in patients with acute myeloid leukemia and advanced myelodysplastic syndrome: association with clinical behavior and outcome of induction therapy. Leukemia. 2007;21:480–488. doi: 10.1038/sj.leu.2404506. [DOI] [PubMed] [Google Scholar]
- 68.Albitar M, Vose JM, Johnson MM, Do KA, Day A, Jilani I, Kantarjian H, Keating M, O'Brien SM, Verstovsek S, Armitage JO, Giles FJ. Clinical relevance of soluble HLA-I and beta2-microglobulin levels in non-Hodgkin's lymphoma and Hodgkin's disease. Leuk Res. 2007;31:139–145. doi: 10.1016/j.leukres.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 69.Cao W, Xi X, Hao Z, Li W, Kong Y, Cui L, Ma C, Ba D, He W. RAET1E2, a soluble isoform of the UL16-binding protein RAET1E produced by tumor cells, inhibits NKG2D-mediated NK cytotoxicity. J Biol Chem. 2007;282:18922–18928. doi: 10.1074/jbc.M702504200. [DOI] [PubMed] [Google Scholar]
- 70.Holdenrieder S, Eichhorn P, Beuers U, Samtleben W, Stieber P, Nagel D, Peterfi A, Steinle A, Salih HR. Soluble NKG2D ligands in hepatic autoimmune diseases and in benign diseases involved in marker metabolism. Anticancer Res. 2007;27:2041–2045. [PubMed] [Google Scholar]
- 71.Pistoia V, Morandi F, Wang X, Ferrone S. Soluble HLA-G: Are they clinically relevant? Semin Cancer Biol. 2007;17:469–79. doi: 10.1016/j.semcancer.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Porcu-Buisson G, Lambert M, Lyonnet L, Loundou A, Gamerre M, Camoin-Jau L, Dignat-George F, Caillat-Zucman S, Paul P. Soluble MHC Class I chain-related molecule serum levels are predictive markers of implantation failure and successful term pregnancies following IVF. Hum Reprod. 2007;22:2261–2266. doi: 10.1093/humrep/dem157. [DOI] [PubMed] [Google Scholar]
- 73.Rebmann V, Schütt P, Brandhorst D, Opalka B, Moritz T, Nowrousian MR, Grosse-Wilde H. Soluble MICA as an independent prognostic factor for the overall survival and progression-free survival of multiple myeloma patients. Clin Immunol. 2007;123:114–120. doi: 10.1016/j.clim.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 74.Tabayoyong WB, Zavazava N. Soluble HLA revisited. Leuk Res. 2007;31:121–125. doi: 10.1016/j.leukres.2006.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Arreygue-Garcia NA, Daneri-Navarro A, del Toro-Arreola A, Cid-Arregui A, Gonzalez-Ramella O, Jave-Suarez LF, Aguilar-Lemarroy A, Troyo-Sanroman R, Bravo-Cuellar A, Delgado-Rizo V, Garcia-Iglesias T, Hernandez-Flores G, Del Toro-Arreola S. Augmented serum level of major histocompatibility complex class I-related chain A (MICA) protein and reduced NKG2D expression on NK and T cells in patients with cervical cancer and precursor lesions. BMC Cancer. 2008;8:16. doi: 10.1186/1471-2407-8-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen XM, Xu XQ, Sun K, Hallett WH, Zhao JD, Zhang DL. NKG2D ligands expression and NKG2D-mediated cytotoxicity in human laryngeal squamous carcinoma cells. Scand J Immunol. 2008;67:441–447. doi: 10.1111/j.1365-3083.2008.02086.x. [DOI] [PubMed] [Google Scholar]
- 77.Fernández-Morera JL, Rodríguez-Rodero S, Lahoz C, Tuñon A, Astudillo A, Garcia-Suarez O, Martínez-Borra J, López-Vázquez A, Rodrigo L, Gonzalez S, López-Larrea C. Soluble MHC class I chain-related protein B serum levels correlate with disease activity in relapsing-remitting multiple sclerosis. Hum Immunol. 2008;69:235–240. doi: 10.1016/j.humimm.2008.01.021. [DOI] [PubMed] [Google Scholar]
- 78.Jinushi M, Vanneman M, Munshi NC, Tai YT, Prabhala RH, Ritz J, Neuberg D, Anderson KC, Carrasco DR, Dranoff G. MHC class I chain-related protein A antibodies and shedding are associated with the progression of multiple myeloma. Proc Natl Acad Sci U S A. 2008;105:1285–1290. doi: 10.1073/pnas.0711293105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tamaki S, Sanefuzi N, Kawakami M, Aoki K, Imai Y, Yamanaka Y, Yamamoto K, Ishitani A, Hatake K, Kirita T. Association between soluble MICA levels and disease stage IV oral squamous cell carcinoma in Japanese patients. Hum Immunol. 2008;69:88–93. doi: 10.1016/j.humimm.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 80.Poggi A, Zocchi MR. Cyclosporin A regulates human NK cell apoptosis induced by soluble HLA-I or by target cells. Autoimmun Rev. 2005;4:532–536. doi: 10.1016/j.autrev.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 81.Poggi A, Zocchi MR. Mechanisms of tumor escape: role of tumor microenvironment in inducing apoptosis of cytolytic effector cells. Arch Immunol The Exp. 2006;54:323–333. doi: 10.1007/s00005-006-0038-7. [DOI] [PubMed] [Google Scholar]
- 82.Carosella ED, Moreau P, Lemaoult J, Rouas-Freiss N. HLA-G: from biology to clinical benefits. Trends Immunol. 2008;29:125–132. doi: 10.1016/j.it.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 83.Bernsen MR, Hakansson L, Gustafsson B, et al. On the biological relevance of MHC class II and B7 expression by tumour cells in melanoma metastases. Br J Cancer. 2003;88:424–431. doi: 10.1038/sj.bjc.6600703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.van Duinen SG, Ruiter DJ, Broecker EB, et al. Level of HLA antigens in locoregional metastases and clinical course of the disease in patients with melanoma. Cancer Res. 1988;48:1019–1025. [PubMed] [Google Scholar]
- 85.Whiteside T, Campoli M, Ferrone S. Tumor Induced Immune Suppression Mechanisms and Possible Solutions. In: Nagorsen D, Marincola FM, editors. Analyzing T Cell Responses How to analyze cellular immune responses against tumor associated antigens. Springer-Verlag; Netherlands: 2005. pp. 43–81. [Google Scholar]
- 86.Puppo F, Contini P, Ghio M, Brenci S, Scudeletti M, Filaci G, Ferrone S, Indiveri F. Soluble human MHC class I molecules induce soluble Fas ligand secretion and trigger apoptosis in activated CD8(+) Fas (CD95)(+) T lymphocytes. Int Immunol. 2000;12:195–203. doi: 10.1093/intimm/12.2.195. [DOI] [PubMed] [Google Scholar]
- 87.Dal Porto J, Johansen TE, Catipovic B, Parfiit DJ, Tuveson D, Gether U, Kozlowski S, Fearon DT, Schneck JP. A soluble divalent class I major histocompatibility complex molecule inhibits alloreactive T cells at nanomolar concentrations. Proc Natl Acad Sci U S A. 1993;90:6671–6675. doi: 10.1073/pnas.90.14.6671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.O'Herrin SM, Slansky JE, Tang Q, Markiewicz MA, Gajewski TF, Pardoll DM, Schneck JP, Bluestone JA. Antigen-specific blockade of T cells in vivo using dimeric MHC peptide. J Immunol. 2001;167:2555–2560. doi: 10.4049/jimmunol.167.5.2555. [DOI] [PubMed] [Google Scholar]
- 89.Xu XN, Purbhoo MA, Chen N, Mongkolsapaya J, Cox JH, Meier UC, Tafuro S, Dunbar PR, Sewell AK, Hourigan CS, Appay V, Cerundolo V, Burrows SR, McMichael AJ, Screaton GR. A novel approach to anti gen-specific deletion of CTL with minimal cellular activation using alpha3 domain mutants of MHC class I/peptide complex. Immunity. 2001;14:591–602. doi: 10.1016/s1074-7613(01)00133-9. [DOI] [PubMed] [Google Scholar]
- 90.Cebecauer M, Guillaume P, Hozak P, Mark S, Everett H, Schneider P, Luescher IF. Soluble MHC-peptide complexes induce rapid death of CD8+ CTL. J Immunol. 2005;174:6809–6819. doi: 10.4049/jimmunol.174.11.6809. [DOI] [PubMed] [Google Scholar]
- 91.Zavazava N, Kronke M. Soluble HLA class I molecules induce apoptosis in alloreactive cytotoxic T lymphocytes. Nat Med. 1996;2:1005–1010. doi: 10.1038/nm0996-1005. [DOI] [PubMed] [Google Scholar]
- 92.Contini P, Ghio M, Merlo A, Brenci S, Filaci G, Indiveri F, Puppo F. Soluble HLA class I/CD8 ligation triggers apoptosis in EBV-specific CD8+ cytotoxic T lymphocytes by Fas/Fas-ligand interaction. Hum Immunol. 2000;61:1347–1351. doi: 10.1016/s0198-8859(00)00212-3. [DOI] [PubMed] [Google Scholar]
- 93.Contini P, Ghio M, Poggi A, Filaci G, Indiveri F, Ferrone S, Puppo F. Soluble HLA-A,-B,-C and -G molecules induce apoptosis in T and NK CD8+ cells and inhibit cytotoxic T cell activity through CD8 ligation. Eur J Immunol. 2003;33:125–134. doi: 10.1002/immu.200390015. [DOI] [PubMed] [Google Scholar]
- 94.Puppo F, Contini P, Ghio M, Indiveri F. Soluble HLA class I molecules/CD8 ligation trigger apoptosis of CD8+ cells by Fas/Fas-ligand interaction. Scientific World Journal. 2002;2:421–423. doi: 10.1100/tsw.2002.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Contini P, Ghio M, Merlo A, Poggi A, Indiveri F, Puppo F. Apoptosis of antigen-specific T lymphocytes upon the engagement of CD8 by soluble HLA class I molecules is Fas ligand/Fas mediated: evidence for the involvement of p56lck, calcium calmodulin kinase II, and calcium-independent protein kinase C signaling pathways and for NF-kappaB and NF-AT nuclear translocation. J Immunol. 2005;175:7244–7254. doi: 10.4049/jimmunol.175.11.7244. [DOI] [PubMed] [Google Scholar]
- 96.Maile R, Wang B, Schooler W, Meyer A, Collins EJ, Frelinger JA. Antigen-specific modulation of an immune response by in vivo administration of soluble MHC class I tetramers. J Immunol. 2001;167:3708–3714. doi: 10.4049/jimmunol.167.7.3708. [DOI] [PubMed] [Google Scholar]
- 97.Goldberg J, Shrikant P, Mescher MF. In vivo augmentation of tumor-spe- cific CTL responses by class I/peptide antigen complexes on microspheres (large multivalent immunogen) J Immunol. 2003;170:228–235. doi: 10.4049/jimmunol.170.1.228. [DOI] [PubMed] [Google Scholar]
- 98.Spaggiari GM, Contini P, Carosio R, Arvigo M, Ghio M, Oddone D, Dondero A, Zocchi MR, Puppo F, Indiveri F, Poggi A. Soluble HLA class I mol- ecules induce natural killer cell apoptosis through the engagement of CD8. Evidence for a negative regulation exerted by CD94/NKG2A complex and KIR2D. Blood. 2002;99:1706–1714. doi: 10.1182/blood.v99.5.1706. [DOI] [PubMed] [Google Scholar]
- 99.Spaggiari GM, Contini P, Dondero A, Carosio R, Puppo F, Indiveri F, Zocchi MR, Poggi A. Soluble HLA class I induces NK cell apoptosis upon the engagement of killer-activating HLA class I receptors through FasL-Fas interaction. Blood. 2002;100:4098–4107. doi: 10.1182/blood-2002-04-1284. [DOI] [PubMed] [Google Scholar]
- 100.Spaggiari GM, Contini P, Negrini S, Dondero A, Carosio R, Ghio M, Puppo F, Indiveri F, Zocchi MR, Poggi A. IFN-gamma production in human NK cells through the engagement of CD8 by soluble or surface HLA class I molecules. Eur J Immunol. 2003;33:3049–3059. doi: 10.1002/eji.200323981. [DOI] [PubMed] [Google Scholar]
- 101.Bellone G, Aste-Amegaza M, Trinchieri G, Rodeck U. Regulation of NK cell functions by TGF-β1. J Immunol. 1995;155:1066–1073. [PubMed] [Google Scholar]
- 102.Zocchi MR, Contini P, Alfano M, Poggi A. Pertussis toxin (PTX) B subunit and the non-toxic PTX mutant PT9K/129G inhibit TAT-induced TGF-βproduc- tion by NK cells and TGF-β-mediated NK cell apoptosis. J Immunol. 2005;174:6054–6061. doi: 10.4049/jimmunol.174.10.6054. [DOI] [PubMed] [Google Scholar]
- 103.Morandi F, Levreri I, Bocca P, Galleni B, Raffaghello L, Ferrone S, et al. Human neuroblastoma cells trigger an immunosuppressive program in monocytes by stimulating soluble HLA-G release. Cancer Res. 2007;67:6433–6441. doi: 10.1158/0008-5472.CAN-06-4588. [DOI] [PubMed] [Google Scholar]
- 104.Rajagopalan S, Fu J, Long EO. Cutting edge: induction of IFN-gamma production but not cytotoxicity by the killer cell Ig-like receptor KIR2DL4 (CD158d) in resting NK cells. J Immunol. 2001;167:1877–1881. doi: 10.4049/jimmunol.167.4.1877. [DOI] [PubMed] [Google Scholar]
- 105.Fons P, Chabot S, Cartwright JE, Lenfant F, L'Faqihi F, Giustiniani J, Herault JP, Gueguen G, Bono F, Savi P, Aguerre-Girr M, Fournel S, Malecaze F, Bensussan A, Plouët J, Le Bouteiller P. Soluble HLA-G1 inhibits angiogenesis through an apoptotic pathway and by direct binding to CD160 receptor expressed by endothelial cells. Blood. 2006;108:2608–2615. doi: 10.1182/blood-2005-12-019919. [DOI] [PubMed] [Google Scholar]
- 106.Groh V, Steinle A, Bauer S, Spies T. Re- cognition of stress-induced MHC molecules by intestinal epithelial gammadeltaT cells. Science. 1998;279:1737–1740. doi: 10.1126/science.279.5357.1737. [DOI] [PubMed] [Google Scholar]
- 107.Jinushi M, Takehara T, Tatsumi T, Kanto T, Groh V, Spies T, Kimura R, Miyagi T, Mochizuki K, Sasaki Y, Hayashi N. Expression and role of MIC-A and MICB in human hepatocellular carcinomas and their regulation by retinoic acid. Int. J. Cancer. 2003;104:354–361. doi: 10.1002/ijc.10966. [DOI] [PubMed] [Google Scholar]
- 108.Poggi A, Zocchi MR, Carosio R, Ferrero E, Angelini D, Galgani S, Caramia D, Bernardi G, Borsellino G, Battistini L. Transendothelial migratory pathways of Vdelta1+TCRgammadelta+ and Vdelta2+TCRgamm-adelta+ T lymphocytes from healthy donors and multiple sclerosis patients: involvement of phosphatidylinositol 3 kinase and calcium calmodulin-dependent kinase II. J Immunol. 2002;168:6071–6077. doi: 10.4049/jimmunol.168.12.6071. [DOI] [PubMed] [Google Scholar]
- 109.Hodi FS, Dranoff G. Combinatorial cancer immunotherapy. Adv Immunol. 2006;90:341–368. doi: 10.1016/S0065-2776(06)90009-1. [DOI] [PubMed] [Google Scholar]
- 110.Jinushi M, Hodi FS, Dranoff G. Therapy-induced antibodies to MHC class I chain-related protein A antagonize immune suppression and stimulate antitumor cytotoxicity. Proc Natl Acad Sci U S A. 2006;103:9190–9195. doi: 10.1073/pnas.0603503103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lefterova P, Märten A, Buttgereit P, Weineck S, Scheffold C, Huhn D, Schmidt-Wolf IG. Targeting of natural killer-like T immunologic effector cells against leukemia and lymphoma cells by reverse antibody-dependent cellular cytotoxicity. J Immunother. 2000;23:304–310. doi: 10.1097/00002371-200005000-00003. [DOI] [PubMed] [Google Scholar]