

Abstract
We use finite-difference time-domain calculations to show that aluminum nanoparticles are efficient substrates for metal-enhanced fluorescence (MEF) in the ultraviolet (UV) for the label-free detection of biomolecules. The radiated power enhancement of the fluorophores in proximity to aluminum nanoparticles is strongly dependent on the nanoparticle size, fluorophore-nanoparticle spacing, and fluorophore orientation. Additionally, the enhancement is dramatically increased when the fluorophore is between two aluminum nanoparticles of a dimer. Finally, we present experimental evidence that functionalized forms of amino acids tryptophan and tyrosine exhibit MEF when spin-coated onto aluminum nanostructures.
Fluorescence is widely used in biology and medicine. However, low radiative emission rates limit the use of the intrinsic fluorescence of biomolecules, resulting in the need for external chemical labeling. The use of external labels requires chemical modification and additional steps which can perturb the functionality of ligand–receptor interactions. In many cases, selective fluorescence labeling of a small number of molecules in a tiny volume like a single cell is cumbersome, adding expense and complexity to the analysis.1–5
Because of the problems noted above, there is interest in label-free detection methods6–8 including surface plasmon resonance9,10 and Raman scattering.11–13 Direct measurement of native fluorescence by proteins is also being pursued.14–18 Proteins exhibit intrinsic absorption maxima in the ultraviolet (UV) around 280 nm.19 We20–23 and others24–26 have been investigating metallic nanostructures for improved fluorescence detection. Metallic structures can substantially modify spontaneous emission rates and the directionality of the emission, leading to metal-enhanced fluorescence (MEF). In the case of a flat metal film on a glass substrate, emission from a fluorophore near the film can excite surface plasmon polaritons on the film, which radiate back into the glass in a highly directional manner, a process termed surface plasmon-coupled emission (SPCE).
At present MEF and SPCE are obtained with mostly silver structures,20,22,23,27–29 with occasional use of gold,30,31 and with relatively little attention being given to other metals such as aluminum. However, aluminum has low absorption at wavelengths ≤400 nm, and a recent article demonstrated that aluminum nanodisks have distinct plasmon resonances that extend into the UV.32 Aluminum nanostructures have also been used for surface enhanced Raman spectroscopy (SERS).33 These facts suggest that aluminum can potentially be used as a substrate for MEF specifically in the UV. In fact we successfully demonstrated SPCE on flat aluminum films34 and MEF on roughened aluminum surfaces21 using fluorophores with fluorescence maxima in the upper UV–visible limit (370–450 nm).
Motivated by the above results, we use finite-difference time domain (FDTD) calculations35–39 to explore enhancing the intrinsic fluorescence of proteins with aluminum nanoparticles. The FDTD method is a rigorous computational electrodynamics method38 that can accurately describe plasmonic effects. We also present experimental results for films containing functionalized forms of the amino acids tryptophan and tyrosine that are spin-coated on a particulate aluminum film. We find significant increases in fluorescence intensity and decreases in lifetime compared to results without the aluminum film.
MATERIALS AND METHODS
FDTD Computational Details
We assume the excitation stage has occurred and consider fluorophore emission as resulting from a radiating dipole source. Three-dimensional FDTD simulations are performed with FDTD Solutions from Lumerical Solutions, Inc. (Vancouver, Canada).35–39 Further details are in the Supporting Information (Figure S-1) and in previous work on MEF in silver nanoparticles.35,36 The FDTD program employs auxiliary differential equations37,39 to implement a realistic frequency-dependent, lossy dielectric model for aluminum (Figure 1a, inset). We calculate the total radiated power enhancement as Prad/P0, where Prad is the integral of the Poynting vector over a surface enclosing the fluorophore and metal nanoparticle(s), and P0 is the result of this integral with only the fluorophore present.35,36,40 This enhancement can be equated with , where γrad is the radiative decay rate of the dipole in proximity of the metal nanoparticle(s) and is the radiative decay rate of an isolated dipole (in air):40
Figure 1.

Results for a d = 20 nm aluminum nanoparticle. (a) Extinction, scattering, and absorption efficiencies. Inset: complex dielectric constant of aluminum. (b) Radiated power enhancement for dipoles spaced s = 5 and 10 nm from the nanoparticle calculated with the FDTD method (solid curves), and with analytical theory (dashed curves). The dipoles are oriented perpendicular to the metal surface.
| (1) |
Equation 1 implies that an enhancement in the total radiated power is indicative of a corresponding increase in the relative radiative decay rate of the system.
Experimental Details
Samples and experimental procedures are discussed in detail in the Supporting Information.
RESULTS AND DISCUSSION
Figures 1 and 2 give results for single aluminum nanoparticles with diameters d = 20 and 80 nm respectively. Figures S-2 and S-3 (Supporting Information) give results for diameters d = 40 and 100 nm, respectively. In these figures, the upper panel has extinction, scattering, and absorption efficiencies [optical cross sections normalized by π(d/2)2], and the lower panels contain radiated power enhancements when a fluorophore (radiating dipole) is placed near the nanoparticle. Two different fluorophore-metal surface distances, s = 5 and 10 nm are considered.
Figure 2.

Results for a d = 80 nm aluminum nanoparticle: (a) Extinction, scattering, and absorption efficiencies; (b) radiated power enhancement for dipoles spaced s = 5 and 10 nm from the nanoparticle. The dipole is oriented perpendicular to the metal surface; (c) similar to (b) but now the dipoles are oriented parallel to the metal surface.
For a d = 20 nm aluminum nanoparticle, the extinction peaks are at ≈150 nm (Figure 1a). In contrast, similar sized silver nanoparticles have plasmon resonances typically in the 350–375 nm region. The differing plasmon resonances of aluminum and silver are consistent with the small particle surface plasmon resonance condition41 of ε(γSP) = −2, where ε is the metallic dielectric constant. For aluminum we find λSP ≈ 150 nm, and for silver λSP ≈ 354 nm. The extinction for this particle size is dominated by absorption. Figure 1b shows the radiated power enhancement for a fluorophore placed at s = 5 and 10 nm from the nanoparticle. The fluorophores are oriented perpendicular to the aluminum surface. (We define fluorophore orientation to be the oscillation direction of the dipole.) The enhancement in the radiated power peaks at ≈155 nm and parallels the form of the optical cross sections (Figure 1a) indicating the role of surface plasmons. This enhancement is expected because in this orientation the fluorophore’s dipole induces a dipole in the aluminum nanoparticle such that the dipoles align head-to-tail, leading to a larger effective dipole than that for an isolated fluorophore. Note also that the degree of enhancement depends on the fluorophore-metal distance with s = 5 nm showing significantly more enhancement than s = 10 nm.
The problem of a radiating point dipole and a metal sphere can also be solved analytically. Figure 1b includes (dashed curves) our implementation of the exact radiated power enhancement calculated using eq 27 of ref 42. The agreement with the FDTD results is very good, giving us confidence in our FDTD results for systems without exact analytical solutions such as the dimer system. We also considered simpler, more approximate analytical forms for the radiated power enhancement.43–45 A quasistatic (d ≪ λ) limit44,45 expression, eq 6 of ref 43, is qualitatively correct but leads to peak positions that are blue-shifted by ≈10 nm relative to the exact positions and peak heights that are 1.5–2 times larger than the exact ones. An improved version45 of a quasistatic limit model due to Gersten and Nitzan44 is much better but still underestimates by 15–20% the peak enhancement and all enhancements on its blue side.
The radiated power enhancements in Figure 1b show long-wavelength limits of ≈1.6 for the s = 10 nm case and ≈2.6 for the s = 5 nm, that is, the enhancement factor does not approach unity as might be naively expected. In this long-wavelength limit, the metal behaves as a perfect electrical conductor (PEC). PECs do not to support surface plasmons. Nonetheless some enhancement via near-field interactions can still occur. Indeed, we carried out calculations assuming the metal is a PEC rather than aluminum and obtained enhancements for λ > 500 nm that are very close to the aluminum results of Figure 1b. However, the PEC enhancement factors rise only slightly with decreasing wavelength and reach values of just 2–3 in the region where the plasmon resonance dominates the aluminum results of Figure 1b.
The d = 40 nm case is discussed in the Supporting Information (Figure S-2). For the d = 80 nm nanoparticles, the dipolar extinction peak is further red-shifted relative to the d = 40 nm particle to ≈250 nm (Figure 2a). Higher order peaks at ≈170 and 140 nm are also observed. The extinction for this particle size is more dominated by the scattering, although there is a small absorptive component. Figure 2b shows the radiated power enhancement for fluorophores oriented perpendicular to the metal surface. The radiated power enhancements, like the previous cases, have features in common with the optical spectra, although with some red-shifting. The enhancement peaks at ≈310 nm, a 60 nm red shift from the corresponding extinction peak. As with the smaller nanoparticle cases, the s = 5 nm fluorophore-surfce spacing shows significantly more enhancement than s = 10 nm and can show sharper higher order mode features.
Figure 2c shows the radiated power enhancements for a dipole oriented parallel to the metal surface with spacings s = 5 and 10 nm from the d = 80 nm metal surface. The dashed line in Figure 2c is the boundary between fluorescence enhancement and quenching. Quenching clearly dominates aside from a small region between λ = 100–175 nm where there are modest enhancements. In the parallel orientation, the fluorophore’s dipole induces a dipole in the aluminum nanoparticle of the opposite polarity. This causes the dipoles to counteract each other, leading to a smaller effective radiating dipole than in case of the isolated dipole. These results also indicate that for most wavelengths the parallel dipole orientation is not conducive for fluorescence emission enhancements with aluminum nanoparticles.
See Figure S-3 (Supporting Information) for a discussion of d = 100 nm aluminum nanoparticles. In Figures 1, 2, S-2 and S-3, we observe a progressive red-shifting of the radiated power enhancement peak as the nanoparticles become larger. This correlates with the scattering cross sections of the nanoparticles which also show a distinct red-shifting with larger nanoparticles.
Figure S-4a–d in the Supporting Information provides a discussion of the relationship between the wavelength of the extinction maximum peak and the wavelength of the radiated power peak for the various aluminum nanoparticle sizes studied. Our calculations also reveal that for emission wavelengths between 300–350 nm (the primary emission region for amino acids tryptophan and tyrosine, and all DNA bases), aluminum is a much more efficient MEF substrate than silver [Figure S-5a–d and discussion in the Supporting Information].
The wavelength region 300–420 nm is typical for intrinsic fluorescence in biomolecules. A unitless measure of enhancement in this region is the integral of the radiated power enhancement over the region and dividing by the integration range of 120 nm (this ratio is = 1 for the case of an isolated dipole, so any such ratio >1 for a dipole-aluminum nanoparticle system represents an enhancement and vice-versa). We use this enhancement measure to ascertain the effect of nanoparticle size, Figure 3. For these calculations, the dipole-aluminum distance is kept constant at s = 5 nm. Three dipole orientations are represented: (i) perpendicular (P); (ii) parallel (L); (iii) orientation averaged = (P + 2L)/3. Two important trends are as follows: (a) There is a clear dependence of the enhancement with particle size, with d = 80 nm giving the maximum enhancement measure of ≈ 12.5; (b) The perpendicular orientation always leads to enhancement whereas the parallel orientation leads to quenching.
Figure 3.
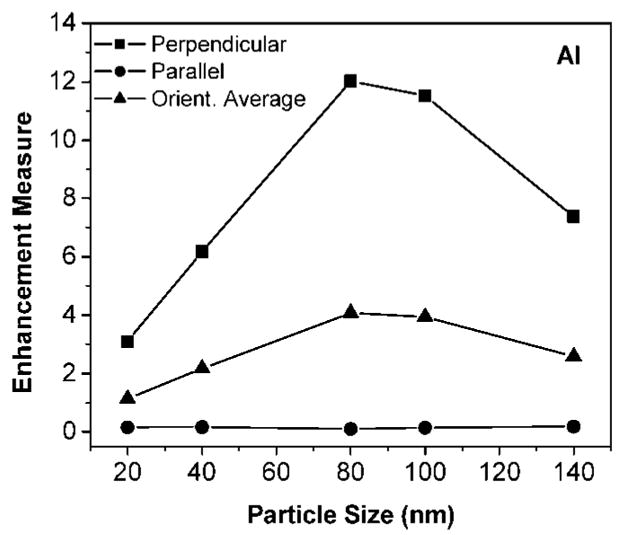
Enhancement measure for emission in 300–420 nm (see text) as a function of aluminum nanoparticle size (d = 20–140 nm). Three dipole orientations are represented: (a) Perpendicular (P); (b) Parallel (L); and Orientation Averaged = (P + 2L)/3.
Figure 4a shows the effect of the fluorophore-metal distance on enhancement for the d = 80 nm aluminum nanoparticle case. Like Figure 3, we consider perpendicular, parallel, and orientationally averaged dipoles and use the same enhancement measure associated with the 300–420 emission region. The largest enhancement measure, ≈15, is obtained for the perpendicular orientation, at the shortest metal-fluorophore distance of 1 nm. This may be understood in terms of the head-to-tail dipole alignment argument concerning Figure 1b and the fact that as s increases the magnitude of the dipole induced in the metal decreases. However, for the parallel case, we see quenching of the radiated power for all metal-fluorophore separations, with the extent of quenching the greatest at s = 1 nm. The unfavorable dipole alignment argument concerning Figure 2c applies here, along with the fact that as s increases, the induced dipole in the metal is weaker and so cancels the fluorophore dipole a little less effectively.
Figure 4.

(a) Enhancement measure for emission in 300–420 nm at various distances (s = 1–20 nm) from the surface of a d = 80 nm aluminum nanoparticle. Three dipole orientations are considered, as in the caption for Figure 3. (b) Enhancement measure for a dipole located in the middle of an 80 nm aluminum nanoparticle dimer with various surface-surface particle spacings (2s = 2–40 nm). Inset: Radiated power enhancement of a perpendicularly oriented dipoledimer system with 2s = 4 nm.
We consider the radiated power enhancement for an 80 nm aluminum nanoparticle dimer system in Figure 4b. The fluorophore is placed between the nanoparticles on the dimer axis. The surface-surface-spacing between particles (Figure S-1, Supporting Information) are in the 2s = 2–40 nm range. Figure 4b shows that for the perpendicular oriented fluorophore: (a) the dimer creates a significant increase in radiated power enhancement, up to ≈3500 with a surface-surface spacing of 2 nm; (b) the degree of enhancement for the dimer decreases with increasing dimer spacing; (c) each dimer system yields a greater enhancement than its corresponding monomer system. For example, dimer particles spaced by 2 nm give larger radiated power enhancements than a single nanoparticle spaced 1 nm from the dipole. This is because the fluorophore’s dipole now induces two dipoles, one in each of the metal nanoparticles. All three dipoles align head-to-tail, leading to a much larger effective radiating dipole. For the parallel oriented dipoles, Figure 4b shows the following: (a) the dimer creates significant quenching of radiated power compared to an isolated dipole, with up over 200-fold quenching for surface-surface spacing of 2 nm; (b) the degree of quenching in the dimers decreases with increasing dimer particle spacing; (c) each of the dimer shows significantly higher quenching of the radiated power than its corresponding monomer system. The large quenching in case of the dimers with parallel dipole orientation is explained by the fluorophore’s dipole inducing two dipoles of the opposite polarity, one in each of the aluminum nanoparticles. This causes the dipoles to strongly counteract each other, thus leading to a much smaller effective radiating dipole than in the single nanoparticle case. See also Table S-1 (Supporting Information). These results are similar to those for silver nanoparticles in the visible range.36 Figure 4b, inset, shows the wavelength-dependent radiation power enhancement of a d = 80 nm aluminum nanoparticle dimer with particles spaced 4 nm apart. The fluorophore is oriented perpendicular and located in the middle of the dimer axis. The major enhancement peak is at ≈405 nm, with higher mode peaks at ≈210 and 170 nm. The enhancements in this case are larger than those in similar figures for single aluminum nanoparticles (Figures 1b, 2b, S-2b, and S-3b).
Figure 5 shows a comparison of the radiated power enhancement for all the 80 nm aluminum particle monomer and dimer systems studied with the perpendicular dipole orientation. It clearly reveals the higher enhancement factors that occur with the dimer systems when compared with single nanoparticles.
Figure 5.
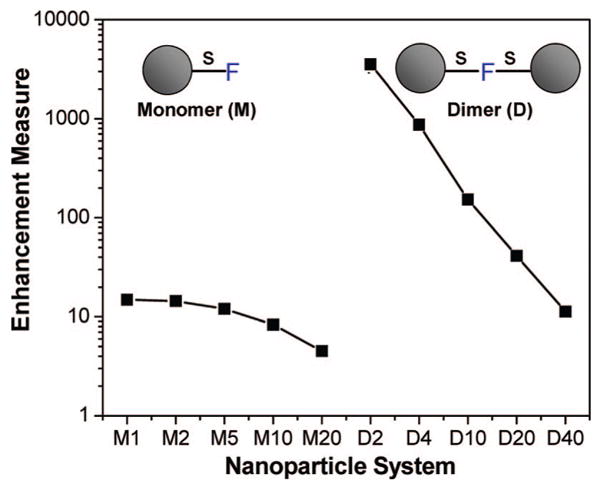
Radiated power enhancement of d = 80 nm aluminum nanoparticle systems with perpendicular fluorophore orientation. The horizontal axis denotes the system where “M” denotes an isolated monomer and “D” denotes a dimer. The number following “M” is the fluorophore-surface spacing, s (nm), and the number after “D” is the surface-surface distance, 2s, between the particles (nm). The fluorophore is at the midpoint of the dimer.
It is interesting to examine the electromagnetic near-field distributions around the aluminum nanoparticles that are created by both excitation light as well as excited-state fluorophores. These fields provide insight into the nature of metal enhanced fluorescence that is interesting from the perspective of applications involving molecular spectroscopy and designing specific fluorophore-metal nanoparticle systems. A description of the effect of the 280 nm excitation plane wave on the near-fields around an 80 nm aluminum nanoparticle is in the Supporting Information (Figure S-6). A wavelength of 280 nm is typical for the excitation of protein fluorescence. All the near-field calculations shown are performed along a single plane, that is, the x-y plane running through the center of the dipole and/or aluminum nanoparticles. Figure S-7 also presents the effect of an excited-state fluorophore emitting at 350 nm on the near-fields around an 80 nm aluminum nanoparticle. To obtain the wavelength-resolved result we keep the fluorophore or dipole oscillating at a fixed frequency corresponding to 350 nm throughout the entire simulation time, and construct a time average of the square of the electric field vector over the last period of evolution. Figures 6a–c show respectively the near-field intensity in the x-y plane around an isolated fluorophore, the near-fields around a d = 80 nm aluminum nanoparticle dimer system with a surface-to-surface distance of 4 nm and a perpendicularly oriented dipole located halfway between the particles, and the near-field enhancement and quenching image that was calculated in an identical manner to Figure S-7c. From Figure 6c we see that the near-field is not enhanced in the gap between the particles, but there are intense field enhancements around all other areas of the particles. The intense near-field enhancement also extends tens of nanometers from the edge of the particles into the free space as observed by the extent of the red areas in the image. The near-field enhancements of Figure 6c are much greater than those of the single aluminum nanoparticle (Figure S-7c). The dark red regions of Figure 6c represent more than an order of magnitude greater enhancements than those of Figure S-7c. It is important to note that the near-fields calculated in Figures 6 and S-7 do not necessarily represent propagating radiation. They could either be propagating fields or localized evanescent fields that are non-propagating.
Figure 6.

Near-field intensities arising from a point dipole radiating at 350 nm: (a) Isolated dipole; (b) Dipole between two 80 nm diameter aluminum nanoparticles with surface-surface particle spacing 2s = 4 nm; (c) Near-field enhancement/quenching obtained by dividing Figure 6b by Figure 6a. Images are on a log scale, and the dipole is oscillating along the x-axis, which corresponds to the perpendicular orientation described in the text.
In Figure 6c, there is only an extremely small area between the two aluminum nanoparticles that shows quenching. The overwhelmingly large portion of the image is bright or dark red which depicts significant near-field enhancements in the region immediately surrounding the dimer which is induced by the excited fluorophore. These near-field enhancements can eventually lead to enhancements in the far-field propagating emission, for example, Figure 5. Hence we see that nanoparticle systems displaying large enhancements in the radiated power also show very strong enhancements in the near-fields. Comparing the near-field distributions around an isolated fluorophore (Figure 6a) and a fluorophore in between two aluminum particles (Figure 6b) suggests a possible mechanism for MEF. Since the intensity of the excited-state fluorophore is actually decreased when it is in between the aluminum nanoparticles (Figure 6c), it suggests that the fluorophore alone is not the entity that is responsible for the enhanced emission. Rather it is the fluorophore coupled with the nearby aluminum nanoparticles, behaving as a single radiating entity, that is the source of the enhanced fluorescence signals. A similar effect is seen in the case of a flurophore in proximity to a single aluminum nanoparticle [Figure S-7c, Supporting Information].
We also performed experiments to corroborate the theoretical predictions above concerning the efficiency of aluminum for MEF-based label-free biological detection. As detailed in the Supporting Information (Figure S-8), we spin-coated a 15 nm layer of polyvinyl alcohol (PVA) containing dissolved NATA and NATA-tyr separately on a thin film of aluminum on a quartz substrate and compared its fluorescence emission intensity, lifetime, and photostability with an identical sample on just the quartz substrate. The aluminum film is rough or nanostructured and so one might expect to see MEF similar to that predicted for the small nanoparticle limit here.
Figure 7a shows that for a 15 nm thick PVA film containing NATA, the 10 nm thick aluminum substrate gives an emission intensity enhancement of approximately 11-fold when compared to the quartz control. The lifetimes of 15 nm thick PVA film containing NATA on quartz and aluminum substrates are shown in the inset of Figure 7a. The solid lines are fits to the experimental decay curves. The intensity decay of NATA on the aluminum surface is faster than that on the quartz control. The intensity-decay of the NATA PVA film on quartz could be fit with a single exponential with a lifetime of 3.2 ns. NATA on the aluminum surface could only be fit with a double-exponential with lifetimes 3.3 ns (8%) and 1.4 ns (92%). The amplitude-weighted lifetime of NATA on aluminum was 1.6 ns. Hence, the intensity decays show that the lifetime was decreased ≈2-fold. In the case of NATA on aluminum, the multiexponential decay may reflect the breadth of the distribution of NATA molecules both in proximity to and distant from the aluminum. The shortening of lifetime on the aluminum nanostructured substrate supports the notion that the increase in observed fluorescence intensity is due to the radiation from the plasmon-fluorophore complex 21,46 that results when excited fluorophores interact with aluminum nanoparticles in the near-field. The near-field images of Figures S-7b,c (Supporting Information), and panels b and c of Figure 6 show it is difficult to differentiate what the origins of the enhanced fields around the aluminum nanoparticle are. They could be either from the fluorophore or from plasmons or both. This lends support to our radiating plasmon model where we believe the enhanced fluorescence emission observed in MEF experiments is due to radiation from the entire excited-state fluorophore-metal nanoparticle complex acting as a single radiating entity.21,46,47 The reduction of the lifetime of NATA on the aluminum surface together with the calculations showing aluminum nanoparticles causing an enhancement in the radiated power of a fluorophore in its proximity suggest an increase in the radiative decay rate of NATA emission due to the interaction of aluminum nanoparticles.46,47 Precise agreement between the increases in intensity and decreases in lifetime cannot be expected. This is because time-domain measurements often result in overweighting of the lifetime by the longer lifetime components in cases involving a heterogeneous decay, especially when the decay of the short components overlaps the instrument response function.
Figure 7.

Fluorescence spectra of functionalized amino acids contained in a 15 nm thick PVA film on top of a 10 nm thick aluminum film and on a quartz control. Insets are the corresponding intensity decays: (a) Tryptophan (NATA); (b) Tyrosine (NATA-tyr). IRF is the Instrument Response Function.
In fluorescence experiments, the photostability of the fluorophore is a factor that governs its detectability. We compare the photostability of NATA on aluminum films and on quartz in the Supporting Information (Figure S-9).
Figure 7b shows that for a 15-nm thick PVA film containing NATA-tyr, the 10 nm thick aluminum film gives emission intensity enhancement of ≈7 compared to the quartz control. The emission spectra of NATA-tyr was collected through a 300 nm long-pass filter to prevent the excitation beam from striking the detector. The inset in Figure 7b shows the intensity decays of 15 nm PVA films containing NATA-tyr on aluminum and quartz substrates. We observe a slightly faster decay for the NATA-tyr PVA film on aluminum when compared to quartz. The intensity-decay of the NATA-tyr PVA film on quartz could be fit with a sum of two exponentials with lifetimes 3.04 ns (32%) and 0.84 ns (68%). NATA-tyr on the aluminum surface also could only be fit with with two lifetimes of 0.62 ns (17%) and 0.93 ns (83%). The amplitude-weighted lifetimes of NATA-tyr on quartz and aluminum were 1.1 and 0.86 ns, respectively. Hence, the intensity decays in Figure 7b (inset) show that the NATA-tyr lifetime on aluminum substrates was decreased by only a modest factor of ≈1.3. The experimental results of Figure 7 and Figure S-9 (Supporting Information) corroborate the validity of our theoretical predictions that aluminum nanoparticles can be used to efficiently enhance the emission of a fluorophore in the ultraviolet region and thus can potentially serve as a valuable tool in implementing a modality for the label free detection of biomolecules in a variety of sensing and imaging platforms.
The more significant theoretical enhancements, relative to the experimental ones discussed above, occur particularly for the dipoles oriented perpendicular to the metal. This is because in this orientation, the fluorophore’s dipole induces a dipole in the aluminum nanoparticle in a configuration that allows the dipoles to align head-to-tail, leading to a much larger effective radiating dipole than in the case of an isolated fluorophore. In the case of the experimental results, the signal observed is ensemble averaged over potentially millions of tryptophan or tyrosine molecules. Since they were spin coated in PVA over the thin aluminum film, their orientation and location could not be precisely controlled. It is expected that in such a system there is a broad distribution of the NATA or NATA-tyr molecules both in proximity to and distant from the aluminum. Additionally, it can be expected that many of these excited molecules are not oriented perpendicular to the metal surface, and might even be oriented parallel to the surface where we expect significant quenching to occur.
CONCLUSIONS
We presented computational and experimental studies showing the effect of aluminum nanoparticles on fluorophore emission in the UV. The excited fluorophore was modeled as a radiating dipole source, and a variety of nanoparticle sizes, fluorophore-particle distances, and fluorophore orientations relative to the aluminum surface were studied.
We saw that spherical aluminum nanoparticles enhance the radiated power of a fluorophore to different degrees for a wide range of wavelengths, 100–450 nm. The peak enhancement wavelength is a function of the nanoparticle size, with larger nanoparticles showing greater enhancements at longer wavelengths. We also saw that the maximum enhancements occur when the fluorophores are oriented perpendicular to the aluminum surface. When the fluorophore is oriented parallel to the metal surface, we observe quenching of the radiation in most cases. The extent of enhancement is a function of the fluorophore-metal distance, with larger separation distances showing lower enhancements. Our results show that the wavelength of maximum radiated power enhancements for aluminum nanoparticles are red-shifted compared to the corresponding extinction spectra maxima, the degree of red-shifting increasing with particle size.
We also observed that, on comparing the wavelength-dependent radiated power enhancement spectra of aluminum to silver for a range of nanoparticle sizes, aluminum consistently shows higher enhancements than silver in the 300–350 nm range, which is relevant for protein (and most other biomolecule) fluorescence. This predicts that aluminum is a more efficient metal than silver for use in MEF applications in the ultraviolet. Silver, however, shows very large increases in radiated power enhancements at λ > 360 nm. This is important because protein emission spectra are quite broad with tails well past λ > 360 nm. As a result we conclude that silver can still be of some use as a substrate for intrinsic protein-MEF applications.
The degree of enhancement in the radiated power increases significantly when the fluorophore is placed in between aluminum nanoparticles in a dimer. In this system we observe maximum enhancements of over 3,500-fold when the fluorophore is oriented perpendicular to the aluminum surfaces, and we observe a maximum quenching of over 200-fold when the fluorophore is oriented parallel to the aluminum surfaces. Inspection of near-field intensity patterns revealed that very specific regions around the nanoparticles experience field enhancements and quenching. This type of result is not easily inferred from far-field observations and is relevant to spatially resolved molecular spectroscopy or detection using fluorescence.
Finally, we presented experimental results showing that thin aluminum films can significantly enhance the emission intensity and photostability of a layer of PVA film containing separately neutral derivatives of tryptophan (NATA) and tyrosine (NATA-tyr). These observations corroborate our theoretical predictions that aluminum nanoparticles are efficient substrates for MEF in the UV.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health-NHGRI (Grant HG002655), and NIBIB (Grant EB006521). S.K.G. was supported by the U.S. Department of Energy, Basic Energy Sciences, under contract No. DE-AC02-06CH11357. We thank Dr. K. Nowaczyk for assistance in making the figures.
Footnotes
SUPPORTING INFORMATION AVAILABLE
Further details are given as noted in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Gershon D. Nat Methods. 2004;1:263. [Google Scholar]
- 2.Young RA. Cell. 2000;102:9. doi: 10.1016/s0092-8674(00)00005-2. [DOI] [PubMed] [Google Scholar]
- 3.Mitchell P. Nat Biotechnol. 2002;20:225. doi: 10.1038/nbt0302-225. [DOI] [PubMed] [Google Scholar]
- 4.MacBearth G. Nat Genet Suppl. 2002;32:526. doi: 10.1038/ng1037. [DOI] [PubMed] [Google Scholar]
- 5.Moreno-Bondi MC, Taitt CR, Shriver-Lake LC, Ligler FS. Biosens Bioelectron. 2006;21:1880. doi: 10.1016/j.bios.2005.12.018. [DOI] [PubMed] [Google Scholar]
- 6.Ramachandran N, Larson DN, Stark PRH, Hainsworth E, LaBaer J. FEBS J. 2005;272:5412. doi: 10.1111/j.1742-4658.2005.04971.x. [DOI] [PubMed] [Google Scholar]
- 7.Yu X, Xu D, Cheng Q. Proteomics. 2006;6:5493. doi: 10.1002/pmic.200600216. [DOI] [PubMed] [Google Scholar]
- 8.Cooper MA. Drug Discov Today. 2006;11:1068. doi: 10.1016/j.drudis.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 9.Willets KA, Van Duyne RP. Annu Rev Phys Chem. 2007;58:267. doi: 10.1146/annurev.physchem.58.032806.104607. [DOI] [PubMed] [Google Scholar]
- 10.Phillips KS, Cheng Q. Anal Bioanal Chem. 2007;387:1831. doi: 10.1007/s00216-006-1052-7. [DOI] [PubMed] [Google Scholar]
- 11.Stuart DA, Yonzon CR, Zhang X, Lyandres O, Shah NC, Glucksberg MR, Walsh JT, Van Duyne RP. Anal Chem. 2005;77:4013. doi: 10.1021/ac0501238. [DOI] [PubMed] [Google Scholar]
- 12.Sun L, Yu C, Irudayaraj J. Anal Chem. 2007;79:3981. doi: 10.1021/ac070078z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grow AE, Wood LL, Chaycomb JL, Thompson PA. J Microbiol Methods. 2003;53:221. doi: 10.1016/s0167-7012(03)00026-5. [DOI] [PubMed] [Google Scholar]
- 14.Engström HA, Andersson PA, Ohlson S. Anal Biochem. 2006;357:159. doi: 10.1016/j.ab.2006.03.058. [DOI] [PubMed] [Google Scholar]
- 15.Schüttpelz M, Müller C, Neuweiler H, Sauer M. Anal Chem. 2006;78:663. doi: 10.1021/ac051938j. [DOI] [PubMed] [Google Scholar]
- 16.Schulze P, Ludwig M, Kohler F, Belder D. Anal Chem. 2005;77:1325. doi: 10.1021/ac048596m. [DOI] [PubMed] [Google Scholar]
- 17.Sluszny C, Yeung ES. Anal Chem. 2004;76:1359. doi: 10.1021/ac035336g. [DOI] [PubMed] [Google Scholar]
- 18.Engström HA, Andersson PA, Ohlson S. J Immunol Methods. 2005;297:203. doi: 10.1016/j.jim.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 19.Lakowicz JR. Principles of Fluorescence Spectroscopy. 3. Springer; New York: 2006. [Google Scholar]
- 20.Zhang J, Fu Y, Chowdhury MH, Lakowicz JR. Nano Lett. 2007;7:2101–2107. doi: 10.1021/nl071084d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ray K, Chowdhury MH, Lakowicz JR. Anal Chem. 2007;79:6480. doi: 10.1021/ac071363l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ray K, Badugu R, Lakowicz JR. J Phys Chem C. 2007;111:7091. doi: 10.1021/jp067635q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ray K, Badugu R, Lakowicz JR. J Am Chem Soc. 2006;128:8998. doi: 10.1021/ja061762i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sokolov K, Chumanov G, Cotton TM. Anal Chem. 1998;70:3898. doi: 10.1021/ac9712310. [DOI] [PubMed] [Google Scholar]
- 25.Antunes PA, Constantino CJL, Aroca RF, Duff J. Langmuir. 2001;17:2958. [Google Scholar]
- 26.Ringler M, Schwemer A, Wunderlich M, Nichtl A, Kürzinger K, Klar TA, Feldmann J. Phys Rev Lett. 2008;100:203002. doi: 10.1103/PhysRevLett.100.203002. [DOI] [PubMed] [Google Scholar]
- 27.Gryczynski I, Malicka J, Gryczynski Z, Lakowicz JR. Anal Biochem. 2004;324:170. doi: 10.1016/j.ab.2003.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gryczynski I, Malicka J, Nowaczyk K, Gryczynski Z, Lakowicz JR. J Phys Chem B. 2004;108:12073. doi: 10.1021/jp0312619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ray K, Szmacinski H, Enderlein J, Lakowicz JR. Appl Phys Lett. 2007;90:251116. doi: 10.1063/1.2751125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang J, Lakowicz JR. Opt Express. 2007;15:2598. doi: 10.1364/oe.15.002598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gryczynski I, Malicka J, Gryczynski Z, Lakowicz JR. J Phys Chem B. 2004;108:12568. doi: 10.1021/jp040221h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langhammer C, Schwind M, Kasemo B, Zorić I. Nano Lett. 2008;8(5):1461. doi: 10.1021/nl080453i. [DOI] [PubMed] [Google Scholar]
- 33.Zhou X, Fang Y, Zhang P. Spectrochim Acta A. 2007;67:122. doi: 10.1016/j.saa.2006.06.043. [DOI] [PubMed] [Google Scholar]
- 34.Gryczynski I, Malicka J, Gryczynski Z, Nowaczyk K, Lakowicz JR. Anal Chem. 2004;76:4076. doi: 10.1021/ac040004c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chowdhury MH, Gray SK, Pond J, Geddes CD, Aslan K, Lakowicz JR. J Opt Soc Am B. 2007;24:2259. doi: 10.1364/JOSAB.24.002259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chowdhury MH, Pond J, Gray SK, Lakowicz JR. J Phys Chem C. 2008;112:11236. doi: 10.1021/jp802414k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taflove A, Hagness SC. Computational Electrodynamics: The Finite-Difference Time-Domain Method. 3. Artech House; Norwood: 2005. [Google Scholar]
- 38.Sullivan DM. Electromagnetic Simulation Using the FDTD Method. IEEE Press; Piscataway: 2000. [Google Scholar]
- 39.Reference Guide for FDTD Solutions, Release 5.0. 2007 http://www.lumerical.com/fdtd.
- 40.Kaminski F, Sandoghdar V, Agio M. J Comput Theor Nanosci. 2007;4:635. [Google Scholar]
- 41.Bohren CF, Huffman DR. Absorption and Scattering of Light by Small Particles. Wiley-Interscience; New York: 1983. [Google Scholar]
- 42.Ruppin R. J Chem Phys. 1982;76:1681. [Google Scholar]
- 43.Bharadwaj P, Novotny L. Opt Express. 2007;15:14266. doi: 10.1364/oe.15.014266. [DOI] [PubMed] [Google Scholar]
- 44.Gersten J, Nitzan A. J Chem Phys. 1981;75:1139. [Google Scholar]
- 45.Mertens H, Koenderink AF, Polman A. Phys Rev B. 2007;76:115123. [Google Scholar]
- 46.Lakowicz JR. Anal Biochem. 2001;298:1–24. doi: 10.1006/abio.2001.5377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lakowicz JR. Anal Biochem. 2005;337:171–194. doi: 10.1016/j.ab.2004.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.