

Abstract
This report describes detailed studies of the scope and mechanism of a new Pd-catalyzed oxidation reaction for the stereospecific conversion of enynes into cyclopropyl ketones. Unlike related PdII/0, Au, and Pt-catalyzed cyclopropane-forming reactions, these transformations proceed with net inversion of geometry with respect to the starting alkene. This result, along with other mechanistic data, is consistent with a PdII/IV mechanism in which the key cyclopropane-forming step involves nucleophilic attack of a tethered olefin onto the PdIV–C bond.
1. Introduction
Bicyclo[3.1.0] and [4.1.0] ring systems are important components of biologically active molecules and also serve as key intermediates in the construction of many functionalized organic products. For example, the natural products mycorrhizin A1 and duocarmycin A,2 which exhibit potent antibiotic activity, both contain bicyclo[3.1.0] hexane moieties. In addition, Martin has utilized a bicyclo[3.1.0]hexane as a key intermediate in the total synthesis of cyclopropane-containing natural product ambruticin S.3 Finally, nucleophilic ring opening of bicyclo[3.1.0] and [4.1.0] hexanes has been employed to stereoselectively generate highly functionalized organic molecules, such as alkoxy- and alkyl-substituted lactones and furans.4
One attractive synthetic route to bicyclo[3.1.0] and [4.1.0] ring systems involves transition metal-catalyzed transformations of readily accessible enyne starting materials. As summarized in Schemes 1 and 2, two mechanistically distinct pathways have been reported for the metal-catalyzed construction of bicyclo[3.1.0]hexanes from enynes. In the first, electrophilic Pt and Au catalysts promote the rearrangement of 1,5- or 1,6-enynes to afford bicyclo[3.1.0] and [4.1.0] ring systems, respectively.5,6,7 These transformations are believed to proceed via nucleophilic attack of the alkene on the metal-coordinated alkyne to produce a bicyclic carbocation such as 3. This intermediate rearranges to form a metallocarbene (4), which can then undergo a 1,2-hydride shift/elimination sequence to afford cyclopropane 2. These transformations typically proceed with high levels of stereoselectivity to afford products where the geometry of the cyclopropane substituents (cis versus trans) is the same as that in the alkene starting material.
Scheme 1.

A representative example of AuI-catalyzed cyclization of enynes.
Scheme 2.

Representative example of Pd0/II-catalyzed cyclization of enynes.
An alternative approach to this class of products involves PdII/0-catalyzed reactions of enynes. As shown in a representative example in Scheme 2, the key cyclopropane-forming step of these transformations proceeds via intramolecular syn olefin insertion, which transforms the σ-alkyl PdII intermediate 8 into cyclopropane 9. Complex 9 is then captured with RSnBu3 to generate product 7.8 Importantly, these reactions proceed with high levels of stereoselectivity with retention of the olefin geometry (cis versus trans) in the cyclopropane product.8,9
We sought to design a new, mechanistically distinct route from enynes to cyclopropanes that takes advantage of the PdII/IV couple to promote cyclopropane ring formation. Two potential mechanisms for this new transformation (Mechanisms A and B) are outlined in Schemes 3 and 4. Both involve the key intermediate 12, which is accessed through the well-precedented sequence of alkyne acetoxypalladation followed by syn olefin insertion.10 Prior work has shown that intermediate 12 can be terminated by β-acetoxy elimination, β-hydride elimination, chlorination, carbonylation, or protonolysis to afford a variety of functionalized heterocyclic products. However, based on recent studies from our group,11 we reasoned that this σ-alkyl PdII intermediate could also be intercepted with strong oxidants (e.g., PhI(OAc)2) to generate the PdIV complex 13 (Mechanism A). Such PdIV intermediates are highly susceptible to SN2-type nucleophilic attack at the α-carbon,12 and we reasoned that the intramolecular vinyl acetate moiety of 13 could serve as an effective nucleophile for cyclopropane formation. Notably, related SN2-type cyclopropane forming reactions with nucleophilic olefins are well-precedented in the organic literature.2,13
Scheme 3.

PdII/IV-catalyzed enyne oxidative cyclization via mechanism A.
Scheme 4.

Alternative PdII/IV-catalyzed mechanism for oxidative enyne cyclization (Mechanism B).
An alternative PdII/IV mechanism (Mechanism B) involving Heck-type olefin insertion from 12 would also be possible (Scheme 4). However, these two potential mechanisms should be readily distinguishable through the use of enynes containing 1,2-disubstituted olefins. In Mechanism A, the SN2-type reductive elimination would result in net inversion of olefin geometry in the cyclopropane product, while syn olefin insertion in Mechanism B would lead to retention of the alkene geometry in the final cyclopropane.
This paper describes the development of Pd-catalyzed reactions of enyne substrates with strong oxidants like PhI(OAc)2 to afford cyclopropane products. The development, optimization, substrate scope, and limitations of these transformations are all described in detail. In addition, experiments designed to provide insights into the mechanism of the cyclopropane-forming step are discussed. These studies implicate Mechanism A as the major pathway in these transformations.11a
2. Results and discussion
2.1. Reaction development and optimization
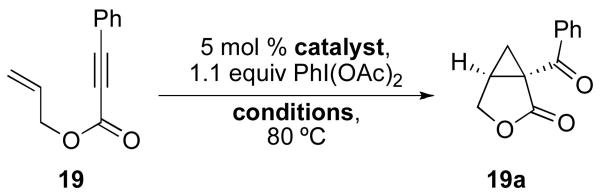
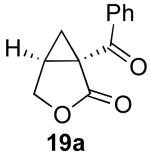
Our initial efforts focused on the cascade cyclization of enyne 19 in the presence of 1.1 equiv of PhI(OAc)2. As discussed above, we anticipated that this hypervalent iodine(III) oxidant would serve to intercept Pd-alkyl intermediate 12, providing either acetoxylated product 20 or cyclopropane 19a. A key requirement for the proposed reaction sequence is that the oxidative functionalization of intermediate 12 must proceed faster than competing β-hydride elimination. As a result, our initial studies focused on a catalyst system composed of 5 mol % of Pd(OAc)2 and 6 mol % of 2,2′-bipyridine (bipy), since Lu has shown that this combination can be important for the cyclization of enynes and can greatly limit competing β-hydride elimination processes.14 We were delighted to find that the reaction of enyne 19 with 1.1 equiv of PhI(OAc)2 in the presence of 5 mol % of Pd(OAc)2, 6 mol % of bipy in AcOH at 80 °C afforded bicyclic β-ketolactone 19a in 79% yield. Interestingly, <5% of compound 20 (the product of oxidatively-induced C–OAc bond-formation from proposed intermediate 12) was observed by GC and 1H NMR spectroscopic analysis of the crude reaction mixture. Further, alkenes 21 and 22, the products of β-hydride elimination/alkene dissociation from putative intermediate 12, were also not observed in the reaction mixture.
We next sought to investigate the effect of the reaction conditions on the yield and distribution of organic products in this transformation. We first examined the influence of ambient water and air on the reaction. A comparison of reactions run using rigorously dried AcOH (Table 1, entry 3) versus those with commercial solvent taken directly from the bottle (Table 1, entry 1) showed that the yield is slightly higher in the latter case. Similar results were also obtained when the reactions were set up under ambient conditions (under air) versus under a nitrogen atmosphere (entries 1 and 2, respectively). These results demonstrate that rigorous purification of the reaction solvent and exclusion of air are not necessary, which enhances the ease of reaction set up and practicality of these transformations.
Table 1.
Optimization of the oxidative cyclization of enyne 19
 | |||
|---|---|---|---|
| Entry | Catalyst | Conditions | Yielda |
| 1 | Pd(OAc)2 with 6 mol % bipy | Ambient | 88% |
| 2 | Pd(OAc)2 with 6 mol % bipy | N2 | 71% |
| 3 | Pd(OAc)2 with 6 mol % bipy | dry AcOH | 83% |
| 4 | Pd(OAc)2bipy | Ambient | 80% |
| 5 | Pd(OAc)2 | Ambient | 93% |
| 6 | Pd(TFA)2 | Ambient | 81% |
| 7 | Pd(Cl)2 | Ambient | 72% |
| 8 | Pd(Cl)2(CH3CN)2 | Ambient | 65% |
| 9 | Pd(Cl)2(PPh3)2 | Ambient | 62% |
| 10 | Pd(Cl)2(PhCN)2 | Ambient | 60% |
| 11 | Na2PdCl4 | Ambient | 51% |
Yields determined by GC relative to an internal standard
We next examined the role of the bipyridine ligand, since it was initially hypothesized to be critical for achieving the desired reaction. As summarized in Table 1, entry 4, the use of 5 mol % of the pre-formed catalyst (bipy)Pd(OAc)2 afforded similar yields to the in situ combination of 5 mol % of Pd(OAc)2 and 6 mol % of bipy. Further studies revealed that bipy was not essential for this transformation, and 93% yield could be obtained with just Pd(OAc)2 as catalyst (entry 5). In addition, other PdII complexes also served as effective catalysts for this transformation in the absence of bipy, although the yields were lower than that with Pd(OAc)2 (entries 6-11). Based on these studies, 5 mol % of Pd(OAc)2 was identified as the optimal catalyst for cyclization of 19, and these reaction conditions provided 88% isolated yield of 19a.
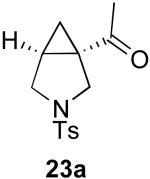
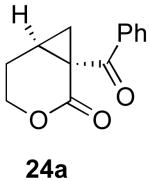
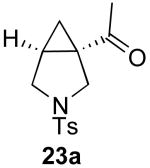
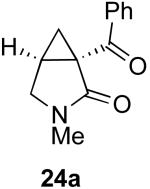
In contrast to the results with substrate 19, the presence/absence of bipy did have a substantial effect on the Pd-catalyzed cyclization of other enynes. As summarized in Table 2, entry 3, with substrate 25, a significant enhancement in yield was observed when 6 mol % of bipy was used (9% versus 54% yield). However, with substrates 23, 24, and 26 (entries 1, 2, and 4), the use of bipy/Pd(OAc)2 led to a decrease in yield relative to just Pd(OAc)2. The origin of these effects is not completely understood and remains under investigation at this time.
Table 2.
Effect of bipyridine on the cyclizations of enynes 23-26a.
| Entry | Starting Materiala | Product | Yield with 6 mol % bipyb | Yield without 6 mol % bipyb |
|---|---|---|---|---|
| 1 | 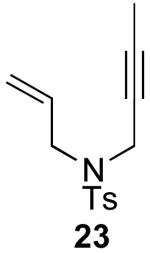 |
 |
61% | 71% |
| 2 | 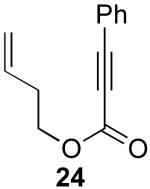 |
 |
9% | 59% |
| 3 | 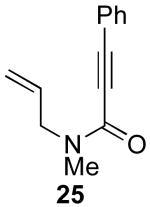 |
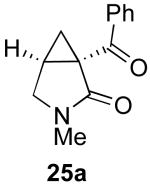 |
54% | 9% |
| 4 | 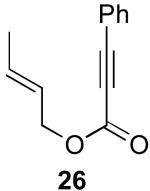 |
 |
29% | 59% |
Reaction Conditions: 5 mol % Pd(OAc)2, 0-6 mol % bipy, 1.1-8 equiv PhI(OAc)2, 60-80 °C, 1-16h.
Yields determined by GC relative to an internal standard.
2.2. Substrate scope
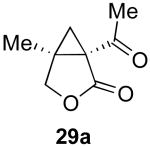
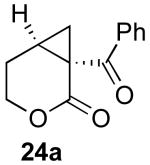
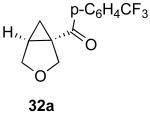
We next investigated the scope of this cyclopropane-forming reaction with a variety of different enynes (Table 3). In general, we found that some optimization of the reaction for each substrate improved the yields substantially. As a result, each was optimized for: (1) the presence/absence of bipy, (2) the amount of oxidant (ranging from 1.1 to 4 equiv), (3) the reaction temperature (between 60 and 80 °C), and (4) the reaction time (between 1 and 16 h). Upon optimization, moderate to good yields of 41-79% were obtained with all of the substrates in Table 3. These transformations led to the generation of bicyclo[3.1.0] and [4.1.0] ring systems containing lactones, tetrahydrofurans, pyrrolidines, and lactams.11a Both alkyl and aryl substituents were tolerated on the alkyne, and both electron donating (p-OMe) and electron withdrawing (p-CF3) groups on the aryl ring were also compatible with the reaction conditions. Furthermore, 1,1-and 1,2-disubstituted enynes were effective substrates. In the latter case, with an ester substituent on the alkene, the product was formed with >20 :1 dr.
Table 3.
Substrate scope of Pd-catalyzed enyne cyclization reactionsa.
| Entry | Substrate | Product | Yieldb |
|---|---|---|---|
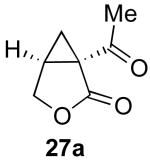
| 1 | 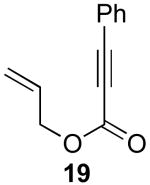 |
 |
79% |
| 2 |  |
 |
55% |
| 3 |  |
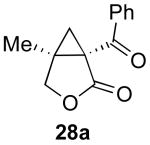 |
78% |
| 4 |  |
 |
66% |
| 5 | 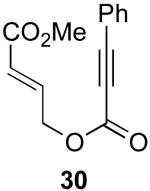 |
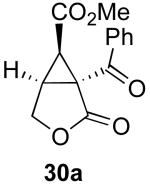 |
70% |
| 6 | 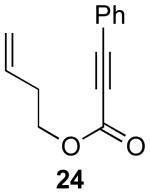 |
 |
55% |
| 7 | 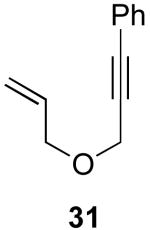 |
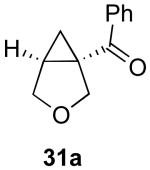 |
48% |
| 8 | 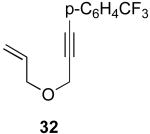 |
 |
41% |
| 9 | 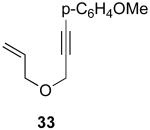 |
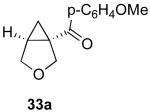 |
44% |
| 10 | 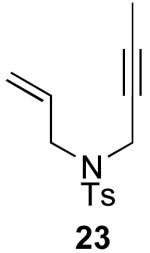 |
 |
71% |
| 11 | 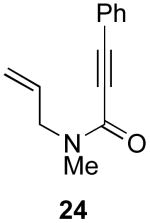 |
 |
47% |
Reaction Conditions: 5 mol % Pd(OAc)2, 0-6 mol % bipy, 1.1-4 equiv PhI(OAc)2, 60-80 °C, 1-16 h.
Isolated yields (average of two runs).
One major limitation of these transformations is that trisubstituted enynes such as 34, 35, and 36 did not react to form cyclopropane products under the standard reaction conditions. Instead, these substrates underwent addition of AcOH across the alkyne, presumably via initial acetoxypalladation followed by protonolysis of the resulting Pd–vinyl intermediate.10 Literature precedent suggests that this undesired side reaction takes place because olefin insertion to form the lactone ring is slow with highly substituted alkene derivatives.15
2.3. Reaction mechanism
We first sought to gain evidence to support a PdII/IV mechanism for these transformations. As such, our initial mechanistic investigations focused on replacing PhI(OAc)2 with a variety of alternative stoichiometric oxidants in the Pd-catalyzed cyclization of substrate 19. As summarized in Table 5, these studies revealed that the use of both Oxone and K2S2O8 as terminal oxidants resulted in significant quantities of the cyclopropane product 19a (Table 4, entries 2 and 3). Importantly, both of these are strong oxidants that have been previously implicated in PdII/IV catalytic cycles.16 In contrast, neither CuII-based oxidants nor benzoquinone or air provided any of the desired product 19a (entries 4-7). With CuCl2, Cu(OAc)2, and benzoquinone, starting material 19a was completely consumed, and a complex mixture of products was formed. With air as the oxidant, a significant quantity of 19 (40%) remained at the end of the reaction, along with a mixture of unidentified products. The requirement for strong two-electron oxidants in this transformation provides initial evidence to support the possibility of PdIV intermediates.
Table 5.
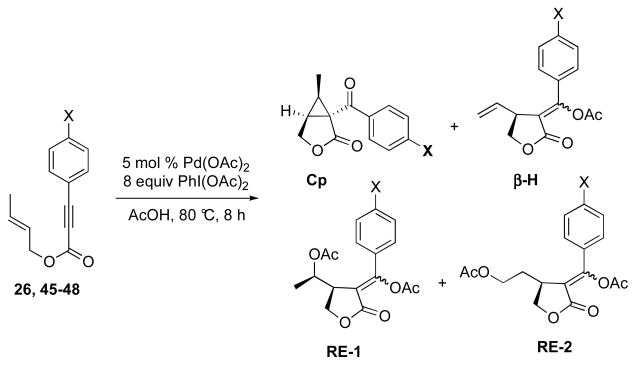
Ratio of Cp : RE-1 as a function of aryl substituent X
 | |||||
|---|---|---|---|---|---|
| X | Cp Yield | β-H Yield | RE-1 Yield | RE-2 Yield | Cp:RE-1 |
| NO2 (45) | 44% | ≪1% | 8% | ≪1% | 5:1 |
| CF3 (46) | 36% | 2% | 5% | 7% | 7:1 |
| H (26) | 55% | <1% | 3% | 12% | 18:1 |
| Me (47) | 56% | 2% | 1% | 5% | 56:1 |
| OMe (48) | 66% | 2% | ≪1% | ≪1% | >200:1 |
Yields determined by HPLC relative to an internal standard
Table 4.
Effect of oxidant on the yield of cyclopropane 19a
 | ||
|---|---|---|
| Entry | Oxidant | Yield of 19aa |
| 1 | PhI(OAc)2 | 94% |
| 2 | Oxone | 50% |
| 3 | K2S2O8 | 27% |
| 4 | CuCl2 | 0% |
| 5 | Cu(OAc)2 | 0% |
| 6 | Benzoquinone | 0% |
| 7 | Air | 0% |
Yields determined by GC relative to an internal standard
A second set of experiments focused on the use of substrates 37 and 26 as mechanistic probes for these reactions. Initial studies of substrate 37 showed that it reacted with 5 mol % of Pd(OAc)2 and 8 equiv of PhI(OAc)2 in AcOH at 80 °C for 2 h to afford a mixture of four major organic products (Scheme 6).11a,17 In addition to the desired cyclopropane product (discussed later in this section), the three side products – acetoxylated isomers 37c and 37d along with alkene 37b – provided significant additional mechanistic insights into these transformations. As shown in Scheme 7, we hypothesize that 37a-d are derived from a common Pd-alkyl intermediate 38. β-Hydride elimination from this intermediate followed by alkene dissociation would afford 37b. Oxidation of 38 with PhI(OAc)2 followed by C–O bond-forming reductive elimination from PdIV would provide acetoxylated product 37c. Finally, reinsertion of alkene 37b into the Pd–H would afford a linear isomer of 38, which could then undergo oxidative functionalization with PhI(OAc)2 to afford 37d. Importantly, C–O bond-forming reductive elimination at unactivated sp3 carbon centers is well-known at PdIV,12b,18 but has not, to our knowledge, been reported at PdII.19 As such, the formation of products 37c and 37d provides additional evidence to support a PdII/IV catalytic cycle.
Scheme 6.

Mixture of products formed in Pd-catalyzed reaction of 37 with PhI(OAc)2.
Scheme 7.

Mechanistic rationale for formation of products 37a-d.
We noted the acetoxylated side product 37c was formed in >95% diastereoselectivity as determined by 1H NMR spectroscopy. However, the relative stereochemistry of the two adjacent stereocenters in the molecule could not be determined conclusively using nOe studies or coupling constant analysis.20 Furthermore, efforts to obtain X-ray quality crystals of 37c were hampered by the fact that this compound is an oil at room temperature. In order to prepare a crystalline product, we replaced the phenyl substituent on the alkyne with a biphenyl group. We anticipated that this modification would alter the physical properties of the product without significantly perturbing the cyclization reaction. The biphenyl-substituted enyne 39 underwent Pd-catalyzed reaction with PhI(OAc)2 to afford acetoxylated product 39c as a single detectable isomer in 7% isolated yield. X-ray crystallographic analysis of 39c definitively established the stereochemical relationship shown in Figure 2.21 Assuming that alkene insertion to form the lactone proceeds in a syn fashion, 39c is the product of C–OAc bond formation with inversion of configuration at carbon. This result provides strong evidence in support of an SN2 mechanism for reductive elimination from the PdIV intermediate in this system. Notably, similar results have been obtained in other C–O bond-forming reactions from PdIV.18,20
Figure 2.
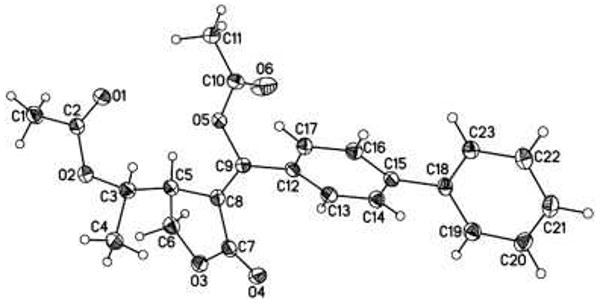
X-ray crystal structure of product 39c22.
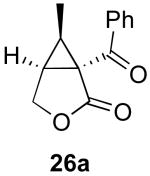
The stereochemical course of cyclopropane formation from substrate Z-37 as well as from isomeric E-26 also provided key insights into the mechanism of these transformations. As outlined in Scheme 9 for substrate Z-37, cyclopropane ring formation via syn olefin insertion into the C–Pd bond (Mechanism B, Scheme 4) would afford cyclopropane 26a. In contrast, cyclopropane formation via nucleophilic substitution (Mechanism A, Scheme 4) would afford the cis-cyclopropane 37a with net inversion of the alkene geometry.
Scheme 9.

Potential mechanisms for cyclopropane formation.
Subjection of E-26 and Z-37 to optimized reaction conditions (5 mol % of Pd(OAc)2 and 8 equiv of PhI(OAc)2 in AcOH at 80 °C) resulted in products 26a and 37a, respectively, with >20:1 dr (Scheme 10). The stereochemistry about the ring in each product was determined by nOe analysis. In addition, the structure of 26a was confirmed by X-ray crystallography.11a
Scheme 10.

Stereochemical experiments with enynes 37 and 26.
In each case, these results correspond to inversion of the starting olefin geometry, which clearly implicates Mechanism A for this transformation. Notably, this result is in contrast to Pd0/II mediated cyclopropanation of enynes, which has been reported to proceed with net retention of olefin geometry (Scheme 2).8,9 Our findings are also distinguished from similar AuI/PtII-catalyzed reactions involving metal carbene intermediates, which also proceed with retention of the initial olefin geometry in the cyclopropane product (Scheme 1).5,6
Based on the data described above, a complete catalytic cycle can be proposed for this transformation. The reaction begins with acetoxypalladation of the alkyne followed by intramolecular olefin insertion to afford intermediate 38. Next, bond rotation followed by oxidation of alkyl palladium complex 38 affords the PdIV intermediate 41. (Note that the order of these two steps could also be reversed). Nucleophilic attack at Cα by the intramolecular vinyl acetate then produces the organic intermediate 42. Importantly, the +4 oxidation state at the Pd center renders it an excellent leaving group; as such, this reaction can be considered analogous to intramolecular organic cyclopropane-forming reactions such as that shown in Scheme 12.13b Finally, hydrolysis of the highly reactive species 42 would provide the observed cyclopropane product.
Scheme 12.

Example of organic cyclopropane-forming reaction with similar mechanism.
A key intermediate in the proposed reaction mechanism is PdIV complex 41. As discussed above, this complex is believed to undergo two competing reactions: (1) intramolecular nucleophilic attack by the vinyl acetate to afford cyclopropane Cp or (2) nucleophilic attack by OAc to afford acetoxylated product RE-1. As such, we hypothesized that the ratio of cyclopropane to acetate products (18 : 1 in the case of substrate 26) should be very sensitive to electronic perturbation of one of these two competing reactions. In particular, we reasoned that p-substitution on the aromatic group conjugated with the alkyne would significantly influence the electronic nature of the vinyl acetate moiety, and thereby affect the rate of cyclopropane formation. However, in contrast, this remote substitution is expected to have minimal impact at the metal center; therefore, the relative rate of C–OAc bond formation should remain essentially unchanged. Based on this analysis, we anticipated that the ratio of cyclopropane (CP) to acetate (RE-1) would increase with more electron donating substituents (which would make the vinyl acetate more nucleophilic) and decrease with more electron withdrawing substituents (which would diminish the nucleophilicity of this moiety).
To probe this hypothesis, we synthesized a series of electronically different p-substituted enynes and submitted them to our optimized conditions (5 mol % of Pd(OAc)2 and 8 equiv of PhI(OAc)2 in AcOH at 80 °C). The reactions were assayed by analytical HPLC with yields calibrated against an internal standard. Interestingly, the reaction time for each electronically different substrate varied substantially from 8 h for 45 (with a p-NO2 substituent) to 10 min for 48 (with a p-OMe group).23 However, it is important to note that both the cyclopropanes (Cp) and the acetoxylated products (RE-1)17 were stable under the reaction conditions over the course of 8 h; therefore, all of the reactions were compared at 8 h for consistency.24
Gratifyingly, we observed a clear trend in the ratio of Cp : RE-1. For example, with electron deficient p-NO2-substituted derivative, a 5 : 1 ratio of Cp : RE-1 was obtained. In striking contrast, the electron donating p-OMe substituent resulted in a >200 : 1 ratio of these two products. This is consistent with a faster relative rate of cyclopropanation for the more electron rich enynes as was hypothesized above. Notably, significant quantities of β-hydride elimination product (β-H)17 and the isomeric acetoxylated product (RE-2)17 were also formed in these reactions, although no clear trends were observed upon variation of the substituent X. However, this was not unexpected, as both of these products are formed by independent pathways that do not involve competing reactivity of intermediate 41.
3. Conclusions
We have demonstrated a new Pd-catalyzed reaction for the stereospecific oxidative cyclization of enynes into bicyclic cyclopropanes. This method allows for the construction of cyclic products from acyclic starting materials with a complementary stereochemical outcome to related Pd0/II AuI and PtII-catalyzed processes. We have shown that the choice of oxidant is crucial to the construction of these products and that with select substrates, bidentate ligands are required for cyclization. This reaction is tolerant of a variety of functional groups and proceeds in moderate to good yield with a broad scope of enynes. The observed stereochemistry as well as detailed electronic studies provide strong evidence supporting the formation of a PdIV intermediate that undergoes competing C–C bond formation (to generate the cyclopropane product) and C–OAc bond formation (to produce an acetoxylated compound). These studies offer insight into a new mode of reactivity for alkyl-PdIV species and should serve as an orthogonal approach to accessing highly functionalized bicyclic cyclopropanes.
4. Experimental
4.1. Instrumentation
NMR spectra were obtained on a Varian Inova 500 (499.90 MHz for 1H; 125.70 MHz for 13C) or a Varian Inova 400 (399.96 MHz for 1H; 100.57 MHz for 13C; 376.34 MHz for 19F) spectrometer. 1H NMR chemical shifts are reported in parts per million (ppm) relative to TMS, with the residual solvent peak used as an internal reference. Multiplicities are reported as follows: singlet (s), doublet (d), doublet of doublets (dd), doublet of doublets of doublets (ddd), doublet of triplets (dt), doublet of quartets (dq), doublet of triplets of doublets (dtd), triplet (t), triplet of triplets (tt), quartet (q), quintet (quin), multiplet (m), and broad resonance (br). IR spectra were obtained on a Perkin-Elmer “Spectrum BX” FT-IR spectrometer. Melting points were obtained on a MEL-TEMP 3.0 from Laboratory Devices Inc., USA.
4.2. Materials and methods
Enyne substrates were prepared according to literature procedures.19c Spectral data for compounds found in Table 1 as well as products 26a-d have been reported in a previous communication.11a PhI(OAc)2 and 2,2′-bipyridine were obtained from Aldrich or Acros and used as received. Pd(OAc)2 was obtained from Pressure Chemical or Frontier Scientific and used as received. The GC yields reported for Table 1, entries 1-11 and Table 4, entries 1-7, are corrected GC yields based on calibration curves against an internal standard (1,3-dinitrobenzene and biphenyl, respectively). Flash chromatography was performed on EM Science silica gel 60 (0.040-0.063 mm particle size, 230-400 mesh) and thin layer chromatography was performed on Merck TLC plates pre-coated with silica gel 60 F254. HPLC separations were performed on a Varian ProStar 210 HPLC using Waters μPorasil® 10 μm silica (19 × 300 mm) columns. HPLC reaction analysis was carried out with a Waters μPorasil® 10 μm silica (3.9 × 300 mm) column with yields based on calibration curves with an internal standard (1,3-dinitrobenzene). HPLC analysis of the products reported herein was carried out using the following method: 97% hexanes/3% ethyl acetate starting eluent, ramp to 50% hexanes/50% EtOAc over 40 min, ramp to 97% hexanes/3% EtOAc over 5 min and hold for 15 min. Gas chromatography was performed on a Shimadzu GC-17A equipped with a Restek Rtx®-5 column (15m, 0.25 mm ID, 0.25 μm df) and an FID detector. GC analysis of all of the products reported herein was carried out using the following method: 100 °C start temperature, ramp 15 °C/min to 240 °C, and hold for 10 min. GCMS analysis was performed on a Shimadzu GCMS QP-5000 equipped with a Restek Rtx®-5 column (30 m, 0.25 mm ID, 0.25 μm df). Control reactions (in the absense of Pd catalyst) were run for each substrate and showed no reaction.
4.3. General procedures
Procedure for synthesis of enynes
The enyne substrates were prepared from the corresponding acid and alcohol according to a literature procedure.19c The carboxylic acids were prepared as follows: A solution of aryl acetylene (1 equiv, 0.14 M) was added to a 2.5 M solution of n-BuLi (0.9 equiv) in hexanes. The reaction was stirred at 0 °C for 30 min, and then CO2 gas was bubbled through the solution for 15 min. The reaction mixture was then washed with 2 M NaOH, the organic layer was discarded, and the aqueous layer was then reacidified with 6 M HCl, and extracted with diethyl ether. The organic layer was dried over MgSO4 and concentrated to a solid. For p-NO2 enyne 45 a slight modification to the above procedure was made: KHMDS (0.95 equiv) was used as the base, and THF was used as the solvent.
Procedure for Pd-catalyzed cyclopropanation of enynes
The following procedure was used for isolation of the products presented in Table 5. In a resealable pressure flask, the enyne substrate (1 equiv), PhI(OAc)2 (8 equiv), and Pd(OAc)2 (5 mol %) were combined in AcOH to make a 0.16 M solution with respect to enyne. The vessel was sealed, and the reaction heated at 80 °C for 10min-8h. The resulting mixture was cooled to room temperature, diluted with EtOAc, and the organic layer was extracted with a saturated solution of NaHCO3. The organic layer was dried over MgSO4, concentrated, and the resulting residue was purified by chromatography on silica gel.
4.4. Characterization data
4.4.1. (E)-but-2-enyl 3-(4-methoxyphenyl)propiolate (48)
Enyne 48 was obtained as a pale yellow oil (Rf = 0.39 in 95% hexanes/5% EtOAc). 1H NMR (500 MHz, CDCl3): δ 7.51 (d, J = 9.0 Hz, 2H), 6.86 (d, J = 9.0 Hz, 2H), 5.85 (dq, J = 15.0, 6.5 Hz, 1H), 5.64 (dt, J = 15.0, 6.5 Hz, 1H), 4.63 (d, J = 6.5 Hz, 2H), 3.81 (s, 3H), 1.73 (d, J = 2.0 Hz, 3H). 13C {1H} NMR (100.6 MHz, CDCl3): δ 161.4, 154.1, 134.9, 132.6, 124.2, 114.2, 111.3, 87.1, 80.0, 66.5, 55.3, 17.7. HRMS (EI): [M]+ calcd for C14H14O3: 230.0943. Found: 230.0952. IR (Thin film): 1699 cm-1.
4.4.2. Pd-catalyzed reaction of 48
Substrate 48 (0.300 g, 1.20 mmol, 1 equiv), PhI(OAc)2 (3.357 g, 10.40 mmol, 8 equiv), and Pd(OAc)2 (14.6 mg, 5 mol %) were combined in AcOH (8.14 mL) and heated at 80 °C for 10 min. After the workup described in the standard procedure above, products 48a and 48b were isolated from this reaction as described below.
4.4.3. 1-(4-methoxybenzoyl)-6-methyl-3-oxabicyclo[3.1.0]hexan-2-one (48a)
Product 48a was purified by gradient column chromatography (gradient = 80% hexanes/20% EtOAc to 50% hexanes/50% EtOAc), and isolated as a pale yellow solid (0.1572 g, 49% yield, mp = 85.0–87.6 °C, Rf = 0.17 in 70% hexanes/30% EtOAc). 1H NMR (500 MHz, CDCl3): δ 7.86 (d, J = 8.5 Hz, 2H), 6.95 (d, J = 8.5 Hz, 2H), 4.54 (dd, J = 10.0, 5.5 Hz, 1H), 4.21 (d, J = 10.0 Hz, 1H), 3.83 (s, 3H), 2.83 (dd, J = 8.0, 5.5 Hz, 1H), 2.09 (dq, J = 8.0, 6.5 Hz, 1H), 1.29 (d, J = 6.5 Hz, 3H). 13C {1H} NMR (125.7 MHz, CDCl3): δ 190.6, 171.4, 163.9, 132.5, 131.8, 113.6, 64.4, 55.4, 41.5, 29.9, 24.6, 7.7. HRMS Electrospray with Na+ added: [M+Na]+ calcd for C14H14O4: 269.0790. Found: 269.0792. IR (Nujol mull): 1756, 1651 cm-1.
4.4.4. (Z)-(4-methoxyphenyl)(2-oxo-4-vinyldihydrofuran-3(2H)-ylidene)methyl acetate (48b)
Product 48b was purified by gradient column chromatography (gradient = 80% hexanes/20% EtOAc to 50% hexanes/50% EtOAc) and isolated as a yellow oil (34 mg, 9% yield, Rf = 0.23 in 70% hexanes/30% EtOAc). 1H NMR (500 MHz, CDCl3): δ 7.50 (d, J = 9.0 Hz, 2H), 6.87 (d, J = 9.0 Hz, 2H), 5.84 (ddd, J = 17.0, 10.0, 9.0 Hz, 1H), 5.21 (dd, J = 17.0, 10.0 Hz, 2H), 4.45 (t, J = 9.0 Hz, 1H), 4.04 (dd, J = 9.0, 5.0 Hz, 1H), 3.89 (td, J = 9.0, 5.0 Hz, 1H), 3.82 (s, 3H), 2.12 (s, 3H). 13C {1H} NMR (100.7 MHz, CDCl3): δ 168.5, 167.4, 161.4, 158.3, 135.7, 130.7, 124.4, 117.1, 115.3, 113.4, 69.2, 55.3, 44.1, 20.8. HRMS Electrospray with Na+ added: [M+Na]+ calcd for C16H16O6: 311.0895. Found: 311.0896. IR (Thin film): 1762, 1755 cm-1.
4.4.5. (E)-but-2-enyl 3-p-tolylpropiolate (47)
Enyne 47 was obtained as a colorless oil (Rf = 0.36 in 95% hexanes/5% EtOAc). 1H NMR (500 MHz, CDCl3) for the E isomer: δ 7.46 (d, J = 8.5 Hz, 2H), 7.15 (d, J = 8.5 Hz, 2H), 5.85 (dq, J = 15.0, 7.0 Hz, 1H), 5.64 (tq, J = 15.0, 7.0 Hz, 1H,), 4.63 (d, J = 7.0 Hz, 2H), 2.36 (s, 3H), 1.73 (d, J = 7.0 Hz, 3H). 13C {1H} NMR (100.6 MHz, CDCl3): δ 154.0, 141.2, 132.9, 132.7, 129.3, 124.2, 116.5, 86.8, 80.2, 66.6, 21.7, 17.6. HRMS (EI) [M]+ calcd for C14H14O2: 214.0994. Found: 214.0996. IR (Thin film): 1707 cm-1.
4.4.6. Reaction with enyne 47
Substrate 47 (0.700 g, 3.27 mmol, 1 equiv), PhI(OAc)2 (8.418 g, 26.1 mmol, 8 equiv), and Pd(OAc)2 (36.6 mg, 5 mol %), were combined in AcOH (20 mL) and heated at 80 °C for 1.5 h. After the workup described in the standard procedure above, products 47a, 47b, 47c and 47d were isolated from this reaction as described below.
4.4.7. 6-methyl-1-(4-methylbenzoyl)-3-oxabicyclo[3.1.0]hexan-2-one (47a)
Product 47a was purified by gradient column chromatography (gradient = 80% hexanes/20% EtOAc to 50% hexanes/50% EtOAc) and isolated as a white solid (345 mg, 46 % yield, mp = 112.0–114.0 °C, Rf = 0.26 in 70% hexanes/30% EtOAc). 1H NMR (500 MHz, CDCl3): δ 7.81 (d, J = 8.5 Hz, 2H), 7.30 (d, J = 8.5 Hz, 2H), 4.60 (dd, J = 10.0, 5.5 Hz, 1H), 4.27 (d, J = 10.0 Hz, 1H), 2.89 (dd, J = 8.0, 5.5 Hz, 1H), 2.44 (s, 3H), 2.20 (dq, J = 8.0, 6.5 Hz, 1H), 1.34 (d, J = 6.5 Hz, 3H). 13C {1H} NMR (100.7 MHz, CDCl3): δ 192.2, 171.3, 144.6, 133.1, 129.5, 129.2, 64.5, 41.8, 30.6, 24.9, 21.8, 7.8. HRMS Electrospray with Na+ added: [M+Na]+ calcd for C14H14O3: 253.0841. Found: 253.0849. IR (Nujol mull): 1760, 1655 cm-1.
4.4.8. (Z)-(2-oxo-4-vinyldihydrofuran-3(2H)-ylidene)(p-tolyl)methyl acetate (47b)
Product 47b was purified by gradient column chromatography (gradient = 80% hexanes/20% EtOAc to 50% hexanes/50% EtOAc) and isolated as a yellow oil (Rf = 0.31 in 70% hexanes/30% EtOAc). Analytically pure material was isolated by further purification by HPLC (85% hexanes/15% EtOAc, 24 mL/min, Waters μ-porasil 19.1 mm). Isolated yield was not determined due to difficulties in purification, but was found to be 7% by calibrated HPLC after 1.5 hours. Independent experiments show this compound decomposes relative to an internal standard under the optimized reaction conditions. 1H NMR (400 MHz, CDCl3): δ 7.46 (d, J = 8.0 Hz, 2H), 7.20 (d, J = 8.0 Hz, 2H), 5.87 (ddd, J = 17.2, 10.0, 6.0 Hz, 1H), 5.25 (dd, J = 17.2, 10.0 Hz, 2H), 4.36 (dd, J = 9.0, 7.2 Hz, 1H), 4.10 (dd, J = 9.0, 2.8 Hz, 1H), 3.93 (m, 1H) 2.38 (s, 3H), 2.34 (s, 3H). 13C {1H} NMR (100.6 MHz, CDCl3): δ 168.3, 167.4, 158.5, 141.0, 135.6, 129.3, 128.9, 128.6, 117.2, 116.1, 69.2, 43.9, 21.5, 20.8. HRMS Electrospray with Na+ added: [M+Na]+ calcd for C16H16O4: 295.0946. Found: 295.0942. IR (Thin film): 1765, 1759 cm-1.
4.4.9. (Z)-1-(4-(acetoxy(p-tolyl)methylene)-5-oxotetrahydrofuran-3-yl)ethyl acetate (47c)
Product 47c was purified by gradient column chromatography (gradient = 80% hexanes/20% EtOAc to 50% hexanes/50% EtOAc) and isolated as a yellow oil (Rf = 0.20 in 70% hexanes/30% EtOAc). Analytically pure material was obtained from further purification by HPLC (85% hexanes/15% EtOAc, 24 mL/min, Waters μ-porasil 19.1 mm). The isolated yield was not determined due to loss during purification. However, HPLC analysis of the crude reaction mixture showed that 47c was formed in 2% yield. 1H NMR (500 MHz, CDCl3): δ 7.48 (d, J = 8.0 Hz, 2H), 7.21 (d, J = 8.0 Hz, 2H), 5.29 (m, 1H), 4.42 (d, J = 10.0 Hz, 1H), 4.28 (dd, J = 10.0, 7.5 Hz, 1H), 3.64 (m, 1H), 2.37 (s, 3H), 2.31 (s, 3H), 2.07 (s, 3H), 1.25 (d, J = 6.5 Hz, 3H). 13C {1H} NMR (100.6 MHz, CDCl3): δ 168.5, 167.4, 161.4, 158.3, 135.7, 130.7, 124.4, 117.1, 115.3, 113.4, 69.2, 55.3, 53.4, 44.1, 20.8. Note that two peaks in the 13C NMR spectrum coincidentally overlap. HRMS Electrospray with Na+ added: [M+Na]+ calcd for C18H20O6: 355.1158. Found: 355.1160. IR (Thin film): 1760, 1738, 1653 cm-1.
4.4.10. (Z)-2-(4-(acetoxy(p-tolyl)methylene)-5-oxotetrahydrofuran-3-yl)ethyl acetate (47d)
Product 47d was purified by gradient column chromatography (gradient = 80% hexanes/20% EtOAc to 50% hexanes/50% EtOAc) and isolated as a yellow oil (Rf = 0.16 in 70% hexanes/30% EtOAc). Analytically pure material was obtained from further purification by HPLC (80% hexanes/20% EtOAc, 24 mL/min, Waters μ-porasil 19.1 mm). The isolated yield was not determined due to loss during purification. However, HPLC analysis of the crude reaction mixture showed that 47d was formed in 6% yield. 1H NMR (500 MHz, CDCl3): δ 7.43 (d, J = 8.0 Hz, 2H), 7.23 (d, J = 8.0 Hz, 2H), 4.38 (dd, J = 9.0, 7.0 Hz, 1H), 4.14 (dd, J = 9.0, 2.5 Hz, 1H), 4.06-4.01 (m, 1H), 3.96 (dt, J = 11.5, 6.0 Hz, 1H), 3.65-3.61 (m, 1H), 2.38 (s, 3H), 2.30 (s, 3H), 1.95 (s, 3H), 1.83-1.77 (m, 2H). 13C {1H} NMR (100.6 MHz, CDCl3): δ 170.6, 168.5, 168.1, 154.0, 141.1, 130.9, 129.5, 127.7, 116.8, 69.7, 61.4, 35.7, 32.0, 21.4, 20.8, 20.7. HRMS Electrospray with Na+ added: [M+Na]+ calcd for C18H20O6: 355.1158. Found: 355.1160. IR (Thin film): 1749, 1649 cm-1.
4.4.11. (E)-but-2-enyl 3-(4-(trifluoromethyl)phenyl)propiolate (46)
Enyne 46 was obtained as a clear oil (Rf = 0.32 in 95% hexanes/5% EtOAc). 1H NMR (500 MHz, CDCl3): δ 7.70 (d, J = 8.5 Hz, 2H), 7.65 (d, J = 8.5 Hz, 2H), 5.90-5.84 (m, 1H), 5.66-5.59 (m, 1H), 4.66 (d, J = 7.0 Hz, 2H), 1.74 (d, J = 7.0 Hz, 3H). 13C {1H} NMR (100.7 MHz, CDCl3): δ 153.4, 133.1, 132.2 (q, 2JCF = 33.2 Hz), 131.1, 125.5 (q, 3JCF = 3.9 Hz), 123.9, 123.5 (q, 1JCF = 272.4 Hz), 123.0, 83.9, 82.2, 66.9, 17.8. 19F NMR (376.3 MHz): δ −63.0. HRMS (EI): [M]+ calcd for C14H11F3O2: 268.0711. Found: 268.0709. IR (Thin Film): 1712 cm-1.
4.4.12. Reaction with enyne 46
Substrate 46 (1.200 g, 3.47 mmol, 1 equiv), PhI(OAc)2 (11.53 g, 35.8 mmol, 8 equiv), and Pd(OAc)2 (50.2 mg, 5 mol %) were combined in AcOH (28 mL) and heated at 80 °C for 6 h. After the workup described in the standard procedure above, products 46a, 46b, 46c, and 46d were isolated from this reaction as described below.
4.4.13. 6-methyl-1-(4-(trifluoromethyl)benzoyl)-3-oxabicyclo[3.1.0]hexan-2-one (46a)
Product 46a was purified by gradient column chromatography (gradient = 80% hexanes/20% EtOAc to 50% hexanes/50% EtOAc) and isolated as a yellow solid (mp = 78.4-79.0 °C, Rf = 0.25 in 70% hexanes/30% EtOAc). Analytically pure material was obtained from further purification by HPLC (85% hexanes/15% EtOAc, 24 mL/min, Waters μ-porasil 19.1 mm). The isolated yield was not determined due to loss during purification. However, HPLC analysis of the crude reaction mixture showed that 46a was formed in 36% yield. 1H NMR (400 MHz, CDCl3): δ 7.96 (d, J = 8.0 Hz, 2H), 7.73 (d, J = 8.0 Hz, 2H), 4.58 (dd, J = 10.0, 5.2 Hz, 1H), 4.27 (dd, J = 10.0, 1.2 Hz, 1H), 3.00 (ddd, J = 8.4, 5.2, 1.2 Hz, 1H), 2.22 (dq, J = 8.4, 6.4 Hz, 1H), 1.36 (d, J = 6.4 Hz, 3H). 13C {1H} NMR (100.6 MHz, CDCl3): δ 192.5, 170.6, 138.5, 134.8 (q, 2JCF = 32 Hz), 129.7, 125.5 (q, 3JCF = 4.0 Hz), 123.5 (q, 1JCF = 271.0 Hz), 64.4, 42.1, 31.2, 26.7, 7.9. 19F NMR (376.3 MHz): δ −63.3. HRMS Electrospray with Na+ added: [M+Na]+ calcd for C14H11F3O3: 307.0558. Found: 307.0547. IR (Nujol mull): 1775, 1680 cm-1.
4.4.14. (Z)-(2-oxo-4-vinyldihydrofuran-3(2H)-ylidene)(4-(trifluoromethyl)phenyl)methyl acetate (46b)
Product 46b was purified by gradient column chromatography (gradient = 80% hexanes/20% EtOAc to 50% hexanes/50% EtOAc) and isolated as a yellow oil (Rf = 0.38 in 70% hexanes/30% EtOAc). Analytically pure material was obtained from further purification by HPLC (90% hexanes/10% EtOAc, 24 mL/min, Waters μ-porasil 19.1 mm). Isolated yield was not determined due to difficulties in purification, but was found to be 2% by calibrated HPLC after 6 hours. 1H NMR (400 MHz, CDCl3): δ 7.63 (s, 4H), 5.83 (ddd, J = 16.8, 10.0, 8.4 Hz, 1H), 5.23 (multiple peaks, 2H), 4.47 (t, J = 8.8 Hz, 1H), 4.06 (dd, J = 8.8, 5.2 Hz, 1H), 3.92 (td, J = 8.4, 5.2 Hz, 1H), 2.11 (s, 3H). 13C {1H} NMR (100.7 MHz, CDCl3): δ 168.4, 167.5, 152.6, 137.0, 135.4, 132.2 (q, 2JCF = 32 Hz), 128.4, 125.5 (q, 3JCF = 3.9 Hz), 123.6 (q, 1JCF = 272.5 Hz), 118.9, 117.8, 70.2, 43.0, 20.7. HRMS Electrospray with Na+ added: [M+Na]+ calcd for C16H13F3O4: 349.0664. Found: 349.0653. IR (Thin film): 1760 cm-1.
4.4.15. (Z)-1-(4-(acetoxy(4-(trifluoromethyl)phenyl)methylene)-5-oxotetrahydrofuran-3-yl)ethyl acetate (46c)
Product 46c was purified by gradient column chromatography, 80% hexanes/20% EtOAc – 50% hexanes/50% EtOAc, and isolated as a yellow oil (Rf = 0.18 in 70% hexanes/30% EtOAc). Analytically pure material was isolated by further purification by HPLC (85% hexanes/15% EtOAc, 24 mL/min, Waters μ-porasil 19.1 mm). The isolated yield was not determined due to loss during purification. However, HPLC analysis of the crude reaction mixture showed that 46c was formed in 5% yield. 1H NMR (400 MHz, CDCl3): δ 7.67 (multiple peaks, 4H), 5.28 (m, 1H), 4.45 (dd, J = 9.6, 2.0 Hz, 1H), 4.31 (dd, J = 9.6, 7.2 Hz, 1H), 3.69 (m, 1H), 2.32 (s, 3H), 2.07 (s, 3H), 1.26 (d, J = 6.4 Hz, 3H). 13C {1H} NMR (100.7 MHz, CDCl3): δ 170.2, 167.9, 167.6, 156.5, 135.1, 132.3 (q, 2JCF = 32.6 Hz), 129.5, 125.0 (q, 3JCF = 3.9 Hz), 123.9 (q, 1JCF = 272.1 Hz), 116.9, 69.3, 65.5, 42.7, 21.1, 20.7, 14.5. 19F NMR (376.3 MHz): δ −63.0. HRMS Electrospray with Na+ added: [M+Na]+ calcd for C18H17F3O6: 409.0875. Found: 409.0865. IR (Thin film): 1775, 1757, 1730 cm-1.
4.4.16. (Z)-2-(4-(acetoxy(4-(trifluoromethyl)phenyl)methylene)-5-oxotetrahydrofuran-3-yl)ethyl acetate (46d)
Product 46d was purified by gradient column chromatography (gradient = 80% hexanes/20% EtOAc to 50% hexanes/50% EtOAc) and isolated as a white solid (mp = 141.5-142.4 °C, Rf = 0.08 in 70% hexanes/30% EtOAc). Analytically pure material was obtained from further purification by HPLC (85% hexanes/15% EtOAc, 24 mL/min, Waters μ-porasil 19.1 mm). The isolated yield was not determined due to loss during purification. However, HPLC analysis of the crude reaction mixture showed that 46d was formed in 7% yield. 1H NMR (400 MHz, CDCl3): δ 7.71 (d, J = 8.4 Hz, 2H), 7.66 (d, J = 8.4 Hz, 2H), 4.42 (dd, J = 9.2, 6.8 Hz, 1H), 4.17 (dd, J = 9.2, 2.0 Hz, 1H), 4.05-3.98 (m, 1H), 3.96-3.90 (m, 1H), 3.57-3.51 (m, 1H), 2.32 (s, 3H), 1.92 (s, 3H), 1.81-1.74 (m, 2H). 13C {1H} NMR (100.7 MHz, CDCl3): δ 170.8, 167.8, 167.5, 155.3, 135.4, 132.0 (q, 2JCF = 32.7 Hz), 129.4, 124.9 (q, 1JCF = 3.8 Hz), 120.2, 123.5 (q, 1JCF = 270.2 Hz), 69.8, 61.6, 36.6, 32.2, 20.9, 20.8. 19F NMR (376.3 MHz): δ −63.0. HRMS Electrospray with Na+ added: [M+Na]+ calcd for C18H17F3O6: 409.0875. Found: 409.0870. IR (Nujol mull): 1763, 1733, 1666 cm-1.
4.4.17. (E)-but-2-enyl 3-(4-nitrophenyl)propiolate (45)
Enyne 45 was obtained as a pale yellow solid (mp = 57.1-58.8 °C, Rf = 0.23 in 95% hexanes/5% EtOAc). 1H NMR (500 MHz, CDCl3): δ 8.23 (d, J = 8.5 Hz, 2H), 7.72 (d, J = 8.5 Hz, 2H), 5.91-5.86 (m, 1H), 5.63 (qd, J = 7.0, 1.5 Hz, 1H), 4.67 (d, J = 7.0 Hz, 2H), 1.74, (d, J = 7.0 Hz, 3H). 13C {1H} NMR (100.7 MHz, CDCl3): δ 153.1, 133.7, 133.4, 126.3, 123.8, 123.7, 122.9, 84.1, 82.9, 67.1, 17.8. HRMS (EI): [M]+ calcd for C13H11NO4: 245.0688. Found: 245.0698. IR (Thin film): 1701 cm-1.
4.4.18. Reaction with enyne 45
Substrate 45 (0.452 g, 1.84 mmol, 1 equiv), PhI(OAc)2 (4.749 g, 14.7 mmol, 8 equiv), and Pd(OAc)2 (20.7 mg, 5 mol %) were combined in AcOH (11.6 mL) and heated at 80 °C for 8 h. After the workup described in the standard procedure above, products 45a and 45b were isolated from this reaction as described below.
4.4.19. 6-methyl-1-(4-nitrobenzoyl)-3-oxabicyclo[3.1.0]hexan-2-one (45a)
Product 45a was purified by gradient column chromatography (gradient = 80% hexanes/20% EtOAc to 50% hexanes/50% EtOAc) and isolated as a clear oil (Rf = 0.16 in 70% hexanes/30% EtOAc). Analytically pure material was obtained from further purification by HPLC (85% hexanes/15% EtOAc, 24 mL/min, Waters μ-porasil 19.1 mm). The isolated yield was not determined due to loss during purification. However, HPLC analysis of the crude reaction mixture showed that 45a was formed in 43% yield. 1H NMR (400 MHz, CDCl3): δ 8.33 (d, J = 9.0 Hz, 2H), 8.02 (d, J = 9.0 Hz, 2H), 4.59 (dd, J = 10.0, 5.6 Hz, 1H), 4.29 (d, J = 10.0 Hz, 1H), 3.07 (ddd, J = 8.0, 5.2, 1.2 Hz, 1H), 2.27 (dq, J = 8.0, 6.0 Hz, 1H), 1.38 (d, J = 6.0 Hz, 3H). 13C {1H} NMR (100.7 MHz, CDCl3): δ 192.4, 170.4, 150.3, 140.6, 130.3, 123.6, 64.36, 42.4, 31.6, 27.7, 8.0. HRMS Electrospray with Na+ added: [M+Na]+ calcd for C13H11NO5: 284.0535. Found: 284.0542. IR (Nujol mull): 1766, 1674 cm-1.
4.4.20. (Z)-1-(4-(acetoxy(4-nitrophenyl)methylene)-5-oxotetrahydrofuran-3-yl)ethyl acetate (45c)
Product 45c was purified by gradient column chromatography (gradient = 80% hexanes/20% EtOAc to 50% hexanes/50% EtOAc) and isolated as a clear oil (Rf = 0.11 in 70% hexanes/30% EtOAc). Analytically pure material was obtained from further purification by HPLC (85% hexanes/15% EtOAc, 24 mL/min, Waters μ-porasil 19.1 mm). The isolated yield was not determined due to loss during purification. However, HPLC analysis of the crude reaction mixture showed that 45c was formed in 8% yield. 1H NMR (400 MHz, CDCl3): δ 8.25 (d, J = 9.2 Hz, 2H), 7.72 (d, J = 9.2 Hz, 2H), 5.27 (dq, J = 6.8, 4.4 Hz, 1H), 4.47 (dd, J = 10.0, 2.0 Hz, 1H), 4.32 (dd, J = 10.0, 7.2 Hz, 1H), 3.72-3.69 (m, 1H), 2.32 (s, 3H), 2.08 (s, 3H), 1.27 (d, J = 6.8 Hz, 3H). 13C {1H} NMR (100.7 MHz, CDCl3): δ 170.3, 168.0, 167.5, 155.4, 148.7, 137.8, 130.2, 123.2, 118.0, 69.3, 65.7, 42.7, 21.1, 20.7, 14.7. HRMS Electrospray with Na+ added: [M+Na]+ calcd for C17H17NO8: 386.0852. Found: 386.0858. IR (Thin film): 1760, 1738, 1664 cm-1.
4.4.21. (Z)-but-2-enyl 3-(biphenyl-4-yl)propiolate (39)
Enyne 39 was obtained as a pale yellow oil (Rf = 0.11 in 95% hexanes/5% EtOAc) along with ∼10% of an unknown impurity. The impurity was removed by further purification via HPLC (99.5% hexanes/0.5% EtOAc, 60mL/min, Waters μ-porasil 30 mm). 1H NMR (500 MHz, CDCl3): δ 7.63 (d, J = 8.5 Hz, 2H), 7.58 (multiple peaks, 4H), 7.44 (t, J = 7.5 Hz, 2H), 7.24 (t, J = 7.5 Hz, 1H), 5.85-5.78 (m, 1H), 5.67-5.61 (m, 1H), 4.82 (d, J = 7.0 Hz, 2H), 1.75 (d, J = 7.5 Hz, 3H). 13C {1H} NMR (125.7 MHz, CDCl3): δ 154.0, 143.3, 139.7, 133.4, 130.7, 128.9, 128.1, 127.1, 127.0, 123.2, 118.2, 86.3, 81.1, 61.4, 13.1. HRMS (EI): [M]+ calcd for C19H16O2: 276.1150. Found: 276.1157. IR (Thin film): 1707 cm-1.
4.4.22. (Z)-1-(4-(acetoxy(biphenyl-4-yl)methylene)-5-oxotetrahydrofuran-3-yl)ethyl acetate (39c)
Substrate 39c (0.790 g, 2.86 mmol, 1 equiv), PhI(OAc)2 (7.362 g, 22.9 mmol, 8 equiv), and Pd(OAc)2 (32.1 mg, 5 mol %) were dissolved in AcOH (18 mL), and heated at 80 °C for 2 h. Product 39c was purified by gradient column chromatography (gradient = 80% hexanes/20% EtOAc to 50% hexanes/50% EtOAc) and isolated as a white solid (mp = 150.0-151.1°C, Rf = 0.22 in 70% hexanes/30% EtOAc). Analytically pure material was obtained from further purification by HPLC (85% hexanes/15% EtOAc, 24 mL/min, Waters μ-porasil 19.1 mm). The isolated yield was not determined due to loss during purification. However, HPLC analysis of the crude reaction mixture showed that 39c was formed in 8% yield. 1H NMR (500 MHz, CDCl3): δ 7.67 (d, J = 8.0 Hz, 2H), 7.61 (multiple peaks, 4H), 7.45 (t, 7.0 Hz, 2H), 7.37 (t, 7.0 Hz, 1H), 5.31 (m, 1H), 4.45 (dd, J = 2.0, 9.5 Hz, 1H), 4.31 (dd, J = 8.0, 10.0 Hz, 1H), 3.68 (m, 1H), 2.35 (s, 3H), 2.08 (s, 3H), 1.27 (d, J = 6.5 Hz, 3H). 13C {1H} NMR (100.6 MHz, CDCl3): δ 170.3, 168.2, 143.7, 140.2, 130.5, 129.5, 128.8, 127.9, 127.2, 126.7, 115.1, 69.6, 65.3, 42.9, 21.2, 20.9, 14.5. Two peaks coincidentally overlap. HRMS (EI): [M]+ calcd for C23H22O6: 394.1416. Found: 394.1417. IR (Nujol mull): 1776, 1758, 1732 cm-1.
Figure 1.

Enyne substrates that did not undergo cyclization.
Scheme 5.

Pd(OAc)2/bipy-catalyzed oxidative cyclization of substrate 19.
Scheme 8.

Reaction of biphenyl-substituted enyne 39.
Scheme 11.

Proposed PdII/IV catalytic cycle for cyclopropane formation.
Scheme 13.

Proposed competing pathways.
Acknowledgments
This work was supported by NIH NIGMS (RO1 GM073836). We also gratefully acknowledge the Camille and Henry Dreyfus Foundation and the Alfred P. Sloan Foundation as well as Abbott, Amgen, AstraZeneca, Boehringer-Ingelheim, Bristol Myers Squibb, Eli Lilly, GlaxoSmithKline, Merck Research Laboratories, and Roche for funding. Additionally, we thank Jeff Kampf for X-ray crystallography and Leilani Welbes for preliminary studies and helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.(a) Trofast J, Wickberg B. Tetrahedron. 1977;33:875. [Google Scholar]; (b) Koft ER, Smith AB., III J Am Chem Soc. 1982;104:2659. [Google Scholar]; (c) Brown RFC, Teo, Peter YT. Tetrahedron Lett. 1984;25:5585. [Google Scholar]
- 2.(a) Ichimura M, Ogawa T, Katsumata S, Takahashi K, Takahashi I, Nakano H. J Antibiotics. 1991;44:1045. doi: 10.7164/antibiotics.44.1045. [DOI] [PubMed] [Google Scholar]; (b) Boger DL, Garbaccio RM, Jin Q. J Org Chem. 1997;62:8875. [Google Scholar]
- 3.Kirkland TA, Colucci J, Geraci LS, Marx MA, Schneider M, Kaelin DE, Jr, Martin SF. J Am Chem Soc. 2001;123:12432. doi: 10.1021/ja011867f. [DOI] [PubMed] [Google Scholar]
- 4.(a) Swain NA, Brown RCD, Bruton G. J Org Chem. 2004;69:122. doi: 10.1021/jo035365r. [DOI] [PubMed] [Google Scholar]; (b) Ok T, Jeon A, Lee J, Lim JH, Hong CS, Lee HS. J Org Chem. 2007;72:7390. doi: 10.1021/jo0709605. [DOI] [PubMed] [Google Scholar]; (c) Clive DLJ, Liu D. J Org Chem. 2008;73:3078. doi: 10.1021/jo702635t. [DOI] [PubMed] [Google Scholar]; (d) Chavan SP, Pasapathy K, Shivasankar K. Synth Commun. 2004;34:397. [Google Scholar]
- 5.(a) Mamane V, Gress T, Krause H, Fürstner A. J Am Chem Soc. 2004;126:8654. doi: 10.1021/ja048094q. [DOI] [PubMed] [Google Scholar]; (b) Harrak Y, Blaszykowski C, Bernard M, Cariou K, Mainetti E, Mouriès, Dhimane A, Fensterbank L, Malacria M. J Am Chem Soc. 2004;126:8656. doi: 10.1021/ja0474695. [DOI] [PubMed] [Google Scholar]; (c) Luzung MR, Markham JP, Toste FD. J Am Chem Soc. 2004;126:10858. doi: 10.1021/ja046248w. [DOI] [PubMed] [Google Scholar]
- 6.For reviews on cycloisomerizations, see: Zhang L, Sun J, Kozmin SA. Adv Synth Catal. 2006;348:2271.Aubert C, Buisine O, Malacria M. Chem Rev. 2002;102:813. doi: 10.1021/cr980054f.Trost BM, Krische MJ. Synlett. 1998:1.
- 7.Intramolecular cyclization of diazo compounds has also frequently been used to access bicyclo[3.1.0] and [4.1.0] ring systems. For examples, see: Doyle MP, Pieters RJ. J Am Chem Soc. 1991;113:1423.Doyle MP, Austin RE, Bailey AS, Dwyer MP, Dyatkin AB, Kalinin AV, Kwan MY, Liras S, Oalmann CJ, Pieters RJ, Protopopova MN, Raab CE, Roos GHP, Zhou Q, Martin SF. J Am Chem Soc. 1995;117:5763.Doyle MP, Peterson CS, Parker DL., Jr Angew Chem, Int Ed. 1996;35:1334.
- 8.Böhmer J, Grigg R, Marchbank JD. Chem Comm. 2002:768. doi: 10.1039/b110890e. [DOI] [PubMed] [Google Scholar]
- 9.For examples, see: Meyer FE, Parsons PJ, Meijere A. J Org Chem. 1991;56:6487.Owczarczyk Z, Lamaty F, Vawter EJ, Negishi E. J Am Chem Soc. 1992;114:10091.Grigg R, Dorrity MJ, Malone JF. Tetrahedron Lett. 1990;31:1343.Brown A, Grigg R, Ravishankar R, Thornton-Pett M. Tetrahedron Lett. 1994:2753.Grigg R, Sridharan V. Tetrahedron Lett. 1992;33:7965.Grigg R, Rasul R, Redpath J, Wilson D. Tetrahedron Lett. 1996;37:4609.Marco-Martínez J, Lòpez-Carrillo V, Buñuel E, Simancas R, Cárdenas DJ. J Am Chem Soc. 2007;129:1874. doi: 10.1021/ja0685598.
- 10.(a) Lu X, Zhu G, Wang Z. Synlett. 1998:115. [Google Scholar]; (b) Wang Z, Zhang Z, Lu X. Organometallics. 2000;19:775. [Google Scholar]; (c) Zhao L, Lu X, Xu W. J Org Chem. 2005;70:4059. doi: 10.1021/jo050121n. [DOI] [PubMed] [Google Scholar]; (d) Zhang Q, Lu X, Han X. J Org Chem. 2001;66:7676. doi: 10.1021/jo0105181. [DOI] [PubMed] [Google Scholar]; (e) Zhang Q, Xu W, Lu X. J Org Chem. 2005;70:1505. doi: 10.1021/jo048414o. [DOI] [PubMed] [Google Scholar]
- 11.For a preliminary account of this work, see: Welbes LL, Lyons TW, Cychosz KA, Sanford MS. J Am Chem Soc. 2007;129:5836. doi: 10.1021/ja071204j.. See also: Tong X, Beller M, Tse MK. J Am Chem Soc. 2007;129:4906. doi: 10.1021/ja070919j.Desai L, Sanford MS. Angew Chem, Int Ed. 2007;46:5737. doi: 10.1002/anie.200701454.Kalyani D, Sanford MS. J Am Chem Soc. 2008;130:2150. doi: 10.1021/ja0782798.Desai L, Hull KL, Sanford MS. J Am Chem Soc. 2004;126:9542. doi: 10.1021/ja046831c.
- 12.For examples, see: Muñiz K, Hövelmann CH, Streuff J. J Am Chem Soc. 2008;130:763. doi: 10.1021/ja075041a.Liu G, Stahl SS. J Am Chem Soc. 2006;128:7179. doi: 10.1021/ja061706h.Bäckvall JE, Bjoerkman EE. J Org Chem. 1980;45:2893.Henry PM, Davies M, Gerguson G, Phillips S, Restivo R. J Chem Soc, Chem Commun. 1974:112.Bäckvall JE. Tetrahedron Lett. 1978;19:163.Bäckvall JE. Acc Chem Res. 1983;16:335.
- 13.For examples, see: Tichenor MS, Trzupek JD, Kastrinsky DB, Shiga F, Hwang I, Boger DL. J Am Chem Soc. 2006;128:15683. doi: 10.1021/ja064228j.Boger DL, Garbaccio RM. J Org Chem. 1999;64:8350. doi: 10.1021/jo991301y.
- 14.(a) Zhang Q, Lu X. J Am Chem Soc. 2000;122:7604. [Google Scholar]; (b) Zhang Q, Lu X, Han X. J Org Chem. 2000;66:7676. doi: 10.1021/jo0105181. [DOI] [PubMed] [Google Scholar]
- 15.(a) Heck RF. Org React. 1982;27:345. [Google Scholar]; (b) Larhed M, Hallberg A. In: Handbook of Organopalladium Chemistry for Organic Synthesis. Negishi E, editor. Wiley-Interscience; New York: 2002. [Google Scholar]; (c) Soderberg BC. In: Comprehensive Organometallic Chemistry II. Hegedus LS, Abel EW, Stone FGA, Wilkinson G, editors. Vol. 12. Pergamon Press; Oxford: 1995. pp. 259–287. [Google Scholar]
- 16.For examples, see: Desai L, Malik HA, Sanford MS. Org Lett. 2006;8:1141. doi: 10.1021/ol0530272.Yang D. Acc Chem Res. 2004;37:497. doi: 10.1021/ar030065h.Muehlhofer M, Strassner T, Herrmann WA. Angew Chem, Int Ed. 2002;124:3824. doi: 10.1002/1521-3773(20020517)41:10<1745::aid-anie1745>3.0.co;2-e.Reddy BVS, Reddy LF, Corey EJ. Org Lett. 2006;8:3391. doi: 10.1021/ol061389j.Tang S, Peng P, Pi SF, Liang Y, Wang NX, Li JH. Org Lett. 2008;10:1179. doi: 10.1021/ol800080w.Wang GW, Yuan TT, Wu XL. J Org Chem. 2008;73:4717. doi: 10.1021/jo8003088.
- 17.The olefin geometry of these side products could not be definitively determined using nOe analysis.
- 18.(a) Desai L, Sanford MS. Angew Chem, Int Ed. 2007;46:5737. doi: 10.1002/anie.200701454. [DOI] [PubMed] [Google Scholar]; (b) Alexanian EJ, Lee C, Sorensen EJ. J Am Chem Soc. 2005;127:7690. doi: 10.1021/ja051406k. [DOI] [PubMed] [Google Scholar]; (c) Li Y, Song D, Dong VM. J Am Chem Soc. 2008;130:2962. doi: 10.1021/ja711029u. [DOI] [PubMed] [Google Scholar]
- 19.Dick AR, Kampf J, Sanford MS. J Am Chem Soc. 2005;127:12790. doi: 10.1021/ja0541940. [DOI] [PubMed] [Google Scholar]
- 20.Reductive elimination from a similar intermediate has been reported to proceed with inversion, as determined by coupling constant analysis (see ref. 11b).
- 21.The olefin geometry of 39c could not be definitively determined in solution via nOe analysis; however, interestingly, the x-ray structure shows a geometry consistent with initial cis-acetoxypalladation of the alkyne. While the literature suggests that trans-acetoxypalladation should predominate under our standard reaction conditions, early studies of alkyne halo- and acetoxypalladation have shown that mixtures of trans and cis products can be formed. For examples, see: Lu X, Zhu G, Ma S. Tetrahedron Lett. 1992;33:7205.Zhu G, Ma S, Lu X, Huang Q. J Chem Soc, Chem Commun. 1995:271.Ma S, Lu X. J Org Chem. 2001;66:7676. doi: 10.1021/jo0105181.Lambert C, Utimoto K, Nazaki H. Tetrahedron Lett. 1984;25:5323.Yanagihara N, Lambert C, Iritani K, Utimoto K, Nozaki H. J Am Chem Soc. 1986;108:2753. doi: 10.1021/ja00279a068.Bäckvall JE, Nilsson YIM, Gatti RGP. Organometallics. 1995;14:4242.. Further investigations are underway to understand the factors controlling cis versus trans acetoxypalladation as well as the effect that this has on the outcome of these cyclopropane-forming transformations.
- 22.Crystallographic data (excluding structure factors) for the structures in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 704193. Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK, (fax: +44-(0)1223-336033 or e-mail: deposit@ccdc.cam.ac.Uk).
- 23.Kinetics studies have shown that these reactions are zero order in oxidant. Therefore, the change in rate with electronically differentiated alkenes does not provide information about the cyclopropane-forming step of this reaction.
- 24.In contrast, significant decomposition of the alkene products (β-H) was observed under these conditions. Many small peaks were observed in the GC and HPLC traces of these reactions after 8 h, suggesting that β-H decomposes to a mixture of unidentified minor products. We believe that this accounts for the poor mass balance in many of these reactions.