

Abstract
Curing HIV-1 infection has remained elusive because of low and fluctuating drug levels arising from poor absorption, the development of viral reservoirs and sanctuary sites, toxicity, and patient nonadherence. The present study addresses the issue of insufficient drug exposure in macrophages. Viral reservoir sites such as macrophages are believed to be responsible for the viral rebound effect observed upon the discontinuation of anti-HIV drug therapy. In our proposed model, a drug can be covalently attached to a nanocarrier in order to facilitate the delivery of therapeutic agents to the site(s) of infection. As an initial step, we propose the covalent attachment of several copies of N-formyl-Met-Leu-Phe (fMLF), a known chemo-attractant for macrophages. In this article, one or more copies of fMLF were conjugated to multifunctional commercially available or novel, peptide-based PEG nanocarriers in which the structure was varied by appending PEGs with average molecular weights of 5, 20, and 40 kDa. U937 cell-specific binding and cellular uptake were analyzed. The results of uptake studies indicate that (i) uptake is energy dependent and mediated by a fMLF receptor, (ii) appending only 2 copies of the targeting ligand to the multifunctional nanocarrier appears sufficient for binding in vitro, and (iii) of the three configurations studied, the nanocarrier with a molecular weight of about 20 kDa, corresponding to a size of 20−60 nm, demonstrated the highest uptake. The results of the current studies demonstrate the feasibility of targeting macrophages and the suitability of using these synthetically versatile peptide–backbone PEG nanocarriers. The convenience, flexibility and possible limitations of this nanocarrier approach are discussed.
INTRODUCTION
Infection by human immunodeficiency virus type 1 (HIV-1) is the cause of acquired immunodeficiency syndrome (AIDS). AIDS is characterized by the severe impairment of immune system functions resulting in a profound decrease in the number of CD4+ T cells. The advent of highly active antiretroviral therapy (HAART), a multiple drug treatment regimen, has led to a dramatic decrease in both the morbidity and mortality of HIV patients (1). Despite this success, curing HIV infection has remained an elusive goal because of many challenges, including low and fluctuating drug concentrations due to poor drug absorption or patient nonadherence (2), the presence of viral reservoirs and sanctuary sites (3), and drug toxicity due to chronic therapy (4).
Macrophages are infected early during HIV infection and play an important role throughout the course of infection (5). Viral strains that establish infection in the new host are macrophage-tropic in over 95% of early asymptomatic individuals (5, 6). HIV enters macrophages via interactions with surface receptor CD4 (cluster designation 4) and coreceptor CCR5 (chemokine C-C motif receptor 5). Individuals bearing a homozygous deletion of CCR5 (CCR5 Δ 32) are highly resistant to HIV-1 infection despite multiple sexual exposures (7, 8). HIV-1 infection of macrophages is productive but noncytopathic, permitting them to serve as a long-lived source of HIV production. Infected macrophages are found in all tissues including the brain, gut, liver, lungs, lymphoid tissues, and spleen. More significantly, they represent major viral reservoirs and are responsible for the relapse of infection and development of resistance upon the discontinuation of treatment. Taken together, macrophages represent a key target and an important HIV cellular reservoir for therapeutic approaches aimed at decreasing the replication of the residual virus that survives HAART (9).
Among the major causes of HIV treatment failure, insufficient drug exposure due to poor penetration into viral reservoirs is one of the areas where little progress has been made, and efforts are generally lacking. Macrophage-specific drug delivery could significantly improve current anti-HIV therapy by improving therapeutic efficacy, minimizing systemic toxicity, and simplifying administration regimens. Targeting may possibly be achieved by covalently attaching a drug to a macrophage-specific ligand via a polymeric nanocarrier. A number of natural ligands for macrophage targeting have been explored. An example of such a ligand is IgG-derived immunomodulating peptide tuftsin (Thr-Lys-Pro-Arg), which has been conjugated to the HIV reverse transcriptase inhibitor 3′-azido-3′-deoxythymidine (AZT) (10). The AZT–tuftsin chimera possessed characteristics of both components, including inhibition of reverse transcriptase and HIV–antigen expression, stimulation of IL-1 release from mouse macrophages, and augmentation of the immunogenic function of the cell. Similarly, acetylated low-density lipoprotein (AcLDL), a ligand for macrophage scavenger receptors, has been conjugated to AZT (11). A 10-fold increase in the uptake of AcLDL–AZT was observed as compared to AZT in murine (J774) and human (U937) macrophage cell lines.
N-formyl peptides are cleavage products of bacterial and mitochondrial proteins and serve as potent chemoattractants for mammalian phagocytic leukocytes. The synthetic peptide N--formyl-methionyl-leucyl-phenylalanine (fMLF) is the first identified and most potent chemoattractant for human macrophages (12). fMLF binds to the formyl peptide receptor (FPR) with high affinity (10−30 nM) and to its variant FPRL1 with low affinity (12). When the free fMLF chemotactic peptide binds to a phagocytic cell, rapid receptor-mediated internalization takes place (13). After binding to the receptors, fMLF activates phagocytic leukocytes through a G-protein-mediated signaling cascade resulting in increased cell migration, calcium mobilization, and the heterologous down-regulation of the expression and function of the two chemokine receptors CCR5 and CXCR4, which are the key fusion coreceptors for HIV-1 (12). fMLF has been used for imaging of infection and inflammation (14).
Previous work by our group showed that increasing the number of fMLF residues (up to eight) attached to a single PEG polymer resulted in enhanced avidity (Kd = 0.18 nM) for neutrophil-like differentiated HL-60 cells relative to free fMLF (Kd = 28 nM) (15). A PEG polymer bearing four fMLF and four digoxigenin residues showed specific enhancement in binding to differentiated HL-60 cells and mouse peritoneal macrophages in situ relative to a polymeric carrier lacking fMLF. Furthermore, increasing copy numbers of fMLF increased avidity more potently than it increased cell activation, suggesting that toxicity due to excessive phagocyte activation might not be a problem. These early results provided conceptual feasibility. However, further studies are required in order to evaluate if biologically compatible polymer conjugates bearing multiple fMLF residues can be used as effective targeted drug delivery systems for phagocytic cells. In general, the successful development of a nanocarrier delivery system requires an understanding of (1) the relationship between cell uptake and the copy number, size, and shape of the constituent PEG chains, (2) the avidity and specificity of the interactions between the ligands appended to the nanocarriers and the cognate receptors on the target cell, and (3) the chemical reactions required for the synthesis of such macromolecules, including the stability of the conjugates prior to administration and disassembly and release of the active drug after reaching the target site.
In the present study, a series of PEG nanocarriers with different numbers of fMLF and various PEG structures were designed, synthesized, and evaluated in vitro for targeting to human U937 cells induced to differentiate into a macrophage phenotype. The relationship between the molecular features of PEG-fMLF nanocarriers (i.e., number of targeting peptide and PEG sizes) and cell uptake was elucidated. The current studies further demonstrate the feasibility of using macrophage-targeted nanocarriers, while also introducing a new nanocarrier that allows for the attachment of multiple drugs and targeting moieties resolving a potentially major limitation of commercially available PEGs.
MATERIALS AND METHODS
Materials
N-formyl-Met-Leu-Phe-Lys-Cys-amide and the backbone peptides acetyl-Cys(thiopyridine)-(β-Ala-β-Ala-Lys)n-amide (n = 2, 4) were synthesized via Fmoc Chemistry by the W.M. Keck Facility (New Haven, CT). N-hydroxysuccinimide (NHS)-PEG-vinyl sulfone (VS; NHS-PEG-VS) (MW ∼5 kDa), mPEG-MAL (MW ∼5.5 kDa), mPEG-(MAL)2 (MW ∼5.5 kDa, 20 kDa, and 40 kDa), mPEG-NH2 (MW ∼5 kDa), and 4-arm PEG-(NH2)4 (MW ∼10 kDa) were obtained from Nektar Therapeutics (Huntsville, AL). N-maleimidobutyryloxysuccinimide ester (GMBS) was obtained from Pierce Biotechnology (Rockford, IL). NHS-carboxyfluorescein was obtained from Sigma-Aldrich (St. Louis, MO). Fluo-4 AM (acetoxymethyl ester) was obtained from Molecular Probes (Eugene, OR). DMF (dimethylformamide), DIEA (diisopropylethylamine), acetonitrile (ACN), trifluoroacetic acid (TFA), ether, and other chemical reagents were purchased from Sigma-Aldrich (St. Louis, MO).
Synthesis and Characterization of PEG-fMLF Nanocarriers
PEG5k-fMLF(fMLFK(FLUORESCEIN)C-mPEG5K) and PEG5K,20K,40K-(fMLF)2 ([fMLFK(FLUORESCEIN)C]2-mPEG5K,20K,40K). mPEG5K-maleimide (11.2 mg, 2 mmol) was dissolved in 1 mL of phosphate-buffered saline (PBS, pH 7.4) at room temperature. To this were added 3 equivalents of fMLFKC (4 mg, 6 mmol). The reaction was stirred overnight at room temperature. For the 2-copy forked conjugates (PEG5K,20K,40K-(fMLF)2 ([fMLFK(FLUORESCEIN)C]2-mPEG5K, 20K, 40K) a similar protocol was used (e.g., 3 equiv 8 mg, 12 mmol fMLFKC was utilized for PEG5K-(fMLF)2 ([fMLFK(FLUORESCEIN)C]2-mPEG5K)). The excess solvent was removed under reduced pressure. The solid PEGylated product was further reacted with 3 equiv of NHS–carboxyfluorescein (3 mg, 6 mmol) and 1% DIEA (5 μL) in 500 μL of DMF. The reaction was stirred for 3 h at room temperature. The final product was recrystallized from cold ether, washed three times to remove impurities, and lyophilized. The dried product was then dissolved in ∼5 mL of ddH2O and subjected to dialysis against ddH2O for 2 days in the dark in order to remove the excess unreacted reagents (NHS–carboxyfluorescein, fMLFKC) and salts, which might have not been removed during the ether precipitation step. The solution was then lyophilized to yield the purified product. [fMLFK(fluorescein)C]2-mPEG20K,40K was synthesized using a similar approach (Scheme 1).
Scheme 1.
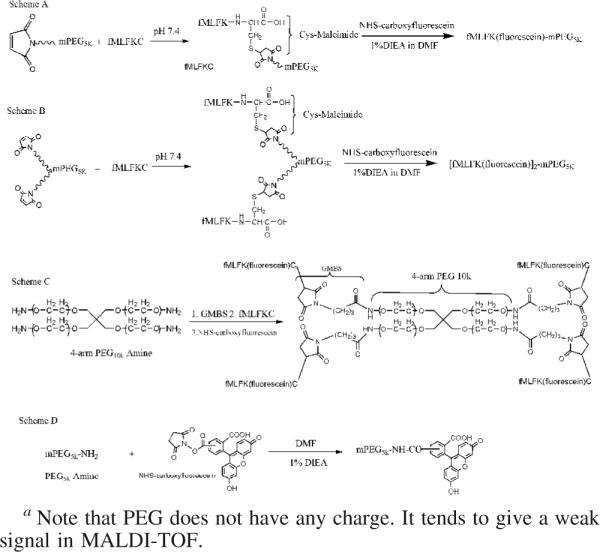
Synthesis of PEG-fMLF Nanocarriers with 1 Copy (PEG5k-fMLF, Scheme A), 2 Copies (PEG5k-(fMLF)2,Scheme B), and 4 Copies (PEG10k-(fMLF)4, Scheme C) of fMLF, and the Fluorescein-Labeled Control PEG5k without fMLF Peptides (Scheme D)a
PEG10k-(fMLF)4[fMLFK(FLUORESCEIN)C]4
PEG10K-(NH2)4 (20 mg, 2 mmol) was first activated with a 3-fold molar excess of the heterobifunctional cross-linker GMBS in DMF to form a maleimide-activated PEG. The reaction was stirred overnight at room temperature. The product was precipitated with cold ether and dried under vacuum to yield the solid PEGylated product. The GMBS linker essentially converts a primary amino group to a maleimide group that can react with a thiol group to form a stable thioether bond. This activated intermediate was reacted with 3 equiv of fMLFKC (16 mg, 24 mmol) in 1 mL of phosphate-buffered saline (PBS, pH 7.4) at room temperature. The reaction was stirred overnight at room temperature. The excess solvent was removed under reduced pressure. The reaction product was further reacted with 3 equiv of NHS–carboxyfluorescein (12 mg, 24 mmol) and 1% DIEA (5 μL) in 500 μL of DMF. The reaction was stirred for 3 h at room temperature. The final product was recrystallized from cold ether and washed three times to remove impurities. Then the dried product was dissolved in ∼5 mL of ddH2O and was subject to dialysis against ddH2O for 2 days in the dark. The solution was lypholyzed to yield the purified product.
PEG5k(mPEG5K-FLUORESCEIN)
The amino groups of mPEG5k-NH2 (5 mg, 1 mmol) were reacted with 3 equiv of NHS–carboxyfluorescein (1.5 mg, 3 mmol) and 1% DIEA (5 μL) in 500 μL of DMF to yield mPEG5k-fluorescein. The reaction was stirred for 3 h at room temperature. The final product was recrystallized from cold ether, washed three times to remove impurities, and dried under vacuum. Then the dried product was dissolved in ∼5 mL of ddH2O and was subject to dialysis against ddH2O for 2 days in the dark. The solution was lyophilized to give the purified product.
Peptide–Backbone PEG-fMLF Nanocarriers
To achieve a branch shape and multiple coupling sites, peptide–backbone PEG nanocarriers were designed (Scheme 2). In order to synthesize fMLF PEG nanocarriers with two copies of fMLF (Scheme 2), the backbone peptide acetyl-Cys(thiopyridine)-β-Ala-β-Ala-Lys-β-Ala-β-Ala-Lys-amide (1.4 mg, 2 mmol) was reacted with 2 equiv of NHS-PEG5k-VS (40 mg, 8 mmol) and 1% DIEA (5 μL) in 500 μL of DMF. The reaction was carried out at room temperature for 3 h. The PEGylated intermediate acetyl-Cys(thiopyridine)-[β-Ala-β-Ala-Lys(PEG5k-VS)]2-amide was purified by size-exclusion chromatography on a TSK Gel-3000pw column. The size of the product in eluted fraction volume was confirmed by MALDI-TOF mass spectrometry, and peak fractions were pooled and dried under vacuum. The PEGylated intermediate was dissolved in 1 mL of phosphate-buffered saline (PBS, pH 7.4) at room temperature. To this, 3 equiv of fMLFKC (8 mg, 12 mmol) was added. The reaction was stirred overnight at room temperature. The product was then treated with a 5 M excess of dithiothreitol (DTT) for another 2 h. The product was dialyzed against ddH2O for 2 days. Then the excess solvent was removed under reduced pressure. The solid PEGylated product was further reacted with 3 equiv of NHS–carboxyfluorescein (6 mg, 12 mmol) and 1% DIEA (5 μL) in 500 μL of DMF. The reaction was stirred for 3 h at room temperature. The final product was recrystallized from cold ether, washed three times to remove impurities, and dried under vacuum. Then the dried product was dissolved in ∼5mL of ddH2O and was subject to dialysis against ddH2O for 2 days in the dark. The solution was dried under vacuum to give the purified product. The PEG nanocarrier with four copies of fMLF and fluorescein was synthesized by a similar scheme (Scheme 2).
Scheme 2.
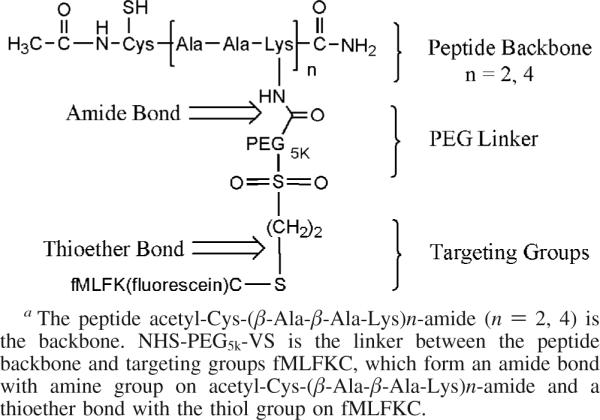
Design of Peptide-Based PEG Nanocarriers with 2 and 4 Copies of fMLF Peptidesa
Size-Exclusion Chromatography (SEC)
Purified nanocarriers were dissolved in ddH2O at a final concentration of ∼1 mg/mL. The solution was loaded into a Waters HPLC equipped with a SEC TSK-GEL G3000PW HPLC column (Tosoh Corp., Japan). The mobile phase was 100% ddH2O, and the flow rate was 1 mL/min. Eluents were monitored for absorbance at 220 nm with a Waters UV detector.
MALDI-TOF Mass Spectrometry
Mass spectrometry was performed using an Applied Biosystems Voyager DE Pro matrix-assisted laser desorption time-of-flight mass spectrometer (MALDI-TOF). The instrument was operated in the linear positive ion mode at 25.0 kV utilizing delayed extraction (100 ns). Data were collected over a mass range of 2000−10,000 M/z with 100 shots per spectrum. Samples were dissolved in 50:50 H2O/ACN with 0.1% TFA at ∼10 mg/mL. The matrix solution was (10 mg/mL) α-cyano-4-hydrocinnamic acid (10 mg/mL) in 50:50 H2O/ACN with 0.1% TFA. A sample (4 μL) was combined with 1 μL of matrix solution. This mixture was then spotted onto the sample plate and allowed to dry. The instrument was externally calibrated by analysis of a three-component peptide mixture over a mass range of ∼1000−10000 m/z. Data analysis consisted of baseline correction and noise reduction utilizing Data Explorer software, version 5.1 (Applied Biosystems). The difference in molecular weight between the final nanocarrier and the unreacted PEG corresponds to the molecular weight additions on the PEG skeleton. The relative intensity for unconjugated PEG is much lower than that of its peptidic core conjugated counterpart since the unconjugated PEG cannot be ionized to the same extent. (Figure 1).
Figure 1.

MALDI-TOF spectra of PEG-fMLF nanocarriers. The starting material mPEG-MAL showed the polydisperse peaks with average molecular weight at about 5518 Da. fMLFK(fluorescein)C-mPEG showed an average peak at 6560 Da, and [fMLFK(fluorescein)C]2-mPEG showed an average peak at 7604 Da.
Amino Acid Analysis
Amino acid analysis of the free fMLF, PEG5k-fMLF, PEG5K-(fMLF)2, PEG10k-(fMLF)4, and peptide-based PEG10k-(fMLF)2 and PEG20k-(fMLF)4 nanocarriers was performed at the Protein Facility of the Iowa State University Office of Biotechnology (Ames, Iowa) to confirm the presence and concentration of fMLF on PEG nanocarriers. The sample was transferred to a hydrolysis tube and dried under vacuum. The tube was placed in a vial containing 6 N HCl and a small amount of phenol, and the samples were hydrolyzed by the HCl vapors under vacuum for 65 min at 150 °C. Following hydrolysis, each sample was dissolved in distilled water containing EDTA. An aliquot containing ∼1 nmol of each amino acid was placed on a sample slide, and derivatization was done under basic conditions for 30 min with phenylisothocyanate (PITC). The resulting phenylthiocarbamyl (PTC) derivatives were separated by reverse-phase chromatography and quantitated by absorbance at 254 nm.
Stability in PBS and Rabbit Plasma
The stability of the fluorescein-labeled PEG-fMLF nanocarriers was tested in 10 mM PBS (pH 7.4) and rabbit plasma at 37 °C for 24 h. The nanocarrier solutions were incubated separately in 10 mM PBS (pH 7.4) or in spiked rabbit plasma at 37 °C. Aliquots were withdrawn at different time points and centrifuged at 14,000g for 90 min with a Microcon filter (molecular weight cutoff = 3,000 Da) (Amicon Inc., Beverly, MA). The free fMLF cleaved from the PEG nanocarrier during the incubation passes through the filter, whereas the fMLF that remains linked is retained. The eluents and retentates resulting from the different incubation time points were withdrawn and subjected to fluorescence detection. Each measurement was done in triplicate.
Cell Lines
The U937 cell line, which had been isolated from a patient suffering from histiocytic lymphoma, was later found capable of terminal differentiation upon induction into a macrophage phenotype. U937 cells were grown in RPMI 1640 DM medium supplemented with 10% FBS (fetal bovine serum) as suspended cells. They differentiated into macrophage-like cells after treatment with 1 mM dibutyryl-cAMP for 48 h, remaining suspended. The U937 cell line is potentially an excellent model system to study formyl peptide receptors because, in the undifferentiated state, these cells express low levels of formyl peptide receptors and showed no movement toward any stimuli (16, 17). A variety of differentiating agents induce U937 cells toward a more mature phenotype in which they express formyl peptide receptors and display migratory properties similar to those of peripheral blood monocytes or neutrophils.
Expression of Formyl Peptide Receptors
The expression of the two relevant human receptors for fMLF, FPR and FPRL1, was evaluated using semiquantitative RT-PCR. Total RNA was extracted from U937 and differentiated U937 cells. RNA (2 μg) was used as a template for RT-PCR. The sense oligonucleotide primer 52-CTGCTGGTGCTGCTGGCAAG-32 and antisense primer 52-AATATCCCTGACCCCATCCTCA-32 based on the sequences flanking the human FPRL1 coding region (1.1 kb) were designed to amplify FPRL1. The sense primer 52-CTCCAGTTGGACTAGCCACA-32 (nucleotide 1639−1658 in exon 2) and antisense primer 52-CCATCACCCAGGGCCCAATG −32 (nucleotide 5341−5359 in the coding region of exon 3) were designed to amplify human FPR. The sense primer 5′-GCTCG TCGTC GACAA CGGCT C-3′ and the antisense primer 5′-CAAAC ATGAT CTGGG TCATC TTCTC-3′ for beta-Actin were used as controls.
A commercially available rabbit polyclonal antibody was used in a Western Blot analysis without success. The antibody should have been able to detect both receptors but produced bands that were not always reproducible or consistent with the expected sizes.
Time-Dependent Cellular Association of PEG-fMLF Nanocarriers in Differentiated U937 Cells
U937 cells in suspension were differentiated into macrophage-like cells after treatment with 1 mM dibutyryl-cAMP for 48 h. The suspended differentiated cells were washed by centrifugation twice with uptake buffer (Hanks Balanced Salt Solutions (HBSS) with 20 mM HEPES to adjust pH to neutral). The washed cells were added to wells of 24-well polycarbonate plates at a density of 1 × 105 cells/well in the presence of 50 nM fluorescein-labeled PEG-fMLF nanocarriers for 1, 2, 4, or 8 h at 37 °C. The concentrations of all fMLF-bearing nanocarriers were normalized based on the fMLF moiety concentration. That is, a nanocarrier bearing one copy of fMLF was used at 50 nM fMLF and 50 nM whole-molecule concentration, a nanocarrier bearing two copies of fMLF was used at 50 nM fMLF and 25 nM whole-molecule concentration, and so forth. At the end of an incubation period, washing the cells twice with ice-cold uptake buffer stopped the uptake. The cells were subsequently resuspended in uptake buffer. Cell-associated fluorescence was analyzed by flow cytometry using a Coulter EPICS PROFILE (Fullerton, CA) equipped with a 25 mW argon laser. For each analysis, 10,000 to 20,000 events were accumulated. The results are shown in Figure 3.
Figure 3.
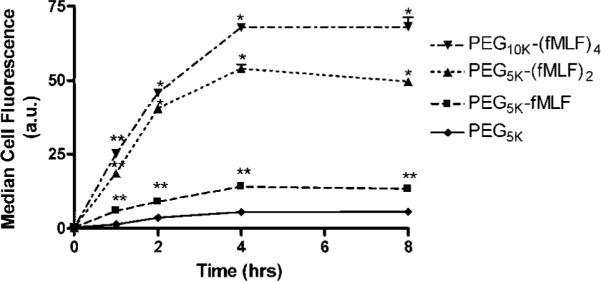
Time course of total cell-associated fluorescein-labeled PEG-fMLF nanocarriers in differentiated U937 cells at 37 °C. Differentiated U937 cells were incubated for the indicated time periods and plotted against the cell-associated nanocarrier amounts. The cell-associated fluorescence was measured by cell flowcytometry and reported as the medium value of intensity distribution. The means ± SD for three independent experiments are shown for each nanocarriers. PEG5K-fMLF enhanced macrophage association about 15-fold (p < 0.05) after 4 h of incubation at 37 °C compared to the control PEG5k. The values were about 45- and 60-fold higher for PEG5k-(fMLF)2 and PEG10k-(fMLF)4, respectively, than for the control PEG5k (p < 0.01). (Statistically significant differences were observed between the PEG-fMLF nanocarriers and the control PEG5k, * p < 0.01, ** p < 0.05.)
U937 Cell Uptake by Undifferentiated and Differentiated U937 Cells
To show that the cellular association of 50 nM PEG-fMLF nanocarriers was based on uptake, the uptake experiments were performed with both undifferentiated and differentiated U937 cells as described above for the time-dependent cellular association. The results are shown in Figure 4.
Figure 4.
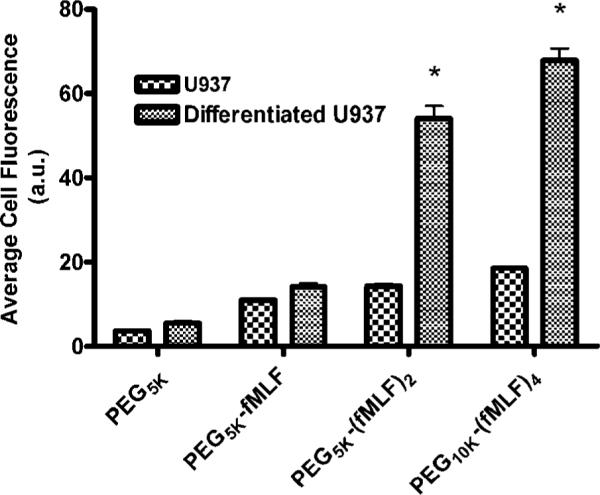
Total cellular association (uptake from now on) of fluorescein-labeled PEG-fMLF nanocarriers in the undifferentiated and the differentiated U937 cells at 37 °C after 4 h of incubation. The means ± SD for three independent experiments are shown for each value. (*, statistically significant difference between undifferentiated and differentiated U937 cells, p < 0.01.)
Temperature-Dependent Uptake of PEG-fMLF Nanocarriers
To further demonstrate that the cellular association of 50 nM PEG-fMLF nanocarriers was based on uptake, differentiated U937 cells were incubated with fluorescein-labeled PEG-fMLF nanocarriers for 4 h at 37 or 4 °C. At the end of an incubation period, the cells were washed, and resuspended, and cell-associated fluorescence was analyzed by flow cytometry as described above. The results are shown in Figure 5.
Figure 5.
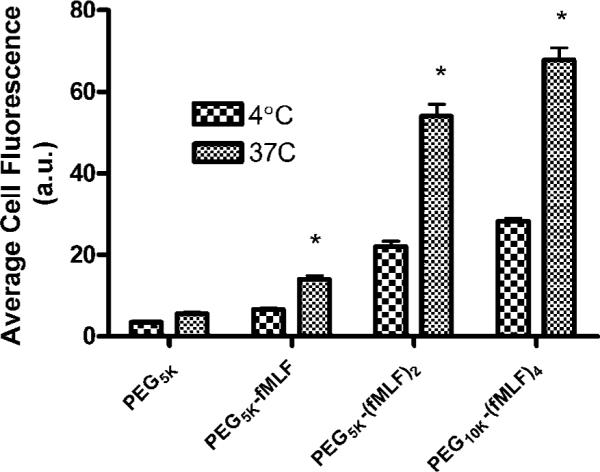
Uptake of fluorescein-labeled PEG-fMLF nanocarriers in differentiated U937 cells at 4 and 37 °C after 4 h of incubation. The means ± SD for three independent experiments are shown for each value. (*, statistically significant difference between 4 and 37 °C treatments, p < 0.01.)
Uptake Inhibition by Free fMLF Peptide
The involvement of formyl peptide receptors in differentiated U937 cell uptake of PEG-fMLF nanocarriers was also investigated using inhibition studies. Free fMLF peptide (5 μM), a competitive substrate of formyl peptide receptor, was coincubated with 50 nM fluorescein-labeled PEG, mPEG-fMLF, mPEG-(fMLF)2, or PEG-(fMLF)4 nanocarriers. Uptake assays were performed as described above for the temperature-dependent uptake. The results are shown in Figure 6.
Figure 6.
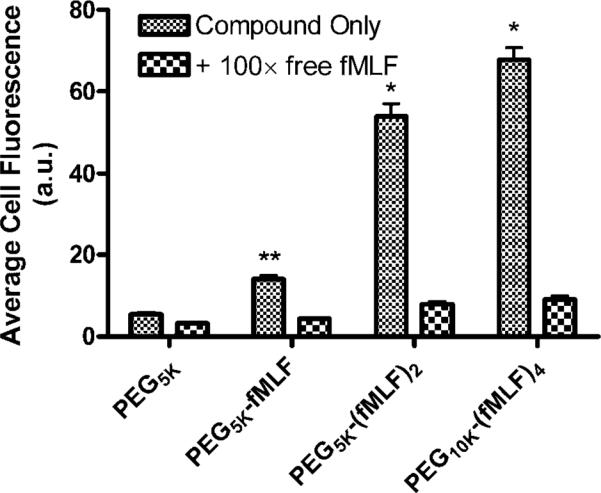
Uptake of fluorescein-labeled PEG-fMLF nanocarriers in differentiated U937 cells at 37 °C after 4 h of incubation in the absence and presence of 5 μM free fMLF (100× in excess to the concentration of the labeled nanocarriers). The means ± SD for three independent experiments are shown for each value. (Statistically significant difference between the differentiated U937 cells incubated with and without free fMLF * p < 0.01, ** p < 0.05.)
Uptake by Fluorescence Microscopy
Differentiated U937 cells were incubated in 24-well microplates with 50 nM fluorescein-labeled PEG, mPEG-fMLF, mPEG-(fMLF)2, or PEG-(fMLF)4 nanocarriers for 4 h. After incubation, the cells were washed twice using centrifugation and resuspended in Hanks Balanced Salt Solutions (HBSS). The stained cell suspension was placed on slides, covered, and observed under a Zeiss Axiostar Plus fluorescence microscope (Thornwood, NY) with an objective magnification of 63× and an Insight digital camera (Sterling, MI) with proper filter sets. Digital images were taken with the Insight digital camera and its companion Spot software.
Calcium Mobilization
Differentiated U937 cells were loaded with 4 μmol/L fluo-4 AM in HBSS for 30 min at 37 °C in the dark. The cells were washed and incubated for 30 min at room temperature to allow the Fluo-4 AM dye to completely de-esterify. The cells were washed, resuspended in HBSS, and treated with 1 μM PEG-fMLF nanocarriers at 37 °C. Fluorescence readings were obtained using a Tecan fluorescence plate reader for 400 at 2 s intervals.
Statistical Analysis
All statistical tests were performed using GraphPad Instat (GraphPad Software, Inc., San Diego, CA). A minimal p-value of 0.05 was used as the significance level for all tests. One-way analysis of variance and the Tukey test were performed on the uptake data. All data are reported as means ± SD of three observations, unless otherwise noted. The graphs were plotted using GraphPad Prism 4.01 (GraphPad Software, Inc., San Diego, CA).
RESULTS
Synthesis and Characterization of PEG-fMLF Nanocarriers
The PEG-fMLF nanocarriers mPEG5K-fMLF, mPEG5K-(fMLF)2, and PEG10K-(fMLF)4 were prepared by coupling the fMLFKC to mPEG-MAL, mPEG-(MAL)2, and PEG-(NH2)4 according to the procedures described in Scheme 1. All nanocarriers were purified by the removal of low molecular weight contaminants using 3 kDa dialysis bags in ddH2O for 2 days. The structures of purified products were confirmed by a combination of SEC, MALDI-TOF, and amino acid analysis. Amino acid analysis of the final nanocarrier showed the presence of methionine, leucine, phenylalanine, lysine, and cysteine in the proper ratios (Table 1). Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry was used to accurately determine the molecular masses of polydisperse polymeric nanocarriers (Figure 1). A comparison of the MALDI-TOF spectra of unreacted mPEG-MAL (MW ∼5518 Da) with that of the fMLFK(fluorescein)C-mPEG (∼6560 Da) and [fMLFK(fluorescein)C]2-mPEG (∼7604 Da) revealed average mass peak shifts of ∼1040 Da and ∼2080 Da, respectively, confirming that the polymeric nanocarriers created were derivatized with either fMLFK(fluorescein)C (1042 Da) or [fMLFK (fluorescein)C]2 (2084 Da). Thus, although the polymer is polydisperse because of variations in the number of ethylene glycol repeats, the fMLFK(fluorescein) content of each preparation is monodisperse.
Table 1.
Results of Amino Acid Analysis of PEG-fMLF Nanocarriersa
| amino acid ratio (normalized by phenylalanine) |
concentration (nM) (expected 50 nM) | ||||
|---|---|---|---|---|---|
| sample name | Met | Leu | Phe | Lys | |
| fMLF | 0.90 | 1.20 | 1.00 | 1.00 | 36.00 |
| PEG-(fMLF) | 0.95 | 1.20 | 1.00 | 1.00 | 37.10 |
| PEG-(fMLF)2 | 0.97 | 1.23 | 1.00 | 0.95 | 37.87 |
| PEG-(fMLF)4 | 0.91 | 1.19 | 1.00 | 1.01 | 42.00 |
| PB-PEG-(fMLF)2 | 0.91 | 1.17 | 1.00 | 2.08 | 40.00 |
| PB-PEG-(fMLF)4 | 0.96 | 1.19 | 1.00 | 1.87 | 37.90 |
The amino acids were normalized relative to phenylalanine. PB: peptide-based.
Novel peptide–backbone nanocarriers were prepared by coupling the peptide backbone, acetyl-Cys(TP)-(β-Ala-β-Ala-Lys-)n-amide (n = 2, 4) to a heterobifunctional PEG, NHS-PEG-VS (Scheme 2). The NHS moiety reacts specifically with the ε-amine group of the backbone peptide, and the VS group is used for the attachment of PEG to the sulfhydryl (SH) moiety of the fMLFKC peptide. The linkages formed amide (for NHS) and thioether (for VS) bonds between PEG and the fMLFKC and the peptide backbone, respectively. Both bonds are highly stable under physiological conditions. The thiol group of the cysteine moiety in the peptide backbone provides a suitable site for the attachment of a drug moiety. This moiety is protected by thiopyridine, which is an excellent leaving group that can be readily reacted with sulfhydryl-containing drugs or peptides. TP is replaced by fMLFKC during synthesis via a reversible thiol linkage. The product was further reacted with DTT in order to remove the reversibly linked fMLFKC peptide. Thus, the targeting moiety fMLFKC in the nanocarrier is attached via stable and nonreversible thioether linkage. The β-alanine moiety in the scaffold serves as a spacer, reducing steric hindrance during the PEGylation reaction. Scaffold (peptide-based PEG carrier) size was varied by using peptide backbones of different lengths, which could be conjugated with different (2 or 4) numbers of copies of NHS-PEG-VS in reactions proceeding to complete peptide derivatization. In Scheme 2, the Cys residue in the backbone peptide is shown with an –SH group. However, during the synthesis reactions, the thiol must be protected such as with a thiopyridine group, which may later be removed for other conjugation reactions, as shown in Scheme 2.
Stability in PBS and Plasma
The stability of the fluorescein-labeled PEG-fMLF nanocarriers was tested in 10 mM PBS (pH 7.4) and rabbit plasma at 37 °C for 24 h. For nonpeptide PEG-fMLF nanocarriers, the linkages formed a thioether bond between fMLFKC and PEG, and a urethane bond between the fMLFKC and the fluorescein. For peptide–backbone PEG nanocarriers, the linkages formed urethane (from NHS) bonds between PEG and the peptide backbone or a thioether (from VS) bond between PEG and fMLFKC. All bonds were highly stable under physiological conditions. Stability was analyzed by the release of the fluorescence tag. The released fluorescence tag was monitored after 24 h of incubation in PBS or plasma at 37 °C. The degraded product was quantitatively detected in the filtrate using ultrafiltration (5 kDa MWCO). The nanocarriers were found to be stable with less than 3% degradation in PBS or plasma during a 24 h incubation at 37 °C.
Expression of Formyl Peptide Receptors
The expression of the two relevant fMLF receptors, FPR and FPRL1, in the undifferentiated and differentiated U937 cells was confirmed using semiquantitative RT-PCR. Differentiated U937 cells expressed high levels of the formyl peptide receptor (FPR, 500 bp RT-PCR product) and formyl peptide receptor-like 1 (FPRL1, 1100 bp RT-PCR product) transcripts, while undifferentiated U937 cells only expressed low levels of FRPL1 and no detectable FPR (Figure 2). Even though an FPRL-1 antibody was available, we had limited success using it because of inconsistency in size and reproducibility. The RT-PCR result is in agreement with known U937 cell phenotype in the literature.
Figure 2.

mRNA expression of formyl peptide receptor (FPR) and formyl peptide receptor-like 1 (FPRL1) in undifferentiated and differentiated U937 by RT-PCR determined using semiquantitative RTPCR. Differentiated U937 cells expressed high levels of FPR mRNA (500 bp product) and FPRL1 mRNA (1100 bp product), while undifferentiated U937 cells only expressed a low level of FPRL1 mRNA and no FPR mRNA.
Time-Dependent Total Cellular Association of PEG-fMLF Nanocarriers in Differentiated U937 Cells
To understand how differentiated U937 cells would interact with PEG-fMLF nanocarriers, we first investigated the time course of total cellular association of 50 nM nanocarriers using flow cytometry. The association of PEG-fMLF nanocarriers with macrophage-like differentiated U937 cells at 37 °C showed a progressive increase from 0 h through 4 h, with steady-state apparently being reached at 4 h (Figure 3). The results are the average of three independent experiments performed on three separate days, and they showed that PEG5k lacking fMLF had very low uptake at all time points during the incubation. In general, the attachment of 1, 2, or 4 copies of fMLF to PEG significantly increased the cellular association at all time points. Specifically, PEG5K-fMLF enhanced macrophage cellular association about 15-fold (p < 0.05) after 4 h of incubation at 37 °C compared to the control PEG5k. PEG5k-(fMLF)2 and PEG10k-(fMLF)4 further increased uptake about 45- and 60-fold (p < 0.01), respectively, compared to the control PEG5k. The correlation between fMLF copy numbers per nanocarrier with the amount of association suggests that the association is mainly mediated by fMLF receptors. Since data below further suggests fMLF receptor-mediated active uptake, from now on we will replace the term total cellular association with the term uptake.
U937 Cell Uptake by Undifferentiated and Differentiated U937 Cells
To show that the uptake of 50 nM PEG-fMLF nanocarriers was mediated by fMLF receptors, a comparison was made between the uptake in undifferentiated U937 cells expressing low levels of FPRL1/no FPR and differentiated U937 cells expressing high levels of FPR and FPRL1 (Figure 4). The results represent the average of three independent experiments performed on three separate days. The uptake of the control PEG5k was low and showed no significant difference between undifferentiated and differentiated U937 cells. In contrast, the uptake of PEG-fMLF nanocarriers with 1, 2, or 4 copies of fMLF was significantly higher in differentiated as compared to undifferentiated U937 cells (p < 0.01). PEG5k-fMLF uptake in differentiated U937 cells increased 1.52-fold as compared to undifferentiated U937 cells. Similarly, PEG5k-(fMLF)2 and PEG10k-(fMLF)4 increased 3.76-fold and 3.65-fold, respectively. Significantly higher uptake in differentiated U937 cells expressing FPR and FPRL1 is consistent with uptake being specifically mediated by formyl peptide receptors.
Temperature-Dependent Uptake of PEG-fMLF Nanocarriers
To confirm the expectation that uptake occurred mainly by an active cellular process, temperature-dependent uptake was investigated. The uptake of PEG-fMLF nanocarriers in differentiated U937 cells after 4 h of incubation at 4 °C showed significantly lower cell uptake than at 37 °C in three separate experiments (Figure 5). Nonderivatized PEG5k showed similar low uptake levels at both temperatures. PEG5k-fMLF uptake at 4 °C was only 46.2% of that at 37 °C, while the ratio of 4:37 °C for PEG5k-(fMLF)2 and PEG10k-(fMLF)4 was 40% and 41.6%, respectively. A substantial portion of cell-associated nanocarriers at low temperature are bound to the cell surface but are not internalized, representing both specific receptor binding as well as binding to other cell-surface components. Thus, the temperature-dependent portion at 37 °C is consistent with a receptor-mediated active uptake mechanism.
Uptake Inhibition by Free fMLF Peptide
The specificity of uptake was further investigated using differentiated U937cells incubated with 50 nM PEG-fMLF nanocarriers at 37 °C in the presence of 5 μM free fMLF, which was 100-fold greater than the 50 nM fMLF moiety nanocarrier concentration (Figure 6; the results are the average of three independent experiments performed on three separate days). The presence of 100-fold excess fMLF significantly inhibited the average uptake of PEG-fMLF nanocarriers in differentiated U937 cells. The inhibition led to a relative decrease in the uptake of PEG-fMLF nanocarriers with 1, 2, and 4 copies of fMLF to 31.5%, 14.8%, and 13.5%, respectively. Excess fMLF did not significantly affect the amount of cell-associated PEG5k. Complete inhibition of uptake may not occur because of the fact that there is always some nonspecific binding that occurs, regardless of the concentration of free fMLF that is used (15). Taken together, these results further suggest that PEG-fMLF nanocarrier uptake was mediated by formyl peptide receptors.
In the presence of free f-MLF, all three f-MLF-bearing PEG nanocarriers still show statistically higher uptake into U937 cells than PEG5k (p < 0.05). PEG-fMLF is higher than PEG5k (p = 0.011), and PEG-(fMLF)2 and PEG-(fMLF)4 are higher than PEG-fMLF (p = 0.018), whereas there is no difference between PEG-(fMLF)2 and PEG-(fMLF)4 (p = 0.254). We roughly estimate that the receptors on the cells are saturated with the 5 μM free fMLF and largely occupied by the 50 nM fMLF-equivalents of fMLF-bearing PEG nanocarriers. Despite receptor saturation by free fMLF and high occupancy by all fMLF-bearing PEG nanocarriers, there is still some correlation between fMLF copy number and the amount of uptake. The reason for this is not clear but could have something to do with the PEG moiety of the fMLF-bearing nanocarriers.
Effect of PEG Sizes on PEG-fMLF Nanocarrier Uptake
To evaluate the effect of PEG size on macrophage uptake, PEG-(fMLF)2 prepared using 5 K, 20 K, or 40 K PEGs were incubated with differentiated U937 cells for 4 h at 37 °C (Figure 7). Increasing PEG size from 5 kDa to 20 kDa slightly enhanced the uptake of PEG-(fMLF)2 by about 14.4% (p < 0.05). However, when PEG size increased to 40 kDa, the uptake of PEG-(fMLF)2 decreased by 38.6% (p < 0.05), suggesting that 20 kDa was the optimal size in this group and that 20−40 kDa should be further explored.
Figure 7.
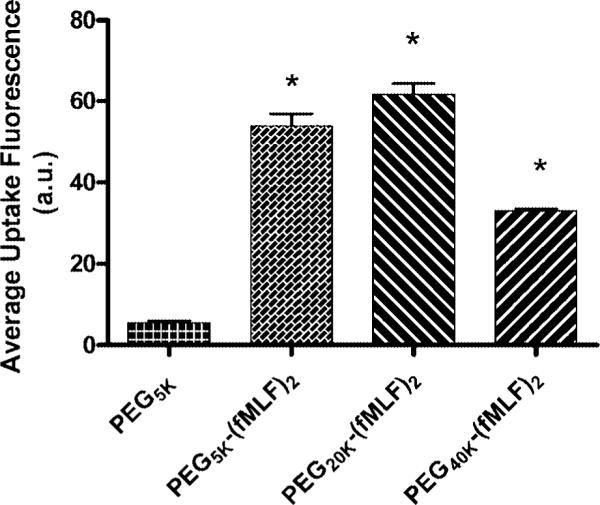
Uptake of fluorescein-labeled PEG-fMLF nanocarriers of different sizes in differentiated U937 cells after 4 h of incubation at 37 °C. The means ± SD for three independent experiments are shown for each value. (*, statistically significant difference between the PEG5k and the derivatized nanocarriers, p < 0.01.)
Peptide-Based PEG Nanocarriers
The PEG-fMLF nano-carriers synthesized from commercially available multifunctional PEGs showed enhanced macrophage uptake. However, the limited ability to produce PEGs with a defined number of cargo moieties, especially in high numbers, limits their use as nanocarriers when multiple targeting or therapeutic moieties are necessary. Thus, a novel peptide–backbone PEG nanocarrier was designed and synthesized to overcome this limitation. The peptide-PEG scaffold comprises the peptide backbone, acetyl-Cys(TP)-[β-Ala-β-Ala-Lys]2,4-amide. PEG is attached to the scaffold via the ε-amine group of lysine. The heterobifunctional PEG (NHS-PEG-VS) is linked to the scaffold by a stable urethane linkage that is the result of the succinimidyl reaction with the ε-amino group of lysine. The targeting moieties are appended to the other end of the PEG. The peptide–backbone acetyl-C-[AAK(PEG5K-(fMLFK(fluorescein)C]2-amide and acetyl-C-[AAK(PEG5K-(fMLFK(fluorescein)C]4-amide demonstrated increased macrophage uptake after 4 h of incubation at 37 °C, about 4.2- and 5.7-fold more than PEG-fMLF (Figure 8), respectively. This is similar to but slightly higher than PEG-(fMLF)2 and PEG-(fMLF)4 derived from the commercially available PEGs. The results indicate that the number of attached targeting groups has much greater impact on the extent of macrophage uptake than other factors such as nanocarrier shape or the geometric distribution of attached groups. Previously, we showed that a linear 30 kDa PEG with 4 conjugated fMLF moieties has a FPR binding constant that is practically indistinguishable from an 8-arm 20 kDa PEG with 4 appended fMLF.
Figure 8.
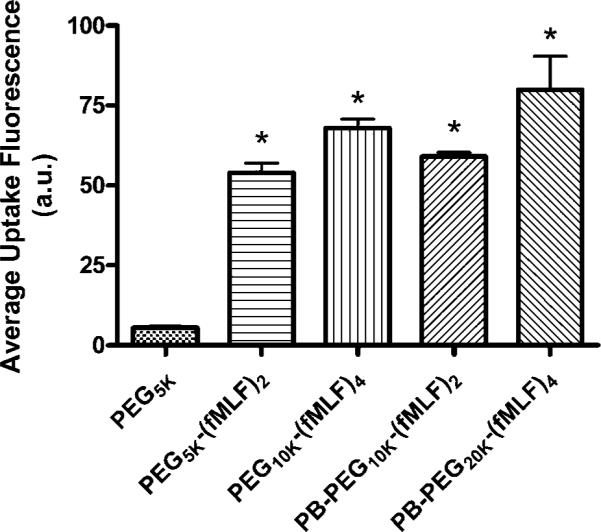
Uptake of fluorescein-labeled PEG-fMLF nanocarriers derived from Nektar PEGs or peptide–backbone (PB-)PEGs in differentiated U937 cells after 4 h of incubation at 37 °C. The means ± SD for three independent experiments are shown for each value. (*, statistically significant difference between the control PEG5k and PEG-fMLF nanocarriers, p < 0.01.)
Fluorescence Microscopy
Cells incubated with the control, fluorescein-labeled PEG5k, lacking fMLF, showed no signal by fluorescence microscopy (Figure 9A). PEG5k-fMLF showed faint fluorescence in cells, suggesting weak uptake (Figure 9B). The presence of punctate fluorescence suggested that the nanocarriers were internalized via endocytosis, which is typical of macromolecular uptake. PEG5k-(fMLF)2 and PEG10k-(fMLF)4 exhibited strong fluorescence throughout the cell, suggesting significantly higher binding/uptake to cells. A representative histogram of flow cytometry results for PEG5k (PEG), PEG5k-fMLF, PEG5k-(fMLF)2, and PEG10k-(fMLF)4 (Figure 9E) confirmed the microscopic observations. Since the same ratio of fluorescein moiety to fMLF moiety was maintained in each nanocarrier particle, and the local fluorescein environment in different particles were similar, the fluorescence intensity per nanocarrier particle was expected to be similar. Therefore, the binding/uptake results could be reasonably compared.
Figure 9.

Fluorescence images of differentiated U937 cells incubated with fluorescein-labeled (A) PEG5k, (B) PEG5k-fMLF, (C) PEG5k-(fMLF)2, and (D) PEG10k-(fMLF)4, all at 50 nM concentrations. (E) Representative histogram of flow cytometry results for PEG5k (PEG), PEG5k-fMLF (PF1), PEG5k-(fMLF)2 (PF2), and PEG10k-(fMLF)4 (PF4).
Calcium Mobilization
All PEG-fMLF nanocarriers showed decreased ability to activate macrophages cells, relative to free fMLF, as measured by transient stimulation of the release of calcium ions from intracellular stores into the cytoplasm. The change of cytoplasmic calcium concentration was monitored by the fluorescent calcium indicator Fluo-4 AM preloaded in the cell cytoplasm. The linkage to PEG decreased calcium release by about 40% in all PEG-fMLF nanocarriers compared to that in the free fMLF peptide (Figure 10). Interestingly, the linkage to PEG not only decreased macrophage activation of fMLF but also delayed the starting time of activation. This time delay and reduced activation by fMLF linked to PEG versus free fMLF was observed in our previous study in differentiated HL-60 cells (15) and may reflect a difference in the molecular pathways for macrophage activation and ligand internalization mediated by the same formyl peptide receptors.
Figure 10.
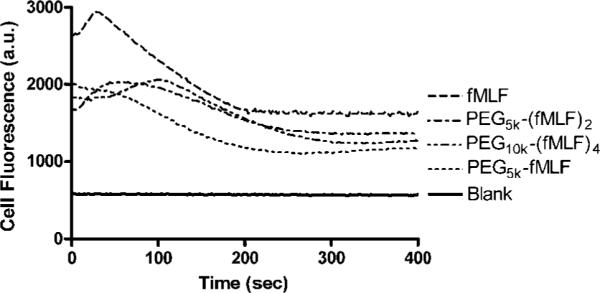
Calcium mobilization of U937 cells after exposure to fMLF and PEG-fMLF nanocarriers. One hundred nanomolars of each compound was tested in differentiated U937 cells preloaded with the calcium indicator Fluo-4 AM, and the calcium flux was monitored in cellular fluorescence at Ex485 nm and Em530 nm for up to 400 s.
DISCUSSION
The clinical potential of anti-HIV agents has been limited by a variety of factors, such as drug toxicity in uninfected cells, and the development of drug resistance, which leads to subtherapeutic drug levels and the formation of viral reservoirs. Better drug delivery and targeting technologies are required to specifically increase target-cell exposure. Therefore, drug-delivery nanocarriers specifically targeted to macrophage cell-surface receptors could potentially improve therapeutic efficacy and minimize the systemic toxicity of anti-HIV drugs.
fMLF was chosen as the targeting moiety for the formyl peptide receptor on macrophages. fMLF resembles the amino terminus of unprocessed eubacterial proteins and binds avidly to the formyl peptide receptors on the surface of macrophages, where it serves as a chemoattractant, triggers cellular activation, and enters the cells to which it binds (18). The potential of using fMLF as a targeting peptide to deliver PEG nanocarriers into macrophages was investigated in this study. The fMLF peptide was chosen as the targeting agent for several reasons. First, formyl peptide receptors are specifically expressed on phagocytic cells, such as macrophages, dendritic cells, and neutrophils. Second, fMLF specifically binds to formyl peptide receptors on macrophages with high affinity. Third, fMLF activates macrophages and down-regulates the coreceptors of HIV entry, CCR5 and CXCR4. The down-regulation of CCR5 coreceptor by fMLF could potentially inhibit viral entry in macrophages, which might potentiate the therapeutic effects of such targeted nanoparticles.
In the current study, multiple copies of fMLF were conjugated to various PEG nanocarriers and evaluated for targeting to human macrophage-like differentiated U937 cells. Since the formyl peptide retains its binding constant for FPR even with the covalent addition of several amino acids to its C-terminus, we were not concerned with a possible reduction in binding activity due to conjugation of the peptide (15, 18). The formyl, methionine and phenylalanine moieties are most crucial for receptor binding (18). Those moieties were specifically left unmodified, but we added a lysine (for fluorescence tagging) and a cysteine for PEG attachment to the C-terminus of the formyl peptide. The number of copies (1, 2, and 4) of fMLFKC conjugated to a single PEG nanocarrier was evaluated in order to determine the effects on avidity and macrophage uptake. Our previous work has shown that increasing the number of fMLF residues (up to eight) attached to a single PEG polymer results in enhanced avidity (Kd = 0.18 nM) for neutrophil-like differentiated HL-60 cells relative to free fMLF (Kd = 28 nM), with less enhancement of activation than of binding by increasing fMLF number (15). In the current study, we show that increasing the number of fMLF residues attached to a single PEG polymer from one to two resulted in significantly enhanced uptake (∼4-fold, p < 0.05) in macrophage-like differentiated U937 cells, while further increasing this number from two to four only resulted in a modest increase in uptake. While the molecular basis of enhanced avidity by multivalency (i.e., multiple ligands attached to a single polymeric carrier molecule) is not proven, it is likely due to multimeric ligands interacting with multiple cellular receptors.
PEG was chosen as the pharmaceutical nanocarrier for the macrophage-targeted drug delivery system because of our long experience with this polymer class. PEGylation, the process of attaching polyethylene glycol (PEG) to drug molecules, has been a highly successful strategy for improving the pharmacokinetics and pharmacodynamics of pharmaceuticals, especially for protein and peptide therapeutics (19-25). In previous reports from our laboratory, we have shown that multicomponent macromolecular conjugates consisting of PEG, saquinavir, and R.I.CK-Tat retained antiviral activity in vitro and showed favorable therapeutic indices (26, 27). Studies of PEG in solution showed that each ethylene glycol subunit is tightly associated with two or three water molecules. The binding of water to PEG makes PEGylated compounds physically behave as though they are 3 to 9 times larger than a corresponding soluble protein of similar molecular weight (28, 29). In drug conjugates, PEG polymers with associated water molecules act like a shield to protect the attached drug from enzymatic degradation, inhibit interactions with cell-surface proteins, and provide increased size to prevent rapid renal filtration and clearance. These stealth properties of PEG have proven to be very valuable in prolonging drug persistence in the systemic circulation. However, the inability of PEG to interact with cell surfaces theoretically limits its use as a biomaterial for cell-surface or intracellular drug delivery and targeting. Therefore, the very property of PEGylation that has made it clinically useful and commercially successful is exactly the opposite of what is needed for promoting effective targeted drug therapy in diseases such as AIDS. In other words, currently used PEGylation approaches that have been enormously successful for treating certain diseases such as cancer may not be appropriate for treating HIV infection in viral reservoirs or sanctuary sites, such as macrophages, the central nervous system, and the testis. Our approach to functionalize PEG in order to shift the site of drug delivery from the blood to target cells is novel and counterintuitive given the current use of PEG. However, the general stealth properties of the PEG moiety may reduce toxicity by reducing nanocarrier binding to cells not expressing FPR and FPRL1.
Experimental studies on targeted drug delivery into cells have identified size as an important factor in the cellular uptake of nanomaterials. It has been shown that particles with radii <50 nm exhibit significantly greater uptake compared with particles >50 nm (30, 31). Aoyama and co-workers (32, 33) reported on the effect of size on receptor-mediated endocytosis in glycoviral gene delivery by excluding potential complications arising from charge effects. They concluded that receptor-mediated endocytosis is strongly size-dependent and that the optimal size is around 25 nm. In our study, increasing PEG size from 5 K to 20 K slightly enhanced nanocarrier uptake, but uptake decreased when the size was further increased to 40 K. The estimated molecular size of a 5 kDa PEG molecule is ∼2 nm, while that of a 20 kDa PEG is 7 nm and 10 nm for 40 kDa (34). The effective or apparent size of PEG nanocarriers is significantly increased by the binding of water to PEG; the hydrodynamic radius of the hydrated PEG nanocarrier is 3 to 9 times higher than that predicted by their molecular weight (34). Therefore, a 20 kDa PEG-(fMLF)2 has an effective molecular size of 20−60 nm. The optimal uptake of this nanocarrier and its size are consistent with the suggestions of Aoyama et al. However, 5 kDa and 40 kDa PEG-(fMLF)2 showed less uptake because their effective sizes are either <25nm or >50 nm. Effective targeting to macrophages residing in tissues depends on multiple factors, including plasma clearance, tissue distribution, receptor binding, and cell uptake. The pharmacokinetics and biodistribution of nanocarriers relate to their size, charge, shape, and rigidity and are primarily controlled by renal glomerular filtration (35). The current results suggest that a delicate balance must be struck between maximizing the circulation half-life of nanocarriers in blood and their penetration into macrophage tissue compartments.
PEGylated systems made from commercially available multifunctional PEGs have a linear or forked shape. However, they have limited capacity to conjugate mulutiple copies of functional groups or drugs. These limitations have affected their potential as pharmaceutical drug carriers. Therefore, we designed and synthesized a prototypical novel peptide-based PEG nanocarrier. The central component of the proposed nanocarriers is the peptide-based PEG scaffold that comprised the peptide backbone, Cys-(β-Ala-β-Ala-Lys-)n-amide (n = 2, 4, or more), with 1 thiol group on the cysteine side chain and amino groups on lysine side chains. The flexibility of this peptide-based PEG nanocarrier is evident. For example, if a higher number of targeting agents is needed, a larger scaffold with more lysine moieties for PEG attachment can be utilized. Drugs can be attached by a disulfide (reversible) or thiosulfonyl (nonreversible) via a vinyl sulfone bond. The disulfide linkage can possibly be cleaved after the entry of the nanocarrier into the cell because of the high concentration of reduced glutathione within the cell. In this scenario, the drug is released from the nanocarrier. The scaffold consisting of a peptide backbone and PEG are the critical central feature of the nanocarrier drug conjugates since (1) it controls body and cellular disposition, and uptake/retention, (2) it allows us to add functional groups (i.e., effectors) to target specific cell types and cause surface binding or uptake, and (3) it allows us to attach exact numbers of multiple drugs or multiple targeting moieties using releasable or nonreleasable bonds. Furthermore, the molecular population of nanocarriers is mono-disperse in its derivatization, a distinct advantage over other PEGylation carriers. Using these features, we could potentially precisely control how, when, and where anti-HIV drugs are able to exert their therapeutic effect.
CONCLUSIONS
The current results demonstrate that PEG nanocarriers with multiple copies of fMLF can specifically target human macrophage-like cells in vitro and that the number of fMLF copies attached determines the extent of uptake. The results suggest that two fMLF residues are sufficient for achieving targeting and improving uptake in vitro. We also demonstrated that 20 kDa (corresponding to 20−60 nm) may be the optimal PEG nanocarrier size for improved macrophage uptake. Since typical commercially available multifunctional PEGs limit the numbers of copies of drugs and targeting groups that may be attached to a nanocarrier, we designed an alternate carrier that retained the targeting and uptake properties of nonpeptide-based PEGs. However, the new nanocarrier allows for far greater flexibility in attaching multiple drugs, copies of drugs, and/or targeting moieties. These results indicate great promise for improving targeted drug delivery to HIV-infected macrophages, which may eventually allow for better therapeutic regimens by reducing dose and improving tolerability and patient compliance with complicated regimens.
ACKNOWLEDGMENT
This work was supported by National Institutes of Health Grants AI 33789 and AI 51214.
LITERATURE CITED
- 1.Vandamme AM, Van Vaerenbergh K, De Clercq E. Anti-human immunodeficiency virus drug combination strategies. AntiViral Chem. Chemother. 1998;9:187–203. doi: 10.1177/095632029800900301. [DOI] [PubMed] [Google Scholar]
- 2.Bangsberg DR, Hecht FM, Charlebois ED, Zolopa AR, Holodniy M, Sheiner L, Bamberger JD, Chesney MA, Moss A. Adherence to protease inhibitors, HIV-1 viral load, and development of drug resistance in an indigent population. AIDS. 2000;14:357–66. doi: 10.1097/00002030-200003100-00008. [DOI] [PubMed] [Google Scholar]
- 3.Schrager LK, D'Souza MP. Cellular and anatomical reservoirs of HIV-1 in patients receiving potent antiretroviral combination therapy. JAMA. 1998;280:67–71. doi: 10.1001/jama.280.1.67. [DOI] [PubMed] [Google Scholar]
- 4.Richman DD. HIV chemotherapy. Nature. 2001;410:995–1001. doi: 10.1038/35073673. [DOI] [PubMed] [Google Scholar]
- 5.Schuitemaker H, Kootstra NA, de Goede RE, de Wolf F, Miedema F, Tersmette M. Monocytotropic human immunodeficiency virus type 1 (HIV-1) variants detectable in all stages of HIV-1 infection lack T-cell line tropism and syncytium-inducing ability in primary T-cell culture. J. Virol. 1991;65:356–363. doi: 10.1128/jvi.65.1.356-363.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roos MT, Lange JM, de Goede RE, Coutinho RA, Schellekens PT, Miedema F, Tersmette M. Viral phenotype and immune response in primary human immunodeficiency virus type 1 infection. J. Infect. Dis. 1992;165:427–432. doi: 10.1093/infdis/165.3.427. [DOI] [PubMed] [Google Scholar]
- 7.Connor RI, Paxton WA, Sheridan KE, Koup RA. Macrophages and CD4+ T lymphocytes from two multiply exposed, uninfected individuals resist infection with primary non-syncytium-inducing isolates of human immunodeficiency virus type 1. J. Virol. 1996;70:8758–8764. doi: 10.1128/jvi.70.12.8758-8764.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R, MacDonald ME, Stuhlmann H, Koup RA, Landau NR. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell. 1996;86:367–377. doi: 10.1016/s0092-8674(00)80110-5. [DOI] [PubMed] [Google Scholar]
- 9.Aquaro S, Calio R, Balzarini J, Bellocchi MC, Garaci E, Perno CF. Macrophages and HIV infection: therapeutical approaches toward this strategic virus reservoir. Antiviral Res. 2002;55:209. doi: 10.1016/s0166-3542(02)00052-9. [DOI] [PubMed] [Google Scholar]
- 10.Fridkin M, Tsubery H, Tzehoval E, Vonsover A, Biondi L, Filira F, Rocchi R. Tuftsin-AZT conjugate: potential macrophage targeting for AIDS therapy. J Pept Sci. 2005;11:37–44. doi: 10.1002/psc.587. [DOI] [PubMed] [Google Scholar]
- 11.Hu J, Liu H, Wang L. Enhanced delivery of AZT to macrophages via acetylated LDL. J. Controlled Release. 2000;69:327–335. doi: 10.1016/s0168-3659(00)00319-9. [DOI] [PubMed] [Google Scholar]
- 12.Le Y, Yang Y, Cui Y, Yazawa H, Gong W, Qiu C, Wang JM. Receptors for chemotactic formyl peptides as pharmacological targets. Int. Immunopharmacol. 2002;2:1–13. doi: 10.1016/s1567-5769(01)00150-3. [DOI] [PubMed] [Google Scholar]
- 13.Niedel JE, Kahane I, Cuatrecasas P. Receptor-mediated internalization of fluorescent chemotactic peptide by human neutrophils. Science. 1979;205:1412–1414. doi: 10.1126/science.472759. [DOI] [PubMed] [Google Scholar]
- 14.van Eerd JE, Boerman OC, Corstens FH, Oyen WJ. Radiolabeled chemotactic cytokines: new agents for scintigraphic imaging of infection and inflammation. Q. J. Nucl. Med. 2003;47:246–255. [PubMed] [Google Scholar]
- 15.Pooyan S, Qiu B, Chan MM, Fong D, Sinko PJ, Leibowitz MJ, Stein S. Conjugates bearing multiple formyl-methionyl peptides display enhanced binding to but not activation of phagocytic cells. Bioconjugate Chem. 2002;13:216–223. doi: 10.1021/bc0100657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischer DG, Pike MC, Koren HS, Snyderman R. Chemotactically responsive and nonresponsive forms of a continuous human monocyte cell line. J. Immunol. 1980;125:463–465. [PubMed] [Google Scholar]
- 17.Pike MC, Fischer DG, Koren HS, Snyderman R. Development of specific receptors for N-formylated chemotactic peptides in a human monocyte cell line stimulated with lymphokines. J. Exp. Med. 1980;152:31–40. doi: 10.1084/jem.152.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Prossnitz ER, Ye RD. The N-formyl peptide receptor: a model for the study of chemoattractant receptor structure and function. Pharmacol. Ther. 1997;74:73–102. doi: 10.1016/s0163-7258(96)00203-3. [DOI] [PubMed] [Google Scholar]
- 19.Kozlowski A, Harris JM. Improvements in protein PEGylation: pegylated interferons for treatment of hepatitis C. J. Controlled Release. 2001;72:217–224. doi: 10.1016/s0168-3659(01)00277-2. [DOI] [PubMed] [Google Scholar]
- 20.Harris JM, Martin NE, Modi M. Pegylation: a novel process for modifying pharmacokinetics. Clin. Pharmacokinet. 2001;40:539–551. doi: 10.2165/00003088-200140070-00005. [DOI] [PubMed] [Google Scholar]
- 21.Kozlowski A, Charles SA, Harris JM. Development of pegylated interferons for the treatment of chronic hepatitis C. BioDrugs. 2001;15:419–429. doi: 10.2165/00063030-200115070-00001. [DOI] [PubMed] [Google Scholar]
- 22.Conover CD, Greenwald RB, Pendri A, Gilbert CW, Shum KL. Camptothecin delivery systems: enhanced efficacy and tumor accumulation of camptothecin following its conjugation to polyethylene glycol via a glycine linker. Cancer Chemother. Pharmacol. 1998;42:407–414. doi: 10.1007/s002800050837. [DOI] [PubMed] [Google Scholar]
- 23.Greenwald RB. PEG drugs: an overview. J. Controlled Release. 2001;74:159–71. doi: 10.1016/s0168-3659(01)00331-5. [DOI] [PubMed] [Google Scholar]
- 24.Greenwald RB, Choe YH, McGuire J, Conover CD. Effective drug delivery by PEGylated drug conjugates. Adv. Drug Delivery Rev. 2003;55:217–250. doi: 10.1016/s0169-409x(02)00180-1. [DOI] [PubMed] [Google Scholar]
- 25.Roberts MJ, Bentley MD, Harris JM. Chemistry for peptide and protein PEGylation. Adv. Drug Delivery Rev. 2002;54:459–476. doi: 10.1016/s0169-409x(02)00022-4. [DOI] [PubMed] [Google Scholar]
- 26.Gunaseelan S, Debrah O, Wan L, Leibowitz MJ, Rabson AB, Stein S, Sinko PJ. Synthesis of poly(ethylene glycol)-based saquinavir prodrug conjugates and assessment of release and anti-HIV-1 bioactivity using a novel protease inhibition assay. Bioconjugate Chem. 2004;15:1322–1333. doi: 10.1021/bc0498875. [DOI] [PubMed] [Google Scholar]
- 27.Wan L, Zhang X, Gunaseelan S, Pooyan S, Debrah O, Leibowitz MJ, Rabson AB, Stein S, Sinko PJ. Novel multi-component nanopharmaceuticals derived from poly-(ethylene) glycol, retro-inverso-Tat nonapeptide and saquinavir demonstrate combined anti-HIV effects. AIDS Res. Ther. 2006;3:12. doi: 10.1186/1742-6405-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harris JM. Poly(Ethylene Glycol) Chemistry: Biotechnical and Biomedical Applications. Plenum Press; New York: 1991. [Google Scholar]
- 29.Sherman MR, Williams LD, Saifer MCP, French JA, Kwak LW, Oppenheim JJ. Conjugation of High, Molecular Weight Poly(ethylene glycol) to Cytokines: Granulocyte-Macrophage Colony Stimulating Factors As Model Substrates. ACS; Washington, DC: 1997. [Google Scholar]
- 30.Desai MP, Labhasetwar V, Walter E, Levy RJ, Amidon GL. The mechanism of uptake of biodegradable microparticles in Caco-2 cells is size dependent. Pharm. Res. 1997;14:1568–1573. doi: 10.1023/a:1012126301290. [DOI] [PubMed] [Google Scholar]
- 31.Prabha S, Zhou WZ, Panyam J, Labhasetwar V. Size-dependency of nanoparticle-mediated gene transfection: studies with fractionated nanoparticles. Int. J. Pharm. 2002;244:105–115. doi: 10.1016/s0378-5173(02)00315-0. [DOI] [PubMed] [Google Scholar]
- 32.Nakai T, Kanamori T, Sando S, Aoyama Y. Remarkably size-regulated cell invasion by artificial viruses. Saccharide-dependent self-aggregation of glycoviruses and its consequences in glycoviral gene delivery. J. Am. Chem. Soc. 2003;125:8465–8475. doi: 10.1021/ja035636f. [DOI] [PubMed] [Google Scholar]
- 33.Osaki F, Kanamori T, Sando S, Sera T, Aoyama Y. A quantum dot conjugated sugar ball and its cellular uptake. On the size effects of endocytosis in the subviral region. J. Am. Chem. Soc. 2004;126:6520–6521. doi: 10.1021/ja048792a. [DOI] [PubMed] [Google Scholar]
- 34.Caliceti P, Veronese FM. Pharmacokinetic and biodistribution properties of poly(ethylene glycol)-protein conjugates. Adv. Drug Delivery Rev. 2003;55:1261–1277. doi: 10.1016/s0169-409x(03)00108-x. [DOI] [PubMed] [Google Scholar]
- 35.Arendshorst WJ, Navar LG. Renal Circulation and Glomerular Hemodynamics. Brown; Boston, MA: 1988. [Google Scholar]