

Abstract
Changes in classical and non-classical HLA class I as well as HLA class II antigens have been identified in malignant lesions. These changes which are described in this paper are believed to play a major role in the clinical course of the disease since both HLA class I and class II antigens are critical to the interaction between tumor cells and components of both innate and adaptive immune system. Abnormalities in HLA antigen expression in malignant cells, which range in frequency from 0-90%, are caused by distinct mechanisms. They include defects in β2-microglobulin (β2m) synthesis, loss of the gene(s) encoding HLA antigen heavy chain(s), mutations which inhibit HLA antigen heavy chain transcription or translation, defects in the regulatory mechanisms which control HLA antigen expression and/or abnormalities in one or more of the antigen processing machinery (APM) components. More recently, epigenetic events associated with tumor development and progression have been found to underlie changes in HLA antigen, APM component, co-stimulatory molecule and TA expression in malignant cells. The types of epigenetic modifications that may occur in normal and malignant cells as well as their role underlying changes in HLA expression by malignant cells have been reviewed. The epigenetic events associated with alterations in HLA antigen expression may be clinically relevant since, in some case, they have been shown to impair the recognition of tumor cells by components of the adaptive immune system. The functional relevance and potential clinical significance of these epigenetic alterations have been addressed. Lastly, unlike genetic alterations, epigenetic modifications can, in some cases, be reversed with pharmacologic agents that induce DNA hypomethylation or inhibit histone deacetylation. Therefore strategies to overcome epigenetic modifications underlying changes in HLA expression in malignant cells have been discussed.
Keywords: acetylation, antigen processing machinery, cancer, classical HLA class I antigen, epigenetic, immune escape, immunotherapy, iRNA, HLA class II antigen, histone, methylation, NK cell activating ligand, non-classical HLA class I antigen
I. Introduction
The revival of the cancer immunosurveillance theory [1] has emphasized the role of escape mechanisms in tumor growth [2-5]. As a result, tumor immunologists have been focusing their investigations on the identification and molecular characterization of the mechanisms by which tumor cells evade immune recognition and destruction [2-6]. Among the many escape mechanisms identified, alterations in classical and non-classical HLA class I as well as HLA class II expression by tumor cells are of particular interest to tumor immunologists and clinical oncologists because of the critical role they play in the generation of tumor antigen (TA)-specific immune responses [7-10], as well as their ability to modulate the interactions of natural killer (NK) cells [10] and T cell subpopulations [7-10] with target cells. The potential clinical relevance of these escape mechanisms is suggested by the statistically significant association of changes in classical HLA class I expression [6], as well as of induction of non-classical HLA class I [11-13] or of HLA class II [14,15] expression with the clinical course of the disease in certain malignancies.
As reviewed in [6] and shown in figure 1, the molecular mechanisms underlying changes in HLA expression in malignant cells include structural gene abnormalities as well as defective regulation of HLA gene transcription and translation. More recently, epigenetic events associated with tumor development and progression have been shown to impair the recognition of tumor cells by components of the adaptive immune system [16-20]. In this regard, epigenetic alterations play a critical role in modifying HLA antigen [6,16,21,22], APM component [6,18,20], co-stimulatory molecule [16,21,23] and TA [24-26] expression in malignant cells. This finding is not surprising since, compared to normal cells, the constitutive patterns of DNA methylation in almost all solid and hematopoietic human malignancies demonstrate global hypomethylation with concomitant, localized, hypermethylation [17,19]. It is noteworthy that, unlike genetic alterations, epigenetic modifications can, in some cases, be reversed with pharmacologic agents that induce DNA hypomethylation or inhibit histone deacetylation. As a result functional HLA expression and recognition of malignant cells by components of the adaptive immune system can be increased [16-20]. Therefore studies focused at defining the molecular mechanisms underlying epigenetic modifications of HLA expression may provide us with improved strategies for the treatment of patients with malignant disease.
Figure 1. Molecular mechanisms underlying the abnormal HLA class I antigen phenotypes identified in malignant cells.
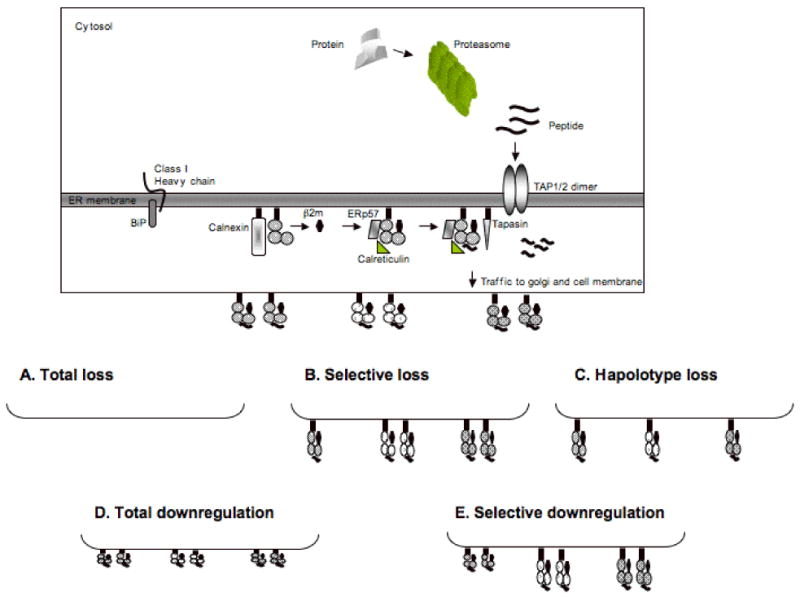
Intracellular protein antigens, which are mostly endogenous, are marked for ubiquitination within the cytosol and subsequently degraded into peptides by proteasomal cleavage. Once generated, peptides are transported into the endoplasmic reticulum through the dimeric transporter associated with antigen processing, TAP1 and TAP2. Nascent, HLA class I heavy chains are synthesized in the ER and associate with the chaperone immunoglobulin heavy chain binding protein (BiP), a universal ER chaperone involved in the translation and insertion of proteins into the ER. Following insertion into the ER, the HLA class I heavy chain associates with antigen processing machinery components (APM), i.e., calnexin, ERp57, calreticulin and tapasin. The trimeric HLA class I‐b2m‐peptide complex is then transported to the plasma membrane where it plays a major role in the interactions between target cells and T cells. (A) Total HLA class I (HLA-A, -B and -C) loss is caused by loss of β2m expression and/or function; (B) Selective HLA class I allospecificity loss is caused by loss of the gene(s) which encode(s) the lost HLA class I allele(s) or by mutations which inhibit their transcription or translation; (C) Total loss of one HLA class I haplotype is caused by total or partial loss of one copy of chromosome 6 which encodes the genes for HLA class I heavy chains; (D) Total HLA class I downregulation is caused by loss or downregulation of APM components; (E) Selective HLA class I locus downregulation may be caused by locus-specific defects in HLA class I gene transcription.
In this review, we will first succinctly review the types of epigenetic modifications that may occur in normal and malignant cells. Interested readers are referred to more in-depth reviews on the topic [17,19]. Second, we will summarize the available information about the frequency of classical and non-classical HLA class I and HLA class II antigen expression in malignant lesions. Third, we will discuss the role of epigenetic modifications underlying changes in HLA antigen expression by malignant cells. It is noteworthy that epigenetic alterations also play a significant role in the deregulation of TA expression and thereby functional HLA class I as well as class II – peptide complex expression [19]. The latter changes are also likely to play a significant role in the escape of tumor cells from immune recognition and destruction. Interested readers are referred to more in-depth reviews on the topic [24-26]. Fourth, we will address the functional relevance and potential clinical significance of abnormalities in HLA expression by malignant cells. Lastly, we will discuss strategies to overcome epigenetic modifications underlying changes in HLA expression in malignant cells.
II. Epigenetic modifications in normal and malignant cells
Epigenetics refers to changes in gene expression that cannot be accounted for by changes in primary DNA coding sequence [17,19]. As summarized in figure 2, epigenetic regulation of gene expression includes both transcriptional [17,19] and post-transcriptional [17,19] mechanisms. These mechanisms, which have been mostly characterized in cell lines, have been documented also in normal tissues as well as in malignant lesions.
Figure 2. Epigenetic modulations underlying abnormal gene expression identified in malignant cells.
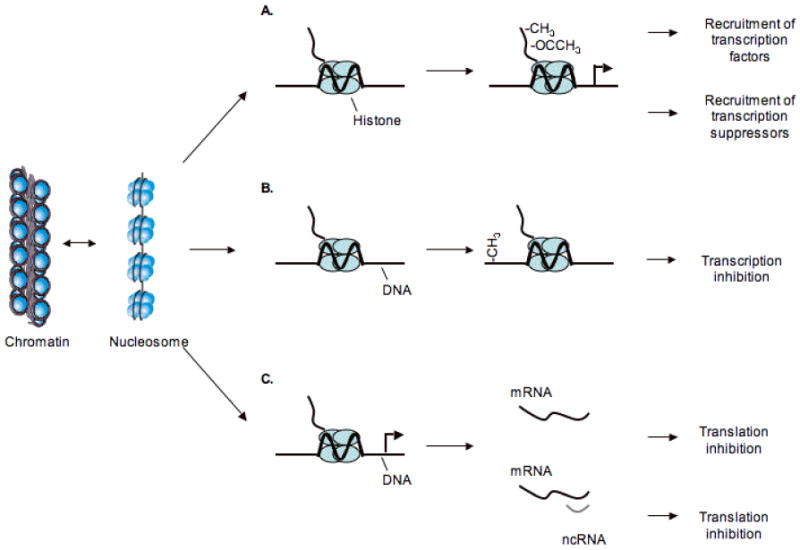
In the nucleus the DNA double helix is wrapped around histone octamers to form nucleosomes, the basic structure of DNA inside the nucleus. (A) Modification of histones through methylation (-CH3) or acetlylation (-OCCH3) affects gene expression by targeting various protein complexes to DNA, resulting either in an open chromatin structure ready for expression or in a closed chromatin configuration that is impermeable to transcription factors and associated with gene silencing. (B) DNA methylation refers to the enzymatic addition of a methyl group to the 5 position of cytosine incorporated into DNA. In mammals, methylation is largely limited to cytosines that are part of the symmetrical dinucleotide CpG. A switch from unmethylated to methylated CpG island results in permanent loss of gene expression. (C) RNA interference (RNAi) is a post-transcriptional mechanism whereby gene expression is suppressed by RNA degradation, triggered by short stretches of complementary RNA. This process is a prominent mechanism of epigenetic regulation in plants and other organisms, where it plays roles varying from genome defense to chromosomal structure.
One of the primary epigenetic modifications of the human genome is methylation of cytosine residues within the context of the CpG dinucleotides [17,19]. Generalized DNA hypomethylation, gene specific DNA hypomethylation, i.e., glioma pathogenesis-related 1 gene, nuclear factor of activated T cell 1 (NFATC1) and N-acetyltransferase 1, as well as tumor-suppressor gene hypermethylation, i.e., RAS, p16(INK4a), fragile histidine triad (FHIT), hMLH1, hMSH2, may be involved in the development and progression of malignant lesions [17,19,27-29]. In this regard, malignant cells display global loss of 5-methylcytosine content (affecting primarily repetitive elements and pericentromeric DNA), which has been linked to the generation of chromosomal instability [19,30]. On the other hand, malignant cells also demonstrate concomitant gains in methylation of promoter-associated CpG islands with silencing of the associated genes, including tumor-suppressor genes, such as p16INK4a, BRCA1 or hMLH [19,31]. The profiles of CpG island hypermethylation of tumor-suppressor genes, which are maintained in established human cancer cell lines, have been shown to vary according to the tumor type [19]. Aberrant hypermethylation of CpG islands, associated with transcriptional silencing of selected genes, can affect multiple cellular functions, including cell growth and differentiation, cell cycle control, and DNA repair, as well as angiogenesis, cell adhesion, invasion and apoptosis [17,19,27-31]. It should be noted that the enzymes that catalyze DNA methylation, i.e., DNA methyltransferases (DNMTs), also interact directly with histone de-acetylases (HDAC) [27] and can be recruited by gene-specific transcriptional repressors to promoters to silence transcription [28].
Histones also play a large part in the control of gene expression and chromatin structure and closely interact with the DNA methylation machinery. Epigenetic modifications of histones, either through targeting by acetylation [31-33] or DNA methylation [17,19,34-37], participate in tumor-suppressor gene silencing, either in conjunction or without CpG island hypermethylation. As an example, hypoacetylation of histones, which is frequently associated with CpG island hypermethylation, results in a compact structure of chromatin and represses gene transcription [17,19]. Furthermore, recent studies have demonstrated that human tumors demonstrate an overall loss of monoacetylation of lysine 16 and trimethylation of lysine 20 in the tail of histone H4 [38]. These unique histone modifications have been considered as almost universal epigenetic markers of malignant transformation [38]. Furthermore, modifications can occur on both histone tails and on core histone residues and changes on one histone can require a modification on another histone. These histone modifications affect the interactions between individual histone proteins and ultimately alter the structure of chromatin and thereby gene transcription. It is noteworthy that all the epigenetic alterations currently recognized on histones are reversible and separate sets of unique enzymes responsible for reversing these alterations, i.e., acetylases, de- acetylases, methylases, and de-methylases, have been identified. It is the balance between the opposing activities of enzymes that add and remove each of the epigenetic marks that determine local changes in chromatin structure at the gene level and gene expression patterns.
An additional type of epigenetic modification that occurs in malignant cells involves the interaction of small, 20-22 nucleotide, non-coding RNA transcripts (ncRNA) with target messenger RNAs (mRNAs) [17,19,39,40]. This interaction results in the post-transcriptional regulation of mRNA translation and thereby protein expression [17,19]. Moreover, targeting of nuclear RNA transcriptional complexes by ncRNA promotes histone methylation and may mediate DNA methylation in addition to chromatin silencing [41-43]. ncRNA, which are typically excised from larger RNA precursors, are involved in crucial biological processes such as development, differentiation, proliferation and apoptosis [17,19,39,40]. Alterations in the level of ncRNA expression have been detected in many types of human tumors [17,19,44,45]. ncRNAs have been proposed to contribute to tumor development and progression because they can function as tumor promoters. In this regard, in human tumors, specific ncRNAs are preferentially located at fragile sites that are common break-point regions involved in malignant transformation [17,19,44,45]. Moreover, chronic B cell leukemia has been shown to have a distinct signature of ncRNAs suggesting that ncRNA may be involved in the pathogenesis of this malignant disease [44]. The activity of ncRNA can be influenced by chromosomal rearrangements, genomic amplifications or deletions as well as mutations in coding DNA. At present the mechanism(s) that underlies changes in the function of ncRNAs in malignant cells appears to be related to aberrant gene expression, characterized by abnormal ncRNA expression compared with the corresponding normal tissues. To the best of our knowledge, this mechanism has not been investigated yet as a potential cause of changes in HLA expression in malignant cells. However the role of ncRNA in the modulation of HLA expression is suggested by the recently described modulation of the NK cell activating ligand MICA in malignant cells [46].
III. HLA expression in malignant lesions
Beginning in the 80's and continuing today, a large number of malignant lesions have been tested with classical HLA class I and HLA class II antigen-specific monoclonal antibodies (mAb) [6,14,15]. More recently, these studies have been extended to the analysis of the expression of the non-classical HLA class I antigens, such as HLA-G [11-13,47-56], since evidence accumulated during the last few years has convincingly shown that these molecules may provide tumor cells with escape mechanisms [11-13,47-56]. However the available information regarding non-classical HLA class I expression is still limited, since the field is in an early stage. Furthermore progress in this area is hindered by the lack and/or limited availability of non-classical HLA class I antigen-specific mAb which are suitable for immunohistochemical (IHC) studies.
As reviewed elsewhere [6], with the exception of liver carcinoma, leukemia and lymphoma, abnormalities in classical HLA class I expression have been described in all the types of tumors analyzed. The frequency of HLA class I loss and/or downregulation has been found to range from 16% to 80% of the lesions tested in the tumors for which more than 100 lesions have been analyzed (Fig. 3A). The highest frequency has been found in bladder, breast and prostate carcinoma, and the lowest in renal cell carcinoma (RCC) and in melanoma.
Figure 3. Frequency of classical HLA class I downregulation and of non-classical HLA class I antigen and HLA class II antigen expression in malignant lesions of different embryological origin.
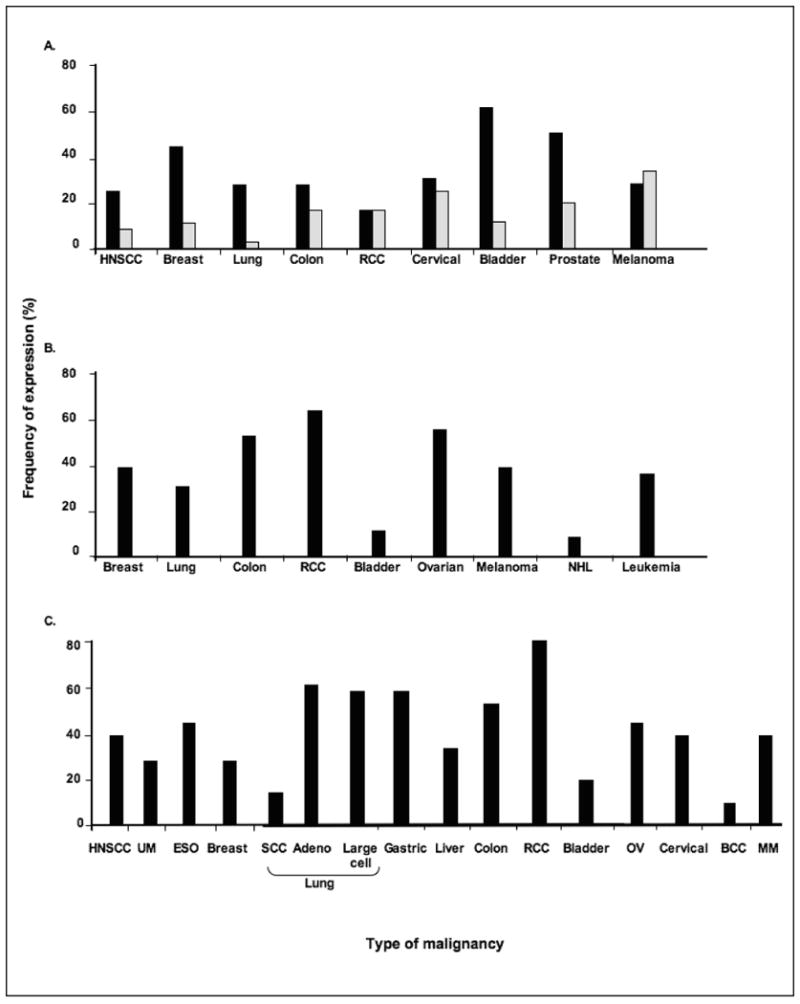
The most common types of solid tumors for which more than 50 primary lesions have been analyzed for (A) classical, (B) non-classical HLA class I and (C) HLA class II expression are shown. Data has been adapted from [6,11-15,47-62]. BCC, cutaneous basal cell carcinoma, ESO, esophageal carcinoma; HNSCC, head and neck squamous cell carcinoma; MM, malignant melanoma; RCC, renal cell carcinoma; SCC, squamous cell carcinoma; UM, uveal melanoma. (■) Indicates total HLA class I antigen loss and/or downregulation; ( ) indicates selective HLA class I allospecificity loss.
) indicates selective HLA class I allospecificity loss.
Among the non-classical HLA class I antigens, HLA-G expression has been studied the most extensively as more than 150 surgically removed lesions have been analyzed for HLA-G expression utilizing binding assays with HLA-G-specific antibodies, immunochemical assays, RT-PCR and IHC assays [11-13,47-56]. Although the results in the literature conflict, there is a general agreement that malignant transformation of cells may be associated with the appearance of HLA-G. In this regard, HLA-G expression has been convincingly demonstrated in glioma, retinoblastoma, carcinomas of the breast, mesothelia, colon, kidney, bladder, endometrium, ovary and cervix as well as in B cell chronic lymphocytic lymphoma (CLL) and in non-Hodgkin B and T cell lymphoma (Fig. 3B) [11-13,47-56].
In contrast to HLA-G, less information is available regarding HLA-E and HLA-F expression by malignant cells [6,55,57]. HLA-E expression has been investigated in a panel of 37 cell lines derived from different types of tumors as well as a limited number of surgically removed malignant lesions [6,55,57]. All the 37 cell lines express HLA-E mRNA, while only 5 (13%) of them express HLA-E proteins on the cell surface [6,55,57]. Three of the latter 5 cell lines had been derived from leukemia. In surgically removed malignant lesions HLA-E expression is increased in glioblastoma, carcinomas of colon and ovary, lymphoma and melanoma [6,55,57]. Analysis of over 30 cell lines derived from transformed normal cells and malignant lesions has demonstrated that HLA-F is primarily localized in the cytoplasm of cells [57]. Only cell lines derived from EBV-transformed lymphoblastoid, monocytic, glioblastoma, liver carcinoma and transitional cell bladder cells have been found to express HLA-F on the cell surface [57]. Recently, we have found that HLA-E and HLA-F are expressed in 4 and 2, respectively, of the 8 breast carcinoma cell lines tested [6]. In 2 cell lines both HLA-E and HLA-F are expressed. Upon IFN-γ stimulation, both HLA-E and HLA-F expression was induced or enhanced on 7 of the 8 cell lines analyzed.
To date, a large number of malignant lesions have been tested with HLA class II antigen-specific mAb in IHC reactions [14,15]. These studies have shown that HLA class II antigens are often up-regulated on malignant cells [14,15]. The most common types of solid tumors for which more than 100 surgically removed primary lesions have been analyzed include carcinomas of the head and neck, esophagus, lung (i.e., adeno, large cell and squamous), breast, colon, liver, kidney, bladder, ovary, and cervix, as well as cutaneous and uveal melanoma. The frequency of HLA class II expression has been found to range from 0% to 100% of the various types of tumors stained with mAb recognizing monomorphic determinants shared by HLA-DR,-DQ and -DP antigens (Fig. 3C). RCC demonstrates the highest frequency and cutaneous basal cell, lung (i.e., squamous) and bladder carcinoma demonstrate the lowest frequency [14,15]. It is noteworthy that in some tumors such as breast [15], colon [58-61], and cervical [62] carcinoma, HLA class II expression is not restricted to cells that have undergone malignant transformation.
Only limited information is available about selective losses of classical HLA class I and HLA class II. Differential HLA-A and –B expression has been observed in head and neck squamous cell carcinoma (HNSCC), colorectal carcinoma and melanoma, and differential HLA-DR, -DQ and -DP expression has been observed in malignant cells derived from carcinoma of breast [15], liver [60], and colon [58-61] as well as leukemic cells [64,65] and primary and metastatic cutaneous melanoma lesions [14,63]. The analysis of HLA allospecificity expression has only been performed for HLA class I, most likely because of the need of this information to interpret the results of T cell based immunotherapy. As observed for total HLA class I defects, the frequency of selective HLA class I abnormalities varies among different types of tumors; it has been found to be higher in primary cervical carcinoma, prostate carcinoma and cutaneous melanoma lesions than in primary HNSCC, breast carcinoma, lung carcinoma, RCC and colon carcinoma lesions [6].
Multiple reasons are likely to contribute to the differences in the frequency of HLA changes in various types of tumors. Some of them are technical in nature such as sensitivity of the IHC reaction used, characteristics of the mAb used in the IHC reactions and subjective evaluation of IHC staining. Furthermore, HLA expression on endothelial and/or lymphoid cells present in tissue sections, which is used in the majority of cases as a standard for evaluating the degree of expression of these molecules by malignant cells, does not represent the most appropriate control, when tumor cells being evaluated are of a different lineage.
Among the biologic variables, which generally have not been taken into account in the design and analysis of previous studies, but are likely to influence the expression of HLA antigens are heterogeneity of the patient populations investigated in terms of clinical stage, previous therapy, and length of the follow-up periods. The complexity of malignant disease, which is not a single disease entity, can also play a role, since the frequency of HLA expression varies among histologic subtypes of breast, lung, colon and ovarian carcinoma as well as uveal and cutaneous melanoma [6,14,15]. These results emphasize the need to stratify tumors according to their histotype when analyzing HLA expression. Moreover, the assessment of the degree of downregulation of HLA antigens on malignant cells suffers from the scant information about their level of expression in normal tissues. Consequently, it is difficult to determine what constitutes a normal or a pathological expression level.
The time length between onset of tumor and diagnosis, along with the level of exposure to T cell selective pressure [4], is also likely to play a role in the variable frequency of HLA expression amongst the varies tumors. In this regard, the lower frequency of classical HLA class I defects in leukemic and lymphoma cells may reflect the time interval between the onset of the disease and the diagnosis. This time is likely to be shorter than that in most solid tumors and thereby results in a decreased level of exposure to T cell selective pressure. Moreover, because of their hematogeneous route of primary tumor and metastasis formation, leukemic and lymphoma cells are targeted by NK cells which are often distributed in the peripheral blood as well as in secondary and tertiary lymphoid sites (i.e., lung, liver, spleen, bone marrow and lymph nodes) [67]. The role of NK cells in controlling leukemic cells is supported by the association between a decrease in NK cell function and progression of chronic myelogenous leukemia (CML) from chronic phase to blast crisis [68-70]. Therefore, the high HLA class I expression found in leukemia and lymphoma cells may result from the outgrowth of tumor cells with high HLA class I expression because of their reduced susceptibility to NK cell-mediated lysis. This possibility is supported by the selective HLA-A and HLA-Bw6 allospecificity downregulation which has been found in leukemic cells; this phenotype does not increase the susceptibility of target cells to NK cell-mediated lysis [71].
In the case of liver carcinoma, normal hepatocytes, which do not express or express very low levels of the transporter associated with antigen processing (TAP), a component of the HLA class I APM, and of classical HLA class I, acquire the expression of these antigens during malignant transformation [6]. The lack of defects in classical HLA class I expression identified in liver carcinoma cells may reflect the type of immune selective pressure imposed on tumor cell populations. In this regard, many NK cells reside in the liver [67]. The decrease in their activity in patients with chronic liver diseases and in those with liver carcinoma may reduce the selective growth of malignant hepatocytes with HLA class I defects [72,73]. Furthermore, expression of the NK cell activating ligands MICA/B by liver carcinoma cells has been suggested to play an important role in the susceptibility of liver carcinoma cells to NK cell-mediated cytolysis [74]. Therefore, the acquisition of classical HLA class I expression by liver cells during malignant transformation might reflect the selection of cells which are resistant to NK cell-mediated lysis.
IV. Epigenetic mechanisms underlying altered HLA expression in malignant cells
During the last few years, epigenetic alterations have been shown to play a role in HLA changes associated with malignant transformation of cells. It is noteworthy that the epigenetic alterations, which will be reviewed in the next few sections, have mostly been documented in cell lines derived from malignant lesions. Therefore the in vivo relevance of these data remains to be assessed.
IV.a. Classical HLA class I antigens
The notion that epigenetic alterations modulate HLA changes in malignant cells was first suggested by studies showing that HLA expression can be recovered by treatment with DNA methylation and HDAC inhibitors [6,16,21,22]. More recently, hypermethylation of the IFN-regulatory factor 1 (IRF-1) gene has been found to bring about inhibition of IFN-γ-mediated HLA class I expression in two melanoma cell lines [75]. In addition, hypermethylation of the HLA-A, B and C gene promoter regions and/or altered chromatin structure of the HLA class I heavy chain gene promoters have been implicated as a major mechanism for transcriptional inactivation of HLA class I genes in esophageal squamous cell carcinoma lesions [76] and for total HLA class I downregulation in cutaneous primary and metastatic melanoma lesions [77,78]. It is noteworthy that epigenetic alterations do not have to be restricted to genes encoding HLA class I heavy chains to affect the expression of these antigens. One mechanism which causes lack of HLA class I cell surface expression is represented by post-transcriptional regulation of β2m, a critical component of the HLA class I-β2m-peptide complex, which has been described in a drug resistant breast carcinoma cell line [79]. Moreover, changes in HLA class I antigen expression may also reflect alterations in transcriptional regulation of the APM components. The latter, play a critical role in the assembly and presentation of functional HLA class I antigens [6,18,20]. In this regard, with the exception of some rare examples of TAP1, tapasin and LMP subunit mutations found in some small cell lung carcinoma, cervical carcinoma, neuroblastoma and cutaneous melanoma cell lines, structural alterations in APM component genes appear to be a rare event [80,81]. Based on the low frequency of sequence abnormalities, it has been suggested that APM components are mainly regulated at the epigenetic, transcriptional and/or posttranscriptional level in human tumors. The latter is supported by the markedly heterogeneous APM component promoter activities detected in a number of human tumor cell lines of different histotype suggesting a transcriptional regulation of APM component expression [80]. Methylation of the taspasin and/or TAP2 promoter has been demonstrated in melanoma and RCC cell lines and treatment with the DNA demethylating agent 5-aza-2′-deoxycytidine (5-AC) results in the enhancement and/or reconstitution of not only tapasin and TAP2 but also TAP1 transcription and translation [80]. In addition, treatment of some esophageal squamous cell carcinoma, colon carcinoma, RCC and melanoma cell lines with the DNA demethylating agent 5-aza-2′-deoxycytidine and/or HDAC results in the induction of APM component transcription and translation [80,81]. Along the same lines, hypoacetylation of the H3 histone leads to deficient TAP-1 expression, a critical APM component, and ultimately reduced HLA class I expression in a lung carcinoma cell line [20]. Similar studies have also demonstrated that specific APM components, including LMP7, TAP1, TAP2, and tapasin, can be epigenetically regulated in certain tumors [18]. It is noteworthy that HDAC inhibitors also enhance the expression of classical MHC class I and class II as well as the co-stimulatory molecule CD40 in mouse tumor cells, suggesting that epigenetic mechanisms play a role across species [16]. Whether these findings reflect direct epigenetic control of APM component expression and/or indirect control through yet to be defined transcriptions factors remains to be determined.
IV.b. Non-classical HLA class I antigens
In attempts to resolve the issue of the mentioned different results about HLA-G expression between in vivo and in vitro conditions [6], the mechanism(s) by which HLA-G expression is modulated in malignant cells have been investigated. A number of studies have demonstrated that stress proteins as well as cytokines such as GM-CSF, IFNs, IL-10 and LIF up-regulate HLA-G protein expression by tumor cells [11,12,48-51]. However, cytokines have no effect on the induction of HLA-G gene transcription in tumor cells in which the HLA-G gene is repressed. In this context, epigenetic mechanisms, such as DNA methylation and histone hypoacetylation have been found to underlie repression of HLA-G gene expression [11,12,48-51]. Specifically, in all tumor cell lines tested the HDAC inhibitor trichostatin A (TSA) and the DNA demethylating agent 5-AC can induce HLA-G gene transcription, and in some cases translation, i.e., increased intracellular and cell surface HLA G expression. These results are not limited to the human systems but have also been described in mouse cells [11,12,48-51]. It should be noted that only a limited number of tumor cell lines have been examined thus far for HLA-G expression after exposure to TSA and/or 5-AC. Thus, whether HLA-G induction by 5-AC represents a general rule in tumor cells remains to be determined. Furthermore, it is not known whether demethylation of the HLA-G promoter is the cause for HLA-G induction in tumor cells in vitro because the methylation status of the HLA-G promoter before and after 5-AC treatment has not been examined and 5-AC can modulate the expression of genes utilizing mechanisms different from demethylation. Lastly, it is not known whether the role of in vitro promoter hypomethylation in HLA-G induction in tumor cells has an in vivo counterpart. If the in vitro findings are not paralleled by in vivo data, it is tempting to speculate that the HLA-G promoter becomes methylated in tumor cells following adaptation to tissue culture. This possibility is supported by the methylation of multiple gene promoters, including MyoD1, α-globin, Major Histocompatabilty Class (MHC) I, and triophosphate isomerase when cells are adapted to tissue culture [18]. It is worth noting that 5-AC causes DNA damage, which may act as a stress factor to induce HLA-G expression in tumor cells. Clearly, additional experiments are required to determine the role of HLA-G promoter hypomethylation in the induction of HLA-G expression by tumor cells.
IV.c. HLA class II antigens
The genes encoding HLA class II antigens, i.e., HLA-DR, -DQ, and -DP, as well as those encoding the accessory molecules involved in HLA class II presentation, i.e., the invariant chain (Ii) and HLA-DM, are coordinately regulated and their expression, at the transcriptional level, is primarily controlled by the Class II transactivator protein (CIITA) [61,82,83]. Four promoters regulate different isoforms of CIITA expression in a cell-specific manner [61,82,83]. Constitutive CIITA expression in dendritic cells (DCs) is controlled by promoter I (D-CIITA), while CIITA expression in B-lymphocytes is driven by promoter III (B-CIITA) [82]. Not surprisingly, epigenetic mechanisms have also been found to underlie defects in HLA class II expression in malignant cells. Both hypermethylation of the CIITA promoter IV [84,85] and/or modification of chromatin structure by histone deacetylation [84,85] may result in defective CIITA expression in tumor cells, causing loss of IFN-γ–inducible HLA-DR expression. It is thought that CpG dinucleotide methylation of CIITA promoter IV DNA as well as histone deacetylation severely impair recruitment of transcription factors such as IRF-1, Stat-1 and USF-1 to CIITA-PIV, thereby reducing CIITA transcription [86]. The above mechanism has been documented in cell lines derived from carcinoma of breast [15], stomach [87,88], colon [22,88], cervix [85], cutaneous epithelia [89] as well as from T cell leukemia [90], neuroblastoma [91], teratocarcinoma [18], choriocarcinoma [18], and uveal and cutaneous melanoma [14]. More recent studies have suggested that Blimp-1, a zinc-finger DNA-binding protein, recruits a co-repressor complex containing HDAC to the CIITA promoter and this may be responsible for the failure of the plasma cells and related tumors to express HLA class II [92]. HDAC inhibitors can induce CIITA expression and enhance HLA class II expression on plasma cell tumors [93]. It should be noted that in several human malignant cell lines and in normal mouse kidney epithelial cell cultures, HDAC inhibitors can induce HLA class II expression through an apparent CIITA-independent pathway [22].
V. Functional relevance of changes in classical and non-classical HLA class I and in HLA class II expression in malignant cells
Convincing evidence indicates that to evade the host's immune response tumor cells may utilize multiple escape mechanisms. In this regard, tumor cells may exploit, in addition to structural mutations, epigenetic repression of genes encoding HLA antigens and accessory molecules to evade immune recognition and destruction (figure 4). In this section we discuss how changes in HLA expression in tumor cells may provide them with escape mechanisms.
Figure 4. Molecular mechanisms underlying the functional properties of HLA expression by malignant cells.
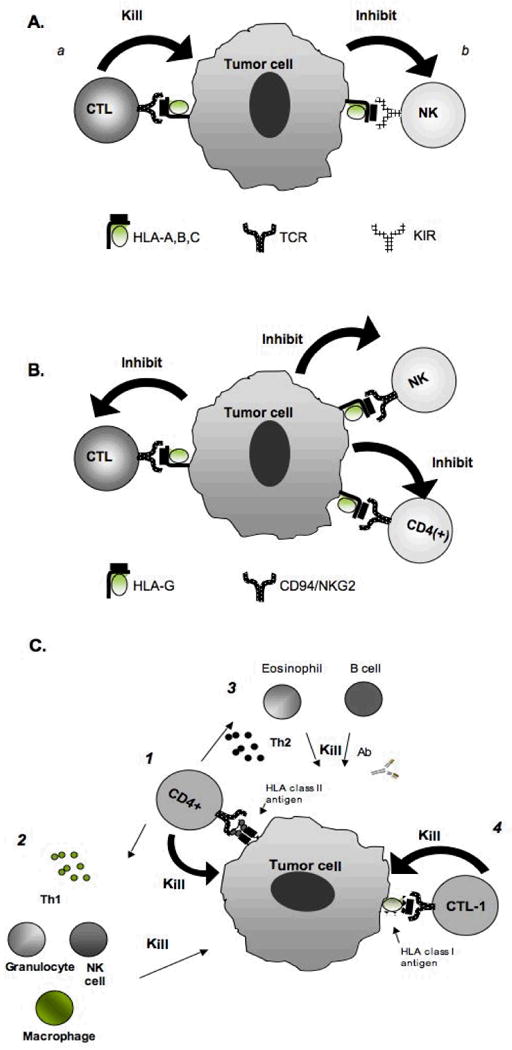
(A) Once the classical HLA class I-β2m-peptide complex is transported to the plasma membrane it plays a major role in the interactions between target cells and (a) activation of peptide-specific CTL through TCR; and (b) inhibition of T cell subpopulations through inhibitory receptors KIR. (B) In contrast to classical HLA class I, the non-classical HLA class I, HLA-G, inhibits CTL, CD4(+) T cells and NK cells through its interaction with the NK cells receptor CD94/NKG2. (C) HLA class II expression by tumor cells may be potentially beneficial to TA-specific immune responses through their interaction with CD4(+) T cells, resulting in the activation of (1) CD4(+) T cell-mediated killing, (2) macrophage through release of Th1 cytokines; (2) B cells and eosinophils through release of Th2 cytokines, and (4) CTL through release of Th1 cytokines.
V.a. Classical HLA class I antigens
The role of HLA class I in the recognition and destruction of tumor cells by HLA class I-restricted, TA-specific cytotoxic T lymphocytes (CTL) implies that defects in HLA class I expression in malignant lesions may have a negative impact on the clinical course of malignant diseases if the latter is influenced by a TA-specific CTL-mediated immune response. Furthermore, defects in HLA class I expression in malignant lesions are expected to counteract the potential beneficial effects of T cell-based immunotherapy, since they may provide tumor cells with a mechanism to evade CTL recognition. It is likely that the interplay of genetic and epigenetic modifications in tumor cells influences not only developmental processes of carcinogenesis but also selective pressures involved in immune escape. The potential involvement of immune selective pressure in these events is suggested by several lines of clinical and experimental evidence, all of which have in common the possibility that tumor cells with genetic and/or epigenetic defects in HLA class I may be out-selected by T cell-mediated selective pressure present in the tumor microenvironment [4,6,94,95]. This notion is supported by the association of HLA class I downregulation with a reduction in disease free interval and survival in HNSCC, breast carcinoma, small cell lung carcinoma, bladder carcinoma, cervical carcinoma and cutaneous melanoma [6]. Furthermore, in prostate carcinoma, when the degree of tumor differentiation is taken into account, the disease free interval and survival of patients with prostate carcinoma, whose lesions demonstrate HLA class I abnormalities, is shorter than those of patients whose lesions have a similar degree of tumor differentiation and do not demonstrate detectable HLA class I abnormalities [6]. These clinical findings have been suggested to reflect resistance of malignant cells to lysis by CTL through lack of TA derived peptides presented properly in the context of a HLA class I allospecificity. The lack of HLA class I antigen-TA peptide complex expression may also account for the association of TAP1, a critical APM component, downregulation with tumor grading, tumor staging and reduction in patients' survival in breast carcinoma, SCLC, cervical cancer and cutaneous melanoma [6]. In this regard, alterations in TAP1 expression lead to defective peptide loading of HLA class I antigens and their ultimate expression on the cell surface. Interestingly, TAP1 expression has been suggested to represent an independent prognostic marker for patients with cutaneous melanoma, since its downregulation in primary lesions has been found to correlate with metastasis development and prognosis better than the traditional prognostic marker, i.e. tumor thickness [6]. If these findings are confirmed, the analysis of TAP1 expression in primary cutaneous melanoma lesions may represent a useful molecular marker in order to evaluate melanoma patients' prognosis. Whether this conclusion applies also to other types of malignancies remains to be determined.
V.b. Non-classical HLA class I antigens
Although there is less information regarding the role of non-classical HLA class I expression by malignant cells in immune escape, the available clinical and experimental evidence suggests, although does not prove that selective pressure is imposed on tumor cells which have spontaneously arisen in a host or have been transplanted to a host, not only by CTL but also by NK cells. Thus the association between poor prognosis and HLA class I loss in malignant lesions described in several malignant diseases [6] as well as the increased ability to metastasize of tumor cells with HLA class I [6] defects raise the possibility that NK cells cannot control tumor growth because the tumor cell population has been immunoselected for non-classical HLA class I expression and/or lack of NK cell activating ligand expression by NK cell selective pressure. A mentioned above, HLA-G expression has been documented in several different types of malignant lesions and its expression in carcinomas of the lung [13], stomach [12] and colon [11] is associated with poor prognosis. Moreover, elevated soluble HLA-G (sHLA-G) protein levels have been demonstrated in sera of patients with carcinomas derived from breast and ovarian cells as well as melanoma, glioma and lymphoproliferative disorders [51,54]. The HLA-G serum level was inversely correlated with survival in glioblastoma multiforme [54] and was directly correlated with advanced disease stage and with tumor load in melanoma [54]. These findings suggest that tumor cell expression of HLA-G expression may represent a mechanism of resistance to NK cell-mediated killing. This mechanism may parallel the manner in which HLA-G protects against NK cell-mediated rejection of fetal tissues in the placenta [96]. Moreover, HLA-G may provide tumor cells with additional resistance from immune recognition and destruction through their ability to inhibit the cytotoxic activity and/or the proliferative ability of CTL and CD4+ T helper cells, respectively, as well as to induce the generation of a new type of regulatory T cells, either CD4+ or CD8+, through transfer of membrane HLA-G from antigen presenting cells (APC) to activated T cells (“trogocytosis”) [55]. It should be noted that NK cell-mediated killing of tumor cells involves a complex interplay between classical and non-classical HLA class I as well as NK cell activating ligands. Therefore, aberrant HLA-G expression may not be required for tumor cell escape in diseases in which NK cells play a major role in controlling tumor growth, since classical HLA class I have a strong inhibitory capacity through their interaction with NK inhibitory receptors [3]. Similarly, other non-classical HLA class I molecules such as HLA-E might also contribute to NK tumor cell escape. It is well documented that HLA-E is widely transcribed in most tissues, and that this molecule is expressed when it combines with the leader peptides of other HLA class I molecules including HLA-G [52,55]. It is thought that NK cells, through interaction with the CD94/NKG2A inhibitory receptor, may utilize HLA-E, to monitor the level of classical HLA class I expression on target cells [52,55].
V.c. HLA class II antigens
Conflicting information is available among different malignant diseases as well as within in the same malignant disease regarding the functional and clinical significance of HLA class II expression in malignant lesions [6,14,15]. The results range from association of HLA class II expression with favorable or unfavorable clinical course of the disease to lack of association. The conflicting results are likely to reflect the role of the technical and biological variables we have previously mentioned to explain the different frequencies of HLA class II expression in malignant lesions. In addition we believe that the conflicting information regarding the clinical significance of HLA class II expression in malignant cells, is due, in part, that the analysis has only been restricted to HLA-DR, -DQ, and -DP expression on the cell membrane without taking into consideration other molecules that are likely to greatly affect the functional properties of HLA class II such as HLA class I, APM components and co-stimulatory molecules. All of these components are required for a cell to efficiently present HLA class II-peptide complexes to CD4(+) T cells and the importance of their coordinated expression is highlighted by several in vitro and in vivo observations. In vitro binding of CD4(+) T cells to HLA class II on tumor cells in the absence of co-stimulatory molecules can lead to an immune suppressed or anergic state of the CD4(+) T cell [97,98]. Moreover, the clinical relevance of the co-expression of HLA class II and co-stimulatory molecule as well as HLA class I is indicated by the association of HLA class II expression in cutaneous metastatic melanoma lesions with a favorable prognosis only when it is co-expressed with the co-stimulatory molecule B7.1 and with HLA class I [99]. In this regard, it should be noted that epigenetic regulation of co-stimulatory molecules has been reported in several human and murine malignant cell lines, and surface expression of CD40, and B7-1/2 co-stimulatory molecules can be induced by treatment with HDAC inhibitors [16].
The role of HLA class II APM component expression in the clinical course of the disease is underscored by at least three lines of evidence. First, the repertoire of peptides displayed by HLA class II is altered and possibly CD4(+) T cell anergy is induced when HLA class II positive malignant cells with display low basal levels of a γ-interferon (IFN)-inducible lysosomal thiol reductase (GILT), a HLA class II APM component [82]. Second, in neuroblastoma, IFN-γ induced HLA-DO expression alters the repertoire of peptides presented by HLA class II to CD4(+) T cells and can lead to CD4(+) T cell anergy [100]. Third, over-expression of Ii, is associated with poor clinical course of the disease in patients with gastric [101] and colorectal [102] carcinoma. Whether alterations in other accessory molecules occur in malignant diseases and play a role in their clinical course remains to be determined. Nevertheless the available information suggests that the analysis of HLA class II expression should be combined with that of other APM components.
In the appropriate setting malignant cells may act as effective APC for HLA class II-restricted, antigen-specific CD4(+) T cells. Normally interaction of CD4(+) T cells with HLA class II-peptide complexes and co-stimulatory molecules expressed on typical APC, leads to i) the production of a spectrum of cytokines that facilitates the growth and differentiation of CD8(+) T cells and ii) activation of APC through CD40-CD40L interactions which leads to enhanced antigen-presentation. Optimal induction of CTL benefits from the presence of activated CD4(+) T cell and APC. More recently, CD4(+) T cells have been shown to exert direct HLA class II-restricted, antigen-specific cytotoxic effects on malignant cells in vitro through Fas-dependent and -independent mechanisms, i.e., granzyme and perforins [97]. Nonetheless, there is only indirect evidence of HLA class II -restricted, TA-specific CD4(+) T cell killing of HLA class II antigen-bearing tumor cells in vitro. Whether and/or how HLA class II expression by tumor cells plays a role in the activation, maintenance, or effector phase of a TA-specific cellular immune response is not known at present.
HLA class II-restricted, TA-specific CD4(+) T cells may also exert cytotoxic effects on HLA class II bearing and non-bearing tumor cells through indirect mechanisms. In this regard, the recognition of HLA class II-TA derived peptide complexes on tumor cells by HLA class II-restricted, antigen-specific CD4(+) T cells may lead to release of cytokines that ultimately results in killing of tumor cells via activated eosinophils, macrophages and neutrophils as well as type 1 cytokines, i.e., interferons (IFNs) [97]. In the latter case, the type 1 cytokine IFN plays an important role in regulating tumor growth in vivo by both the innate and adaptive immune system as well as by inhibiting tumor bed angiogenesis [97]. In contrast to their beneficial role in TA-specific immune responses, HLA class II expressed by tumor cells may have a negative impact on the immune response through their interaction with a subpopulation of CD4(+) T cells known as T regulatory (Treg) cells [103,104]. Two groups of Treg cells have been identified, natural Treg cells (nTreg), which are believed to be selected in the thymus as an anti-self repertoire, and induced Treg cells (iTreg), which are believed to be induced in the periphery. Both play a major role in maintaining self-tolerance by regulating immune responses [103,104]. While nTreg cells require TCR ligands and co-stimulation for functional activation, iTreg cells need only TCR ligands [103,104]. Once activated, both nTreg and iTreg are powerful non-specific suppressors of TA-specific immunity by inhibiting IL-2 transcription in target CD4(+) T and NKT cells, thereby preventing expansion of effector and helper T cell populations [103,104]. HLA class II expression by malignant cells may therefore lead to Treg cell activation and possibly downregulation of an ongoing TA-specific immune response through the secretion of immunosuppressive cytokines like IL-10 or TGF-β in a contact-independent manner or through a contact-dependent manner via CTLA-4 [103,104].
VI. Immune escape and epigenetic therapeutics
The lack of effective treatment for advanced stage malignant disease has highlighted the need to develop alternative therapeutic strategies. Among them, immunotherapy has attracted much attention because of the potential role played by immunological events in the clinical course of malignant disease [105]. In view of the well-recognized immune escape mechanisms utilized by malignant cells [5] as well as their genetic instability, the disappointing clinical response rates, observed in a majority of the active-specific immunotherapy trials conducted to date [105], are likely to reflect the ability of malignant cells to escape immune recognition and destruction and/or their capacity to interfere with the differentiation, function and/or survival of immune effector cells. In the remaining part of this section we will review several potential strategies that may counteract the multiple epigenetic-based escape mechanisms utilized by tumor cells and provide us with improved strategies for the treatment of patients with malignant disease.
The available evidence we have summarized in a previous section suggests that epigenetic alterations may underlie not only changes in HLA antigen, but also APM component and co-stimulatory molecule expression in malignant cells. The latter result in the ability of malignant cells with HLA antigen defects to avoid immune recognition and destruction. At variance with structural gene mutations, epigenetic changes can generally be reversed in cells with various pharmacological agents. Therefore, pharmacological modulation of the epigenetic profile of malignant cells, such as histone acetylation/deacetylation, histone methylation and, in particular, DNA methylation, may allow the development of improved approaches toward the implementation of active-specific immunotherapy for patients with malignant disease.
Among the currently available pharmacological agents 5-AC has been shown to be a suitable agent to alter methylation profiles in malignant cells [106]. At high doses, 5-AC induces apoptotic cell death triggered by the production of DNA adducts and consequent DNA synthesis arrest, whereas at low doses it is incorporated into DNA and inhibits DNMT leading to global hypomethylation [106]. Treatment of malignant cells with 5-AC results not only in the induction of HLA class I and class II expression, each of which are required for an effective TA-specific immune response, but also in the induction of genes involved in the control of apoptosis, proliferation, DNA repair and cellular senescence, i.e. p21waf, p16 and p27 and downregulation of Cyclin A, Cyclin D, CDK4 and dephosphorylation of pRb [18,107]. The upregulation of classical MHC class I expression on malignant cells after treatment with 5-AC has been shown to enhance the recognition of tumor cells by CTL in animal models and human malignant cell lines [18,108-112].
Both 5-AC and its derivatives,i.e., Decitabine, have been introduced into the clinic with encouraging results in hematopoietic disorders such as myelodysplastic syndrome (MDS), acute myeloid leukemia (AML) and CML [113-116] as well as solid tumors including lung and esophageal cancers [117]. In each case treatment with Decitabine was shown to induce or up-regulated CIITA expression in malignant cells. Along this line, recent results of a phase I clinical study of the combined treatment with decitabine plus high-dose interleukin-2 (IL-2) in melanoma and RCC suggest that decitabine may synergistically act with immunological treatments, i.e. IL-2 therapy, and improved clinical response rates [118].
Similar to 5-AC, HDAC inhibitors may be useful in the treatment of malignant diseases. Upregulation of p21waf, p16 and p27 and downregulation of Cyclin A, Cyclin D, CDK4 and dephosphorylation of pRb, genes necessary for cell cycle arrest and growth inhibition of tumor cells, are common features of HDAC inhibitor treatment and are due to both direct and indirect mechanisms [119-121]. Moreover, HDAC inhibitor treatment of a broad spectrum of malignant cells including carcinomas of the breast, lung, bladder, ovarian, and prostate as well as leukemia, lymphoma and myelomas results in the concentration dependent induction of differentiation, growth arrest and apoptosis [18]. To date, HDAC inhibitors have shown significant anti-tumor effects in pre-clinical models and in phase I/II clinical trials for the treatment of carcinomas of the breast, lung and cervix as well as MDS, AML, CLL, glioma and melanoma [122-128]. More recently the HDAC inhibitor Zolinza™ was approved for use in the treatment of cutaneous T cell lymphoma [129].
Although the precise molecular pathways involved in the anti-tumor effects of DNA-methylation and HDAC inhibitors have not been fully determined, it is likely they can be attributed to their ability to modulate genes necessary for cell cycle arrest and growth inhibition as well as genes involved in tumor immunity. The inhibition of DNA-methylation and histone de-acetylation can enhance classical MHC class I and class II, CD40, B7-1/2, as well as ICAM-1 expression in various human and mouse tumor cell lines [16,18]. Moreover, inhibition of histone de-acetylation in malignant cells can increase the expression of various pro-apoptotic and death inducing pathways, such as Fas, DR4 and DR5 expression causing tumors to become sensitive to killing by FasL and TRAIL, respectively [130]. Alternatively, inhibition of histone de-acetylation can down modulate the anti-apoptotic molecule, cFLIP, in malignant cells rendering them more susceptible to Fas-mediated killing by CTL [122,128]. This is an important reminder that modulation of epigenetic processes in tumor cells may silence nearly as many genes as they activate and may thereby reverse specific gene induction critical to tumor growth and/or immune escape.
Despite the promising data arising from clinical trials, there are several pitfalls regarding the clinical application of demethylating agents as well as HDAC inhibitors. The inherent toxicity of nucleoside DNMT inhibitors might be caused by the formation of covalent adducts between DNA and trapped DNMTs, which may be responsible for bone marrow and other host toxicities [130-132]. Furthermore, the majority of the available drugs that inhibit DNA-methylation are not specific and result in widespread DNA hypomethylation and the production of unwanted effects such as induction of oncogene expression. However optimizing treatment schedules might decrease toxicity, e.g., giving lower doses over longer time periods, thereby exposing more cells to the drug during the S phase. It should be noted that on additional pitfall of the DNMT inhibitor decitabine is the increased expression of multi-drug resitance (MDR) 1, a gene whose expression enhances drug resistance, and uPA, a pro-metastatic gene in non-metastatic breast tumor cells [131]. Additional compounds that directly inhibit DNMTs and do not need to be incorporated in DNA such as non-nucleoside inhibitors and antisense oligodeoxynucleotides are currently being developed and may lead to lower toxicity [134,135]. In general HDAC inhibitors given systemically are generally well tolerated but accumulation of acetylated histones in normal tissues may induce some toxicity depending on the dose, route and specific drug [134,135].
VI. Conclusion
Malignant cells frequently demonstrate alterations in HLA expression which is believed to play a major role in their ability to escape immune recognition and destruction. Epigenetic modulations in gene expression are likely to play a large part in the observed alterations in HLA expression by malignant cells, since epigenetic events are involved in the silencing and/or in the downregulation of genes necessary for cell cycle arrest and growth inhibition as well as genes involved in tumor immunity. The ability of epigenetic drugs to restore the defective HLA antigen, APM component and co-stimulatory molecule expression, and the consequent increase in immune recognition of malignant cells, provides us with new therapeutic tools that may improve the clinical efficacy of active-specific immunotherapy for the treatment of malignant disease. In this regard, the combination of immunization strategies with approaches that counteract tumor cell-induced immune suppression and tumor immune escape, such as DNMT and HDAC inhibitors, may enhance the clinical efficacy of immunotherapy. Nonetheless, to date very little attention has been focused on the regulation of HLA antigens in malignant lesions following treatment with DNMT and HDAC inhibitors. Therefore, future studies should be directed at investigating the ability of epigenetic pharmacologic agents to modulate HLA expression malignant lesions.
List of Abbreviations
- 5-AC
5-aza-2′-deoxycytidine
- AML
Acute myeloid leukemia
- APC
Antigen presenting cells
- APM
Antigen processing machinery
- β2m
β2-microglobulin
- CIITA
Class II transactivator protein
- CLL
Chronic lymphocytic lymphoma
- CML
Chronic myelogenous leukemia
- CTL
Cytotoxic T lymphocyte
- DC
Dendritic cell
- DNMT
DNA methyltransferases
- FHIT
Fragile histidine triad
- GILT
γ-interferon-inducible lysosomal thiol reductase (GILT)
- HDAC
Histone de-acetylases
- HNSCC
Head and neck squamous cell carcinoma
- Ii
Invariant chain
- IFN
Interferon
- IL-2
Interleukin 2
- IRF-1
IFN-regulatory factor 1
- MDR1
Multi-drug resistance
- MDS
Myelodysplastic syndrome
- MHC
Major histocompatability class
- mRNA
Messenger RNA
- ncRNA
Non-coding RNA transcripts
- NFATC1
Nuclear factor of activated T cell 1
- NK
Natural killer
- RCC
Renal cell carcinoma
- sHLA-G
Soluble HLA-G
- TA
Tumor antigen
- Treg
T regulatory cell
- iTreg
Induced Treg
- nTreg
Natural Treg
- TSA
Trichostatin A
References
- 1.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–998. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 2.Campoli M, Ferrone S, Zea AH, Rodriguez PC, Ochoa AC. Mechanisms of tumor evasion. Cancer Treat Res. 2005;123:61–88. doi: 10.1007/0-387-27545-2_3. [DOI] [PubMed] [Google Scholar]
- 3.Chang CC, Ferrone S. NK cell activating ligands on human malignant cells: molecular and functional defects and potential clinical relevance. Semin Cancer Biol. 2006;16:383–92. doi: 10.1016/j.semcancer.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 4.Chang CC, Ferrone S. Immune selective pressure and HLA class I antigen defects in malignant lesions. Cancer Immunol Immunother. 2007;56:227–36. doi: 10.1007/s00262-006-0183-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim R, Emi M, Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology. 2007;121:1–14. doi: 10.1111/j.1365-2567.2007.02587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang CC, Campoli M, Ferrone S. Classical and nonclassical HLA class I antigen and NK cell-activating ligand changes in malignant cells: current challenges and future directions. Adv Cancer Res. 2005;93:189–234. doi: 10.1016/S0065-230X(05)93006-6. [DOI] [PubMed] [Google Scholar]
- 7.Ostrand-Rosenberg S. Animal models of tumor immunity, immunotherapy and cancer vaccines. Curr Opin Immunol. 2004;16:143–150. doi: 10.1016/j.coi.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 8.Knutson KL, Disis ML. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol Immunother. 2005;54:721–728. doi: 10.1007/s00262-004-0653-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marsman M, Jordens I, Griekspoor A, Neefjes J. Chaperoning antigen presentation by MHC class II molecules and their role in oncogenesis. Adv Cancer Res. 2005;93:129–158. doi: 10.1016/S0065-230X(05)93004-2. [DOI] [PubMed] [Google Scholar]
- 10.Moretta L, Bottino C, Pende D, Vitale M, Mingari MC, Moretta A. Human natural killer cells: molecular mechanisms controlling NK cell activation and tumor cell lysis. Immunol Lett. 2005;100:7–13. doi: 10.1016/j.imlet.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 11.Ye SR, Yang H, Li K, Dong DD, Lin XM, Yie SM. Human leukocyte antigen G expression: as a significant prognostic indicator for patients with colorectal cancer. Mod Pathol. 2007;20:375–83. doi: 10.1038/modpathol.3800751. [DOI] [PubMed] [Google Scholar]
- 12.Yie SM, Yang H, Ye SR, Li K, Dong DD, Lin XM. Expression of human leukocyte antigen G (HLA-G) correlates with poor prognosis in gastric carcinoma. Ann Surg Oncol. 2007;14:2721–9. doi: 10.1245/s10434-007-9464-y. [DOI] [PubMed] [Google Scholar]
- 13.Yie SM, Yang H, Ye SR, Li K, Dong DD, Lin XM. Expression of human leucocyte antigen G (HLA-G) is associated with prognosis in non-small cell lung cancer. Lung Cancer. 2007;58:267–74. doi: 10.1016/j.lungcan.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 14.Campoli M, Ferrone S. A fresh look at an old story: revisiting HLA class II antigen expression by melanoma cells. Expert Review of Dermatology. 2006;1:737–755. [Google Scholar]
- 15.Campoli M, Chang CC, Oldford SA, Edgecombe AD, Drover S, Ferrone S. HLA antigen changes in malignant tumors of mammary epithelial origin: molecular mechanisms and clinical implications. Breast Dis. 2004;20:105–25. doi: 10.3233/bd-2004-20112. [DOI] [PubMed] [Google Scholar]
- 16.Magner WJ, Kazim AL, Stewart C, Romano MA, Catalano G, Grande C, et al. Activation of MHC class I, II, and CD40 gene expression by histone deacetylase inhibitors. J Immunol. 2000;165:7017–7024. doi: 10.4049/jimmunol.165.12.7017. [DOI] [PubMed] [Google Scholar]
- 17.Esteller M. Epigenetics provides a new generation of oncogenes and tumour-suppressor genes. Br J Cancer. 2006;94:179–83. doi: 10.1038/sj.bjc.6602918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tomasi TB, Magner WJ, Khan AN. Epigenetic regulation of immune escape genes in cancer. Cancer Immunol Immunother. 2006;10:1159–84. doi: 10.1007/s00262-006-0164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lettini AA, Guidoboni M, Fonsatti E, Anzalone L, Cortini E, Maio M. Epigenetic remodelling of DNA in cancer. Histol Histopathol. 2007;22:1413–24. doi: 10.14670/HH-22.1413. [DOI] [PubMed] [Google Scholar]
- 20.Setiadi AF, David MD, Seipp RP, Hartikainen JA, Gopaul R, Jefferies WA. Epigenetic control of the immune escape mechanisms in malignant carcinomas. Mol Cell Biol. 2007;27:7886–7894. doi: 10.1128/MCB.01547-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khan ANH, Magner WJ, Tomasi TB. An epigenetically altered tumor cell vaccine. Cancer Immunol Immunother. 2004;53:748–754. doi: 10.1007/s00262-004-0513-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chou SD, Khan ANH, Magner WJ, Tomasi TB. Histone acetylation regulates the cell type specific CIITA promoters, MHC class II expression and antigen presentation in tumor cells. Int Immunol. 2005;17:1483–1494. doi: 10.1093/intimm/dxh326. [DOI] [PubMed] [Google Scholar]
- 23.Singh NP, Yolcu ES, Taylor DD, Gercel-Taylor C, Metzinger DS, Dreisbach SK, et al. A novel approach to cancer immunotherapy: tumor cells decorated with CD80 generate effective antitumor immunity. Cancer Res. 2003;63:4067–4073. [PubMed] [Google Scholar]
- 24.Scanlan MJ, Gure AO, Jungbluth AA, Old LJ, Chen YT. Cancer/testis antigens: an expanding family of targets for cancer immunotherapy. Immunol Rev. 2002;188:22–32. doi: 10.1034/j.1600-065x.2002.18803.x. [DOI] [PubMed] [Google Scholar]
- 25.Maio M, Coral S, Fratta E, Altomonte M, Sigalotti L. Epigenetic targets for immune intervention in human malignancies. Oncogene. 2003;22:6484–8488. doi: 10.1038/sj.onc.1206956. [DOI] [PubMed] [Google Scholar]
- 26.Germenis AE, Karanikas V. Immunoepigenetics: the unseen side of cancer immunoediting. Immunol Cell Biol. 2007;85:55–59. doi: 10.1038/sj.icb.7100006. [DOI] [PubMed] [Google Scholar]
- 27.Fuks F, Burgers WA, Godin N, Kasai M, Kouzarides T. Dnmt3a binds deacetylases and is recruited by a sequence-specific repressor to silence transcription. EMBO J. 2001;20:2536–2544. doi: 10.1093/emboj/20.10.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Di Croce L, Raker VA, Corsaro M, Fazi F, Fanelli M, Faretta M, et al. Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science. 2002;295:1079–1082. doi: 10.1126/science.1065173. [DOI] [PubMed] [Google Scholar]
- 29.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. New Engl J Med. 2003;349:2042–2054. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 30.Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143–53. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 31.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–28. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 32.deRuijten AJM, van Gennip AH, Caron HN, Kemp S, van Kuilenburg ABP. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Dave JR, Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 34.Lachner M, O'Sullivan RJ, Jenuwein T. An epigenetic road map for histone lysine methylation. J Cell Sci. 2003;116:2117–2124. doi: 10.1242/jcs.00493. [DOI] [PubMed] [Google Scholar]
- 35.Sims RJ, 3rd, Nishioka K, Reinberg D. Histone lysine methylation: a signature for chromatin function. Trends Genet. 2003;19:629–639. doi: 10.1016/j.tig.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 36.Chuikov S, Kurash JK, Wilson JR, Xiao B, Justin N, Ivanov GS, et al. Regulation of p53 activity through lysine methylation. Nature. 2004;432:353–360. doi: 10.1038/nature03117. [DOI] [PubMed] [Google Scholar]
- 37.Bedford MT, Richard S. Arginine methylation: an emerging regulator of protein function. Mol Cell. 2005;18:263–272. doi: 10.1016/j.molcel.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 38.Fraga MF, Esteller M. Towards the human cancer epigenome: a first draft of histone modifications. Cell Cycle. 2005;4:1377–81. doi: 10.4161/cc.4.10.2113. [DOI] [PubMed] [Google Scholar]
- 39.Berezikov E, Guryev V, van de Belt J, Wienholds E, Plasterk RH, Cuppen E. Phylogenetic shadowing and computational identification of human microRNA genes. Cell. 2005;120:21–24. doi: 10.1016/j.cell.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 40.Sontheimer EJ, Carthew RW. Silence from within: endogenous siRNAs and miRNAs. Cell. 2005;122:9–12. doi: 10.1016/j.cell.2005.06.030. [DOI] [PubMed] [Google Scholar]
- 41.Grewal SI, Moazed D. Heterochromatin and epigenetic control of gene expression. Science. 2003;301:798–802. doi: 10.1126/science.1086887. [DOI] [PubMed] [Google Scholar]
- 42.Kawasaki H, Taira K. Induction of DNA methylation and gene silencing by short interfering RNAs in human cells. Nature. 2004;431:211–217. doi: 10.1038/nature02889. [DOI] [PubMed] [Google Scholar]
- 43.Wassenegger M. The role of the RNAi machinery in heterochromatin formation. Cell. 2005;122:13–16. doi: 10.1016/j.cell.2005.06.034. [DOI] [PubMed] [Google Scholar]
- 44.Calin GA, Liu CG, Sevignani C, Ferracin M, Felli N, Dumitru CD, et al. MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc Natl Acad Sci USA. 2004;101:11755–11760. doi: 10.1073/pnas.0404432101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chan JA, Krichevsky AM, Kosik KS. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005;65:6029–6033. doi: 10.1158/0008-5472.CAN-05-0137. [DOI] [PubMed] [Google Scholar]
- 46.Friese MA, Wischhusen J, Wick W, Weiler M, Eisele G, Steinle A, et al. RNA interference targeting transforming growth factor-beta enhances NKG2D-mediated antiglioma immune response, inhibits glioma cell migration and invasiveness, and abrogates tumorigenicity in vivo. Cancer Res. 2004;64:7596–603. doi: 10.1158/0008-5472.CAN-04-1627. [DOI] [PubMed] [Google Scholar]
- 47.Chang CC, Ferrone S. HLA-G in melanoma: can the current controversies be solved? Semin Cancer Biol. 2003;13:361–9. doi: 10.1016/s1044-579x(03)00027-0. [DOI] [PubMed] [Google Scholar]
- 48.Chang CC, Murphy SP, Ferrone S. Differential in vivo and in vitro HLA-G expression in melanoma cells: potential mechanisms. Hum Immunol. 2003;64:1057–63. doi: 10.1016/j.humimm.2003.08.357. [DOI] [PubMed] [Google Scholar]
- 49.Lin A, Yan WH, Xu HH, Gan MF, Cai JF, Zhu M, et al. HLA-G expression in human ovarian carcinoma counteracts NK cell function. Ann Oncol. 2007;18:1804–1809. doi: 10.1093/annonc/mdm356. [DOI] [PubMed] [Google Scholar]
- 50.Morandi F, Levreri I, Bocca P, Galleni B, Raffaghello L, Ferrone S, et al. Human neuroblastoma cells trigger an immunosuppressive program in monocytes by stimulating soluble HLA-G release. Cancer Res. 2007;67:6433–41. doi: 10.1158/0008-5472.CAN-06-4588. [DOI] [PubMed] [Google Scholar]
- 51.Pistoia V, Morandi F, Wang X, Ferrone S. Soluble HLA-G: Are they clinically relevant? Semin Cancer Biol. 2007;17:469–479. doi: 10.1016/j.semcancer.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rouas-Freiss N, Moreau P, Menier C, Lemaoult J, Carosella ED. Expression of tolerogenic HLA-G molecules in cancer prevents antitumor responses. Semin Cancer Biol. 2007;17:413–421. doi: 10.1016/j.semcancer.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 53.Sebti Y, Le Maux A, Gros F, De Guibert S, Pangault C, Rouas-Freiss N, et al. Expression of functional soluble human leucocyte antigen-G molecules in lymphoproliferative disorders. Br J Haematol. 2007;138:202–12. doi: 10.1111/j.1365-2141.2007.06647.x. [DOI] [PubMed] [Google Scholar]
- 54.Sheu JJ, Shih IM. Clinical and biological significance of HLA-G expression in ovarian cancer. Semin Cancer Biol. 2007;17:436–443. doi: 10.1016/j.semcancer.2007.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wischhusen J, Waschbisch A, Wiendl H. Immune-refractory cancers and their little helpers-an extended role for immunetolerogenic MHC molecules HLA-G and HLA-E? Semin Cancer Biol. 2007;17:459–468. doi: 10.1016/j.semcancer.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 56.Duncker K, Schlaf G, Bukur J, Altermann W, Seliger B, Handke D. Expression and regulation of non-classical HLA-G in renal cell carcinoma. Tissue Antigens. 2007 doi: 10.1111/j.1399-0039.2008.01090.x. Submitted. [DOI] [PubMed] [Google Scholar]
- 57.Lee N, Geraghty DE. HLA-F surface expression on B cell and monocyte cell lines is partially independent from tapasin and completely independent from TAP. J Immunol. 2003;171:5264–5271. doi: 10.4049/jimmunol.171.10.5264. [DOI] [PubMed] [Google Scholar]
- 58.Cabrera T, Ruiz-Cabello F, Garrido F. Biological implications of HLA-DR expression in tumours. Scand J Immunol. 1995;41:398–406. doi: 10.1111/j.1365-3083.1995.tb03584.x. [DOI] [PubMed] [Google Scholar]
- 59.Seliger B, Maeurer MJ, Ferrone S. Antigen-processing machinery breakdown and tumor growth. Immunol Today. 2000;21:455–464. doi: 10.1016/s0167-5699(00)01692-3. [DOI] [PubMed] [Google Scholar]
- 60.Altomonte M, Fonsatti E, Visintin A, Maio M. Targeted therapy of solid malignancies via HLA class II antigens: a new biotherapeutic approach? Oncogene. 2003;22:6564–6569. doi: 10.1038/sj.onc.1206960. [DOI] [PubMed] [Google Scholar]
- 61.Marsman M, Jordens I, Griekspoor A, Neefjes J. Chaperoning antigen presentation by MHC class II molecules and their role in oncogenesis. Adv Cancer Res. 2005;93:129–158. doi: 10.1016/S0065-230X(05)93004-2. [DOI] [PubMed] [Google Scholar]
- 62.Chil A, Sikorski M, Bobek M, Jakiel G, Marcinkiewicz J. Alterations in the expression of selected MHC antigens in premalignant lesions and squamous carcinomas of the uterine cervix. Acta Obstet Gynecol Scand. 2003;82:1146–1152. doi: 10.1046/j.1600-0412.2003.00252.x. [DOI] [PubMed] [Google Scholar]
- 63.Ruiter DJ, Mattijssen V, Broecker EB, Ferrone S. MHC antigens in human melanomas. Sem Cancer Biol. 1991;2:35–45. [PubMed] [Google Scholar]
- 64.Drexler HG, Gignac SM, Brenner MK, Coustan-Smith E, Janossy G, Hoffbrand AV. Differential expression of MHC class II antigens in chronic B-cell disorders. Clin Exp Immunol. 1988;71:217–223. [PMC free article] [PubMed] [Google Scholar]
- 65.Lecchi M, Lovisone E, Genetta C, Peruccio D, Resegotti L, Richiardi P. Gamma-IFN induces a differential expression of HLA-DR, DQ and DP antigens on peripheral blood myeloid leukemic blasts at various stages of differentiation. Leuk Res. 1989;13:221–226. doi: 10.1016/0145-2126(89)90015-5. [DOI] [PubMed] [Google Scholar]
- 66.Malmberg KJ, Levitsky V, Norell H, de Matos CT, Carlsten M, Schedvins K, et al. IFN-gamma protects short-term ovarian carcinoma cell lines from CTL lysis via a CD94/NKG2A-dependent mechanism. J Clin Invest. 2002;110:1515–23. doi: 10.1172/JCI15564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Backström E, Kristensson K, Ljunggren HG. Activation of natural killer cells: underlying molecular mechanisms revealed. Scand J Immunol. 2004;60:14–22. doi: 10.1111/j.0300-9475.2004.01475.x. [DOI] [PubMed] [Google Scholar]
- 68.Verfaillie C, Kay N, Miller W, McGlave P. Diminished A-LAK cytotoxicity and proliferation accompany disease progression in chronic myelogenous leukemia. Blood. 1990;76:401–408. [PubMed] [Google Scholar]
- 69.Pierson BA, Miller JS. CD56+bright and CD56+dim natural killer cells in patients with chronic myelogenous leukemia progressively decrease in number, respond less to stimuli that recruit clonogenic natural killer cells, and exhibit decreased proliferation on a per cell basis. Blood. 1996;88:2279–2287. [PubMed] [Google Scholar]
- 70.Chiorean EG, Dylla SJ, Olsen K, Lenvik T, Soignier Y, Miller JS. BCR/ABL alters the function of NK cells and the acquisition of killer immunoglobulin-like receptors (KIRs) Blood. 2003;101:3527–3533. doi: 10.1182/blood-2002-04-1172. [DOI] [PubMed] [Google Scholar]
- 71.Demanet C, Mulder A, Deneys V, Worsham MJ, Maes P, Claas FH, et al. Down-regulation of HLA-A and HLA-Bw6, but not HLA-Bw4, allospecificities in leukemic cells: an escape mechanism from CTL and NK attack? Blood. 2004;103:3122–3130. doi: 10.1182/blood-2003-07-2500. [DOI] [PubMed] [Google Scholar]
- 72.Chuang WL, Liu HW, Chang WY. Natural killer cell activity in patients with hepatocellular carcinoma relative to early development and tumor invasion. Cancer. 1990;65:926–930. doi: 10.1002/1097-0142(19900215)65:4<926::aid-cncr2820650418>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 73.Ono K, Yamanaga Y, Yamamoto K, Koga SI, Nishimura J, Nawata H. Natural killing activities in chronic liver diseases and hepatocellular carcinoma. J Clin Immunol. 1996;16:41–45. doi: 10.1007/BF01540971. [DOI] [PubMed] [Google Scholar]
- 74.Jinushi M, Takehara T, Tatsumi T, Kanto T, Groh V, Spies T, et al. Expression and role of MICA and MICB in human hepatocellular carcinomas and their regulation by retinoic acid. Int J Cancer. 2003;104:354–361. doi: 10.1002/ijc.10966. [DOI] [PubMed] [Google Scholar]
- 75.Rodríguez T, Méndez R, Del Campo A, Jiménez P, Aptsiauri N, Garrido F, Ruiz-Cabello F. Distinct mechanisms of loss of IFN-gamma mediated HLA class I inducibility in two melanoma cell lines. BMC Cancer. 2007;7:34. doi: 10.1186/1471-2407-7-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nie Y, Yang G, Song Y, Zhao X, So C, Liao J, et al. DNA hypermethylation is a mechanism for loss of expression of the HLA class I genes in human esophageal squamous cell carcinomas. Carcinogenesis. 2001;22:1615–1623. doi: 10.1093/carcin/22.10.1615. [DOI] [PubMed] [Google Scholar]
- 77.Serrano A, Tanzarella S, Lionello I, Mendez R, Traversari C, Ruiz-Cabello F, et al. Reexpression of HLA class I antigens and restoration of antigen-specific CTL response in melanoma cells following 5-aza-2′-deoxycytidine treatment. Int J Cancer. 2001;94:243–251. doi: 10.1002/ijc.1452. [DOI] [PubMed] [Google Scholar]
- 78.Fonsatti E, Sigalotti L, Coral S, Colizzi F, Altomonte M, Maio M. Methylation-regulated expression of HLA class I antigens in melanoma. Int J Cancer. 2003;105:430–431. doi: 10.1002/ijc.11077. [DOI] [PubMed] [Google Scholar]
- 79.Ogretmen B, McCauley MD, Safa AR. Molecular mechanisms of loss of beta 2-microglobulin expression in drug-resistant breast cancer sublines and its involvement in drug resistance. Biochemistry. 1998;3:11679–11691. doi: 10.1021/bi980573c. [DOI] [PubMed] [Google Scholar]
- 80.Seliger B. Molecular mechanisms of MHC class I abnormalities and APM components in human tumors. Cancer Immunol Immunother. 2008 doi: 10.1007/s00262-008-0515-4. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Khan AN, Gregorie CJ, Tomasi TB. Histone deacetylase inhibitors induce TAP, LMP, Tapasin genes and MHC class I antigen presentation by melanoma cells. Cancer Immunol Immunother. 2008;57:647–654. doi: 10.1007/s00262-007-0402-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Creswell P, guest editor. Antigen processing and presentation. Immunol Rev. 2005;207:5–313. doi: 10.1111/j.0105-2896.2005.00320.x. [DOI] [PubMed] [Google Scholar]
- 83.Wright KL, Ting JP. Epigenetic regulation of MHC-II and CIITA genes. Trends Immunol. 2006;27:405–412. doi: 10.1016/j.it.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 84.Reith W, LeibundGut-Landmann S, Waldburger JM. Regulation of MHC class II gene expression by the class II transactivator. Nat Rev Immunol. 2005;5:793–806. doi: 10.1038/nri1708. [DOI] [PubMed] [Google Scholar]
- 85.Wright KL, Ting JP. Epigenetic regulation of MHC-II and CIITA genes. Trends Immunol. 2006;9:405–412. doi: 10.1016/j.it.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 86.van den Elsen PJ, van der SN, Vietor HE, Wilson L, van Zutphen M, Gobin SJ. Lack of CIITA expression is central to the absence of antigen presentation functions of trophoblast cells and is caused by methylation of the IFN-gamma inducible promoter (PIV) of CIITA. Hum Immunol. 2000;61:850–862. doi: 10.1016/s0198-8859(00)00159-2. [DOI] [PubMed] [Google Scholar]
- 87.van der Stoep N, Biesta P, Quinten E, van den Elsen PJ. Lack of IFN-gamma-mediated induction of the class II transactivator (CIITA) through promoter methylation is predominantly found in developmental tumor cell lines. Int J Cancer. 2002;97:501–507. doi: 10.1002/ijc.1623. [DOI] [PubMed] [Google Scholar]
- 88.Satoh A, Toyota M, Ikeda H, Morimoto Y, Akino K, Mita H, et al. Epigenetic inactivation of class II transactivator (CIITA) is associated with the absence of interferon-gamma-induced HLA-DR expression in colorectal and gastric cancer cells. Oncogene. 2004;23:8876–8886. doi: 10.1038/sj.onc.1208144. [DOI] [PubMed] [Google Scholar]
- 89.Kanaseki T, Ikeda H, Takamura Y, Toyota M, Hirohashi Y, Tokino T, et al. Histone deacetylation, but not hypermethylation, modifies class II transactivator and MHC class II gene expression in squamous cell carcinomas. J Immunol. 2003;170:4980–4985. doi: 10.4049/jimmunol.170.10.4980. [DOI] [PubMed] [Google Scholar]
- 90.Morimoto Y, Toyota M, Satoh A, Murai M, Mita H, Suzuki H, et al. Inactivation of class II transactivator by DNA methylation and histone deacetylation associated with absence of HLA-DR induction by interferon-gamma in haematopoietic tumour cells. Br J Cancer. 2004;90:844–852. doi: 10.1038/sj.bjc.6601602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Takamura Y, Ikeda H, Kanaseki T, Toyota M, Tokino T, Imai K, et al. Regulation of MHC class II expression in glioma cells by class II transactivator (CIITA) Glia. 2004;45:392–405. doi: 10.1002/glia.10343. [DOI] [PubMed] [Google Scholar]
- 92.Yu J, Angelin-Duclos C, Greenwood J, Liao J, Calame K. Transcriptional repression by blimp-1 (PRDI-BF1) involves recruitment of histone deacetylase. Mol Cell Biol. 2000;20:2592–2603. doi: 10.1128/mcb.20.7.2592-2603.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ghosh N, Gyory I, Wright G, Wood J, Wright KL. Positive regulatory domain I binding factor 1 silences class II transactivator expression in multiple myeloma cells. J Biol Chem. 2001;276:15264–15268. doi: 10.1074/jbc.M100862200. [DOI] [PubMed] [Google Scholar]
- 94.Johnsen AK, Templeton DJ, Sy M, Harding CV. Deficiency of transporter for antigen presentation (TAP) in tumor cells allows evasion of immune surveillance and increases tumorigenesis. J Immunol. 1999;163:4224. [PubMed] [Google Scholar]
- 95.Lozupone F, Rivoltini L, Luciani F, Venditti M, Lugini L, Cova A, et al. Adoptive transfer of an anti-MART-127–35-specific CD8+ T cell clone leads to immunoselection of human melanoma antigen-loss variants in SCID mice. Eur J Immunol. 2003;33:556. doi: 10.1002/immu.200310032. [DOI] [PubMed] [Google Scholar]
- 96.Hunt JS, Langat DK, McIntire RH, Morales PJ. The role of HLA-G in human pregnancy. Reprod Biol Endocrinol. 2006;4 1:S10. doi: 10.1186/1477-7827-4-S1-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Knutson KL, Disis ML. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol Immunother. 2005;54:721–728. doi: 10.1007/s00262-004-0653-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Whiteside T, Campoli M, Ferrone S. Tumor induced immune suppression mechanisms and possible solutions. In: Nagorsen D, Marincola FM, editors. Analyzing T Cell Responses. How to analyze cellular immune responses against tumor associated antigens. Springer-Verlag; Netherlands: 2005. pp. 43–81. [Google Scholar]
- 99.Bernsen MR, Hakansson L, Gustafsson B, et al. On the biological relevance of MHC class II and B7 expression by tumour cells in melanoma metastases. Br J Cancer. 2003;88:424–431. doi: 10.1038/sj.bjc.6600703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chamuleau ME, Ossenkoppele GJ, van de Loosdrecht AA. MHC class II molecules in tumour immunology: prognostic marker and target for immune modulation. Immunobiology. 2006;211:619–625. doi: 10.1016/j.imbio.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 101.Tamori Y, Tan X, Nakagawa K, Takai E, Akagi J, Kageshita T, Egami H. Clinical significance of MHC class II-associated invariant chain expression in human gastric carcinoma. Oncol Rep. 2005;14:873–877. [PubMed] [Google Scholar]
- 102.Jiang Z, Xu M, Savas L, LeClair P, Banner BF. Invariant chain expression in colon neoplasms. Virchows Arch. 1999;435:32–36. doi: 10.1007/s004280050391. [DOI] [PubMed] [Google Scholar]
- 103.Bluestone JA, Tang Q. How do CD4+CD25+ regulatory T cells control autoimmunity? Curr Opin Immunol. 2005;17:638–642. doi: 10.1016/j.coi.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 104.Chattopadhyay S, Chakraborty NG, Mukherji B. Regulatory T cells and tumor immunity. Cancer Immunol Immunother. 2005;54:1153–1161. doi: 10.1007/s00262-005-0699-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang E, Panelli MC, Marincola FM. Understanding the response to immunotherapy in humans. Springer Semin Immunopathol. 2005;1:105–17. doi: 10.1007/s00281-004-0198-7. [DOI] [PubMed] [Google Scholar]
- 106.Esteller M. Relevance of DNA methylation in the management of cancer. Lancet Oncol. 2003;4:351–358. doi: 10.1016/s1470-2045(03)01115-x. [DOI] [PubMed] [Google Scholar]
- 107.Neumeister P, Albanese C, Balent B, Greally J, Pestell RG. Senescence and epigenetic dysregulation in cancer. Int J Biochem Cell Biol. 2002;34:1475–1490. doi: 10.1016/s1357-2725(02)00079-1. [DOI] [PubMed] [Google Scholar]
- 108.Sigalotti L, Fratta E, Coral S, Tanzarella S, Danielli R, Colizzi F, et al. Intratumor heterogeneity of cancer/testis antigens expression in human cutaneous melanoma is methylation-regulated and functionally reverted by 5-aza-2′-deoxycytidine. Cancer Res. 2004;64:9167–9171. doi: 10.1158/0008-5472.CAN-04-1442. [DOI] [PubMed] [Google Scholar]
- 109.Coral S, Sigalotti L, Colizzi F, Spessotto A, Nardi G, Cortini E, et al. Phenotypic and functional changes of human melanoma xenografts induced by DNA hypomethylation: immunotherapeutic implications. J Cell Physiol. 2006;207:58–66. doi: 10.1002/jcp.20540. [DOI] [PubMed] [Google Scholar]
- 110.Guo ZS, Hong JA, Irvine KR, Chen GA, Spiess PJ, Liu Y, et al. De novo induction of a cancer/testis antigen by 5-aza-2′- deoxycytidine augments adoptive immunotherapy in a murine tumor model. Cancer Res. 2006;66:1105–1113. doi: 10.1158/0008-5472.CAN-05-3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Coral S, Sigalotti L, Covre A, Nicolay HJM, Natali PG, Maio M. 5-aza-2′-deoxycytidine in cancer immunotherapy: a mouse to man story. Cancer Res. 2007;66:1105–1113. doi: 10.1158/0008-5472.CAN-06-2986. [DOI] [PubMed] [Google Scholar]
- 112.Fonsatti E, Nicolay HJ, Sigalotti L, Calabrò L, Pezzani L, Colizzi F, Altomonte M, Guidoboni M, Marincola FM, Maio M. Functional up-regulation of human leukocyte antigen class I antigens expression by 5-aza-2′-deoxycytidine in cutaneous melanoma: immunotherapeutic implications. Clin Cancer Res. 2007;13:3333–3338. doi: 10.1158/1078-0432.CCR-06-3091. [DOI] [PubMed] [Google Scholar]
- 113.Sigalotti L, Altomonte M, Colizzi F, et al. 5-Aza-2′-deoxycytidine (decitabine) treatment of hematopoietic malignancies: a multimechanism therapeutic approach. Blood. 2003;101:4644–4646. doi: 10.1182/blood-2002-11-3458. as comment on. [DOI] [PubMed] [Google Scholar]; Blood. 2002;100:2957–2964. doi: 10.1182/blood.V100.8.2957. [DOI] [PubMed] [Google Scholar]
- 114.Wijermans P, Lubbert M, Verhoef G, Bosly A, Ravoet C, Andre M, et al. Low-dose 5-aza-2′-deoxycytidine, a DNA hypomethylating agent, for the treatment of high-risk myelodysplastic syndrome: a multicenter phase II study in elderly patients. J Clin Oncol. 2000;18:956–962. doi: 10.1200/JCO.2000.18.5.956. [DOI] [PubMed] [Google Scholar]
- 115.Issa JP, Garcia-Manero G, Giles FJ, Mannari R, Thomas D, Faderl S, et al. Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2′-deoxycytidine (decitabine) in hematopoietic malignancies. Blood. 2004;103:1635–1640. doi: 10.1182/blood-2003-03-0687. [DOI] [PubMed] [Google Scholar]
- 116.Sigalotti L, Altomonte M, Colizzi F, Degan M, Rupolo M, Zagonel V, et al. 5-Aza-2′-deoxycytidine (decitabine) treatment of hematopoietic malignancies: a multimechanism therapeutic approach? Blood. 2003;101:4644–4646. doi: 10.1182/blood-2002-11-3458. [DOI] [PubMed] [Google Scholar]
- 117.Schrump DS, Fischette MR, Nguyen DM, Zhao M, Li X, Kunst TF, et al. Phase I study of decitabine- mediated gene expression in patients with cancers involving the lungs, esophagus, or pleura. Clin Cancer Res. 2006;12:5777–5785. doi: 10.1158/1078-0432.CCR-06-0669. [DOI] [PubMed] [Google Scholar]
- 118.Gollob JA, Sciambi CJ, Peterson BL, Richmond T, Thoreson M, Moran K, et al. Phase I trial of sequential low-dose 5-aza-2′-deoxycytidine plus high-dose intravenous bolus interleukin-2 in patients with melanoma or renal cell carcinoma. Clin Cancer Res. 2006;12:4619–4627. doi: 10.1158/1078-0432.CCR-06-0883. [DOI] [PubMed] [Google Scholar]
- 119.Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429:457–463. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 120.Li H, Wu X. Histone deacetylase inhibitor, trichostatin A, activates p21WAF1/CIP1 expression through down regulation of c-myc and release of the repression of c-myc from the promoter in human cervical cancer cells. Biochem Biophys Res Commun. 2004;324:860–867. doi: 10.1016/j.bbrc.2004.09.130. [DOI] [PubMed] [Google Scholar]
- 121.Fang JY, Lu J, Chen YX, Yang L. Methylation on expression of tumor suppressor genes and proto- oncogene in human colon cancer cell lines. World J Gastroenterol. 2003;9:1976–1980. doi: 10.3748/wjg.v9.i9.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bovenzi V, Le NL, Cote S, Sinett D, Momparler LF, Momparler RL. DNA methylation of retinoic acid receptor in breast cancer and possible therapeutic role of 5′-aza-2′CdR. Anticancer Drugs. 1999;10:471–476. doi: 10.1097/00001813-199906000-00007. [DOI] [PubMed] [Google Scholar]
- 123.Lucas DM, Davis ME, Parthun MR, Mone AP, Kitada S, Cunningham KD, et al. The histone deacetylase inhibitor MS-275 induces caspase-dependent apoptosis in B-cell chronic lymphocytic leukemia cells. Leukemia. 2004;18:1207–1214. doi: 10.1038/sj.leu.2403388. [DOI] [PubMed] [Google Scholar]
- 124.Florenes VA, Skrede M, Jorgensen K, Nesland JM. Deacetylase inhibition in malignant melanomas: impact on cell cycle regulation and survival. Melanoma Res. 2004;14:173–181. doi: 10.1097/01.cmr.0000129576.49313.26. [DOI] [PubMed] [Google Scholar]
- 125.Moore PS, Barbi S, Donadelli M, Costanzo C, Bassi C, Palmieri M, et al. Gene expression profiling after treatment with histone deacetylase inhibitor trichostatin A reveals altered expression of both pro- and anti-apoptotic genes in pancreatic adenocarcinoma cells. Biochem Biophys Acta. 2004;1693:167–176. doi: 10.1016/j.bbamcr.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 126.Eramo A, Pallini R, Lotti F, Sette G, Patti M, Bartucci M, et al. Inhibition of DNA methylation sensitizes glioblastoma for tumor necrosis factor-related apoptosis-inducing ligand-mediated destruction. Cancer Res. 2005;65:11469–11477. doi: 10.1158/0008-5472.CAN-05-1724. [DOI] [PubMed] [Google Scholar]
- 127.Insinga A, Monestiroli S, Ronzoni S, Gelmetti V, Marchesi F, Viale A, et al. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nat Med. 2005;11:71–76. doi: 10.1038/nm1160. [DOI] [PubMed] [Google Scholar]
- 128.Natoni F, Diolordi L, Santoni C, Gilardini Montani MS. Sodium butyrate sensitises human pancreatic cancer cells to both the intrinsic and the extrinsic apoptotic pathways. Biochim Biophys Acta. 2005;1745:318–329. doi: 10.1016/j.bbamcr.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 129.Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007;12:1247–1252. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- 130.French LE, Tschopp J. Defective death receptor signaling as a cause of tumor immune escape. Semin Cancer Biol. 2002;12:51–55. doi: 10.1006/scbi.2001.0405. [DOI] [PubMed] [Google Scholar]
- 131.Szyf M. DNA methylation and demethylation as targets for anticancer therapy. Biochemistry (Moscow) 2005;70:533–549. doi: 10.1007/s10541-005-0147-7. [DOI] [PubMed] [Google Scholar]
- 132.Bhalla KN. Epigenetic and chromatin modifiers as targeted therapy of hematological malignancies. J Clin Oncol. 2005;23:3971–3993. doi: 10.1200/JCO.2005.16.600. [DOI] [PubMed] [Google Scholar]
- 133.Chuang JC, Yoo CB, Kwan JM, Li TW, Liang G, Yang AS, et al. Comparison of biological effects of non- nucleoside DNA methylation inhibitors deoxycytidine. Mol Cancer Ther. 2005;4:1515–1520. doi: 10.1158/1535-7163.MCT-05-0172. [DOI] [PubMed] [Google Scholar]
- 134.Acharya MR, Sparreboom A, Venitz J, Figg WD. Rational development of histone deacetylase inhibitors as anticancer agents: a review. Mol Pharm. 2005;68:917–932. doi: 10.1124/mol.105.014167. [DOI] [PubMed] [Google Scholar]
- 135.Villar-Garea A, Esteller M. Histone deacetylase inhibitors: understanding a new wave of anticancer agents. Int J Cancer. 2004;112:171–178. doi: 10.1002/ijc.20372. [DOI] [PubMed] [Google Scholar]