

Abstract
Background
Conventionally, the first step in analyzing the large and high-dimensional data sets measured by microarrays is visual exploration. Dendrograms of hierarchical clustering, self-organizing maps (SOMs), and multidimensional scaling have been used to visualize similarity relationships of data samples. We address two central properties of the methods: (i) Are the visualizations trustworthy, i.e., if two samples are visualized to be similar, are they really similar? (ii) The metric. The measure of similarity determines the result; we propose using a new learning metrics principle to derive a metric from interrelationships among data sets.
Results
The trustworthiness of hierarchical clustering, multidimensional scaling, and the self-organizing map were compared in visualizing similarity relationships among gene expression profiles. The self-organizing map was the best except that hierarchical clustering was the most trustworthy for the most similar profiles. Trustworthiness can be further increased by treating separately those genes for which the visualization is least trustworthy. We then proceed to improve the metric. The distance measure between the expression profiles is adjusted to measure differences relevant to functional classes of the genes. The genes for which the new metric is the most different from the usual correlation metric are listed and visualized with one of the visualization methods, the self-organizing map, computed in the new metric.
Conclusions
The conjecture from the methodological results is that the self-organizing map can be recommended to complement the usual hierarchical clustering for visualizing and exploring gene expression data. Discarding the least trustworthy samples and improving the metric still improves it.
Background
Statistical data analysis usually consists of two successive phases: exploratory and confirmatory. In the first phase, the data is inspected and explored to form hypotheses that are then verified in the second, confirmatory phase.
For data sets measured with microarrays, the exploratory phase is particularly important for two reasons. First, if the number of plausible research hypotheses is very large, it is advisable to narrow them down with thorough exploration. A search for correlates of cancer types is one example. Second, all microarray studies generate a large amount of data as a side product. The database can be explored later for other purposes.
In this paper, we study one of the main tasks of exploratory data analysis: visualization of similarity relationships among high-dimensional data samples. We will focus particularly on the similarities, although the methods may additionally reveal clusters (groups of mutually similar data) and their similarity relationships. Visualizing similarities in high-dimensional (from a few to hundreds of dimensions) data items is a difficult task since the displays can be at most three-dimensional in practice. In particular, it is impossible to project the samples in such a way that all similarity relationships are preserved. Hence, the methods need to make compromises regarding which kinds of relationships to visualize.
On one side of the coin, the visualizations should be trustworthy, in the sense that samples appearing similar (proximate) in the visualization can be trusted to be similar in actuality. The other side of the coin is whether all original proximities become visualized. This dualism is analogous to precision and recall in information retrieval and classification.
We argue that, for data exploration, it is more important that the initial visualizations are trustworthy. The other side of the coin is important but not equally so. The proximities that are visible on the display are salient, and if they are not trustworthy the whole display is misleading. In contrast, if all similar samples cannot be placed proximate, the consequence is only that potentially useful discoveries may be overlooked. Since both goals cannot be achieved simultaneously, we argue that the compromise should initially be made in favor of trustworthiness, which will guarantee that at least a portion of the similarities will be perceived correctly. Afterwards, the potentially overlooked similarities may be hunted for by alternative visualizations.
To our knowledge, studying trustworthiness of visualizations of similarity is a new idea. Projection methods have been compared earlier for other kinds of data [1,2] but the criterion has been the capability of preserving (all) the actual distances instead of the proximities (neighborhoods). This option biases the comparison in favor of methods that directly aim at optimizing the distances. An additional problem is that, in our opinion, the trustworthiness of proximate samples is more important than accurate preservation of all distances, as argued above.
We have designed a measure of how trustworthy the proximate points on a display are. We use it to compare the trustworthiness of three unsupervised methods, hierarchical clustering [3], self-organizing map (SOM) [4], and multidimensional scaling (MDS) [5]. Of these, hierarchical clustering is an extremely popular tool in the bioinformatics community [6-8], and self-organizing maps have been applied as well [9-12].
In the first part of the paper, these unsupervised tools will be applied to functional genomics data measured by DNA microarrays in gene knock-out mutation experiments [8] and in different tissues [13]. Functionally similar genes are sought by visualizing the similarity of the expression profiles of 1410 (after preprocessing) yeast genes, measured in 179 knock-out mutations. Likewise, the similarity of 1600 mouse genes will be studied based on their expression profile over 45 tissues.
In the second part of the paper, we address another major question in visualization of similarity, and as a side note in clustering in general: how to measure similarity. Gene expression measurements in a variety of treatments potentially include valuable information about the function and co-regulation of genes. The important variation is, however, hidden within all the biological and measurement noise in the high-dimensional expression space.
For the knock-out mutation data, the question is which mutations to select, and how to weight the mutations so that the functionally meaningful variation is emphasized and irrelevant variation suppressed. Moreover, the weighting should be different for different genes, that is, at different locations of the expression space.
The learning metrics principle [14,15] is a new approach to finding important aspects of data, and expressing them in a way usable by standard data analysis and data mining methods. In general, the learning metrics principle refers to using certain differential-geometric methods for deriving metrics to data spaces, based on the interrelationship between the (primary) data set and auxiliary data. The metrics are called "learning metrics" because they are learned from the two data sets.
In this paper, metrics will be learned in two case studies to measure differences between gene expression profiles, and used in visualizing similarities of the profiles in the yeast data. In the first experiment, the auxiliary data is selected to be functional classes of the genes, and in a second experiment the activity of the genes in the tissues of another organism. The crucial assumption underlying the learning is that differences in primary data (gene expression) are assumed important if they cause changes in the auxiliary data. The metric of the primary data space is adjusted locally to measure only the important differences, as illustrated in Figure 1. The adjustment may be different in different locations of the space.
Figure 1.

Schematic illustration of the change of metric by functional classes of genes. The expression of three genes, x, y, and z, has been measured in two treatments. The ellipses are kinds of contour lines; compared to the point x, on each line the distribution of functional classes differs by the same amount. In the new metric that takes the contour lines into account (on the right), y is much closer to x. A gene at y is more likely to have a similar functional classes than at z, which is expressed by the new metric.
Formally, in the first case study with the knock-out mutation data [8], we have an expression data matrix containing measurements in n = 179 different treatments (columns) for N = 1400 different genes (rows). We know the functional class for each of the N genes that we will analyze. The method is generally applicable in such set-ups, assuming n is not too large compared to N, to avoid overfitting of the metric. Technical details of how to estimate the metric are given in the Methods section.
We will use the new metric to find the set of genes for which the new metric is the most different from the usual similarity measure (correlation or Euclidean); their visualizations and clusterings with the usual metric are possibly misleading. Finally, the similarities of all genes in the new metric will be visualized.
The methods are, additionally, briefly validated with another data set. The primary data are the gene expression profiles of human genes measured in different tissues [13], and the auxiliary data are the activities of the homologous mouse genes in a set of tissues. The abstract setting is almost identical to that in the yeast study: each human gene belongs to one or multiple classes that correspond to the mouse tissues. If the homologous mouse gene is expressed in the tissue number i, the human gene belongs to the class number i.
The necessary condition for applying the learning metrics principle is that a suitable auxiliary data set is available. This is the case when learning metrics-based exploratory analysis of the primary data is used to complement supervised learning (regression, classification). When classifying genes to different functional groups or tissues to disease types, for example, learning metrics-based visualizations can reveal relationships among the groups, highlight outliers, or even help the discovery of new groups. These kinds of auxiliary data are typically constructed manually, and hence are costly. Alternatively, the auxiliary data can be constructed automatically to summarize another data set, for example the mouse gene expressions in this paper. Then the aspects of the primary data that are related to the auxiliary data will become emphasized in the visualizations.
In summary, this paper (i) compares visualizations generated by a set of commonly used methods with a new criterion, trustworthiness; and (ii) presents a method for adjusting the metric to further improve the visualizations.
Results
Trustworthiness of the visualizations
We consider a projection onto a display trustworthy if all samples close to each other after the projection can be trusted to have been proximate in the original space as well. Measuring such trustworthiness requires specifying what is meant by 'proximate', and how to quantify possible non-trustworthiness of the proximate samples.
The details of the measures and their motivation are given in the Methods Section. In summary, we use simple non-parametric definitions to avoid biases in favor of any of the projection methods. The k nearest samples will be regarded 'proximate', and results will be reported for several values of k. If the proximate samples are not also neighbors in the original space, their rank distance from the neighborhood will be measured to quantify the magnitude of error. Our trustworthiness measure M1 (Eq. 3) is essentially the average trustworthiness over all data.
We compared the trustworthiness of four visualization methods: Sammon's projection, non-metric MDS, SOM, and hierarchical clustering (see the Methods Section). All were applied in the standard textbook way. Sammon's mapping and non-metric MDS were selected to represent MDS methods since they have beneficial properties; Sammon's mapping emphasizes the preservation of short distances which are the focus of our trustworthiness measure as well. Non-metric MDS tries to preserve rank orders of distances, which is the error measure we use. For hierarchical clustering, there are lots of variants; we compared all variants available in the Cluster program by Eisen [16]: centroid linkage, complete linkage, and single linkage. Complete linkage gave clearly better results than the other variants and is the only one included in the results below.
All methods used the same inner product (correlation) metric, which is the most commonly used metric for gene expression data sets. Additional justification for the choice is that correlation metric works well for classification of the specific yeast dataset (preliminary studies). It is imperative to use the same metric for all methods to keep the results comparable. In principle, the whole study could be repeated for different metrics. However, it is unlikely that the conclusions would change; in an earlier experiment [17] on Euclidean metrics for non-biological data sets, the conclusions were the same.
Trustworthiness
The results are shown in Figure 2. We focus on trustworthiness of relatively small neighborhoods, of the order of some tens of genes, which are perceived to be most saliently proximate in displays such as Figure 8. In this range, hierarchical clustering is the best for the smallest neighborhoods (k < 10), and SOM after that. The excellent performance of hierarchical clustering at very small neighborhood sizes was to be expected as it explicitly connects the closest points first.
Figure 2.

Trustworthiness of the visualized similarities (neighborhoods of k nearest samples). Sammon: Sammon's mapping, NMDS: non-metric multidimensional scaling, SOM: self-organizing map, HC: hierarchical clustering, with the ultrametric distance measure and with the linear distance measure. RP: Random linear projection is the approximate worst possible practical result (the small standard deviation over different projections, approximately 0.01, is not shown). The theoretical worst case, estimated with random neighborhoods, is approximately M1 = 0.5. a) Yeast data. b) Mouse data.
Figure 8.

Visualization of the similarity relationships of the yeast genes with a SOM in learning metrics. The names of the genes have been marked on the SOM units (every second hexagon) onto which they have been mapped, and the units having no genes have been marked with dots. The hexagons in between each pair of SOM units are so-called U-matrix units (see the Methods Section) whose gray shades indicate clusteredness in the region (light: cluster; dark: sparse area). The 202 genes for which the metric is the most different from the usual inner product metric have been underlined. Three sample areas analyzed briefly in the text have been circled and numbered.
Preservation of the original neighborhoods
As discussed in the Background Section, all methods make a compromise between trustworthiness and preservation of the original proximities. The latter kinds of errors result from discontinuities in the projection; we measured them by how well neighborhoods of data points in the original data space were preserved. Non-parametric measures were again used to avoid biases. The neighborhood of size k of an expression profile is defined as those k profiles that have the smallest distance (here, strongest correlation) from the profile. If a profile becomes projected away from the neighborhood, the error is quantified by rank distances on the display. The measure M2 (Eq. 4; for details, see the Methods Section) summarizes the errors for all expression profiles. For these data sets, the SOM and multidimensional scaling (Sammon and non-metric MDS) are the best for preserving small (k < 50) original neighborhoods (Fig. 3). Hierarchical clustering is by far the worst.
Figure 3.

Capability of the visualizations to preserve the similarities (the neighborhoods of size k) of the original data space. Sammon: Sammon's mapping, NMDS: non-metric multidimensional scaling, SOM: self-organizing map, HC: hierarchical clustering, with the ultrametric distance measure and with the linear distance measure. RP: Random linear projection is the approximate worst possible practical result (the small standard deviation over different projections, about 0.01, is not shown). The theoretical worst case, estimated with random neighborhoods, is approximately M2 = 0.5. a) Yeast data. b) Mouse data.
Improving the trustworthiness
Trustworthiness can be improved by discarding the least trustworthy data samples and analyzing them separately. Figure 4 shows the increase of trustworthiness as the number of discarded samples is increased. It is striking that although the performance of most of the other methods increases rapidly, they do not reach even the starting point of the SOM before nearly one third of the data set has been discarded. The ultrametric measure (see the Methods Section) of similarity for hierarchical clustering has the smallest improvement rate.
Figure 4.

Improvement of trustworthiness of the yeast data visualizations when q least trustworthy genes are discarded from the visualization. Initially, the size of the neighborhood was k = 20 but was gradually decreased to keep its ratio to the number of the remaining data points constant. Sammon: Sammon's mapping, NMDS: non-metric multidimensional scaling, SOM: self-organizing map, HC: hierarchical clustering, with the ultrametric distance measure and with the linear distance measure. The sudden steps in the trustworthiness of hierarchical clustering coincide with the changes of the neighborhood radius k.
Visualization of functional similarity by learning metrics
A main problem in comparing gene expression profiles is to choose which properties to compare, that is, how to define the similarity measure or, equivalently, the metric. When comparing knock-out mutation profiles of genes, the relevant mutations need to be selected and scaled suitably for each gene.
There is not enough prior knowledge to do this manually, and our goal is to learn automatically the proper metric from interrelationships between the expression data set and another data set that is known to be relevant to gene function: the functional classification of the genes. In an additional study, the primary data are the gene expression profiles of human genes measured in different tissues, and the auxiliary data used to guide the learning are the activities of the homologous mouse genes in a set of tissues [13].
Details on how to learn the metrics are described in the Methods Section [14,15]. In summary, the metric is such that functional classes change uniformly in the new metric. If some of the knock-out mutations have only a weak correlation with the functional classes, they contribute only weakly in the measured similarity among expression profiles. The similarity measure focuses on those differences that are relevant for the functional classes.
The metric is defined as a local scaling of the expression space, which makes it very general; the contributions of the knock-out profiles to the similarities may be different for different genes.
We applied the new metric to one of the visualization methods, the SOM, and compared the results with the same method in the standard correlation metric. For technical details of combining of the SOM and the learning metrics, see the Methods Section.
We began by measuring quantitatively whether SOMs in learning metrics represented the functional classes better than those in the standard inner product metric. In short, a standard estimator is used to predict the (probability) distribution of functional classes for each SOM unit, and when a new expression profile is projected to the SOM, the accuracy of the prediction is computed. A standard accuracy measure, the log-likelihood, was used. The prediction is derived from the same probability estimator that is used for computing the learning metrics (cf. the Methods Section). The estimator has a free parameter called 'kernel width'; the value that produced the best results was selected for the subsequent experiments. The results shown in Figures 5 and 6 confirm that the new metric yielded more accurate results for the two data sets for a wide parameter range.
Figure 5.

Accuracy of SOMs of knock-out yeast gene expression data in representing the functional classes of the genes. Technically, the goodness measure is the log-likelihood of the estimator of the conditional probability density of the classes at the closest SOM unit for each data point. The horizontal axis is the 'smoothness' of the density estimator. Dashed line: SOM in learning metrics, dotted line: SOM in inner product metrics, solid line: the approximate upper limit, i.e. the estimate computed at the data point instead of at the SOM unit.
Figure 6.

Accuracy of SOMs of human gene expression data in representing expression of homologous mouse genes. Technically, the goodness measure is the log-likelihood of the estimator of the conditional probability density of the classes at the closest SOM unit for each data point. The horizontal axis is the "smoothness" of the density estimator. Dashed line: SOM in learning metrics, dotted line: SOM in inner product metrics, solid line: the approximate upper limit, i.e. the estimate computed at the data point instead of at the SOM unit.
We finally used the SOM in learning metrics to visualize similarity relationships of the knock-out expression profiles of yeast genes, and picked up sample findings as demonstrations. To make the display as trustworthy as possible, 10% of the least trustworthy genes were discarded. If desired, the genes most similar to them can be later sought by directly comparing expression profiles. The entire analysis process is summarized in diagram form in Figure 7.
Figure 7.

Summary of the process of constructing a SOM in learning metrics for yeast genes.
The learning metrics SOM is shown in Figure 8. The display visualizes similarity relationships; if two genes are proximate in it, they can be reasonably well trusted to behave similarly. Clusteredness of the data is shown by the U-matrix visualization of the SOM (Figure 8. for details see the Methods Section), revealing several lighter areas with mutually relatively similar genes, and darker areas in between, where the genes are relatively more different.
The novelty in the display, compared with standard SOM displays of gene expression data, is in the metric. Proximate genes both behave similarly in the mutation experiments, and are likely to have similar functional classes. Knowledge about the functional classes has been incorporated in the theoretically justified method described in the Methods Section, such that the display still shows correctly similarities among the expression profiles. The main thing that has changed is that the mutation experiments are weighted to bring forth better the differences related to gene function.
We will next analyze as demonstrations three sample findings from the SOM. There is an interesting small group or subcluster of nine genes (number 1 in Fig. 8) associated with mitochondria. Four of the genes are additionally associated with branched-chain amino acid biosynthesis (YJR016C, YHR208W, YLR355C, YCL009C). These were grouped with three genes related to fermentation and carbohydrate utilization (YOL059W, YER073W, YKL120W), and genes involved in the threonine and lysine, as well as leusine biosynthesis (YDR234W, YER086W). All the genes with a known function in this cluster were located to mitochondria.
Assuming the new metric is more informative than the standard correlation metric, it is particularly interesting to know for which genes the metric has changed the most. All old analyzes with the standard correlation metric have potentially yielded misleading results. Such genes were sought by comparing how many of the closest neighbors were different in the two metrics, and emphasized by underlining the gene names in Figure 8.
The area number 3 is an example where the metric has changed. The analysis of functional classifications and annotations of the area revealed that 8 out of the 17 genes were associated with transcription and DNA repair (YMR179W, YAR007C, YBR088C, YDR501W, YDL101C) or cell cycle (YAL024C, YHR153C, YMR198W). Two genes are protein tyrosine kinases (YDL101C, YGL179C), and as the former is involved in DNA damage response, it is possible that also YGL179C mediates similar kind of function. The gene YJL196C may be an outlier in this area; it is associated with fatty acid elongation. The rest of the genes in this cluster have an unknown function.
Finally, we sought the display for groups of genes belonging to known pathways. Some of these groups occurred proximate on the SOM. The genes involved in purine biosynthesis occurred together on the area number 2. These and other proximate genes had been classified under nucleotide metabolism. In addition, the gene YDL241W, whose function is unknown, was located nearby. Hence, it might be worthwhile to examine closer whether this gene is also related to nucleotide metabolism or the purine biosynthesis pathway.
Conclusions
Comparison of unsupervised methods for visualizing similarity of gene expression profiles revealed that the self-organizing map (SOM) was the most trustworthy except for the most similar gene expression profiles, where hierarchical clustering was the best. The less important other side of the coin is whether there are discontinuities in the mapping. In this latter regard, the relative goodness of the methods depends on the data (see also [17]). In the comparisons with gene expression data, the SOM has hitherto performed well.
The learning metrics principle was then applied to derive a new location-specific metric based on functional classes of the genes. The resulting metric measures changes in gene expression but weights the changes according to how much they contribute to changes in the functional classes. The metric is a step toward a more comprehensive picture of the functional similarity of the genes, incorporating prior biological knowledge in the measurements.
The basic learning metrics method considers the auxiliary data as a classification of the primary data into mutually exclusive classes. This restriction can be easily relaxed by considering multi-class data as samples from the class distribution at the point. Generalizations to hierarchical classifications and other more general types of auxiliary data are also possible and will be considered in later work.
Methods
Data
Three different gene expression data sets were used in the experiments.
The first data set was provided by Hughes et al. [8]. It consists of expression measurements for all yeast (Saccharomyces cerevisiae) genes in 300 knock-out mutations. They had derived error estimates based on replicated measurements, and tested whether the expression of the genes differed significantly from noise. We selected a subset of the data containing saliently expressed genes and mutations that induced expressions. Only genes and mutations with at least two measurements that differed significantly from noise (P < 0.01) and were expressed over 2-fold when compared to the control were selected, resulting in a data set of the size of 1410 genes measured for 179 mutations.
We have (similarly as in [18]) compared different preprocessing methods (normalization of measurement error, standard deviation, length and/or mean), with the classification error of a k-nearest neighbor classifier as the performance measure. For this data, the following alternative gave the best performance: the data was preprocessed by dividing each measurement by its estimated measurement error, and then the standard deviation of each mutation was normalized. Finally, all gene expression profiles were normalized to unit length.
The auxiliary data for the first gene expression data set was selected from the MIPS functional classification [19] for yeast. The classification consists of over two hundred classes at different levels of hierarchy. Many of the functional classes are known to correlate with gene expressions, although some classes undoubtedly are very heterogeneous at the level of gene expression. A set of 46 classes were selected from the various levels of functional classification, in order to obtain non-hierarchical and a priori as coherently behaving classes as possible.
The second data set [13] was used to confirm the findings on trustworthiness. Expression of over 13000 mouse genes had been measured in 45 tissues. We selected an extremely simple filtering method, similar to that originally used in [13]. Of the mouse genes significantly (average difference in Affymetrix chips, AD > 200) expressed in at least one of the 45 tissues, a random sample of 1600 genes was selected, preprocessed as described above, and visualized based on their profile of expression in the tissues. The variance in each tissue was normalized to unity.
The third data set was created for an additional validation of the learning metrics. The data were taken from the same publication as the second one [13], but now consisted of over 13000 human genes measured in 46 tissues. From these genes, a set of genes with known homologues in mouse and expressed (AD > 200) at least in one human tissue, were selected, resulting in 3724 genes. The comparison of different preprocessing methods (logarithm, normalized tissue variances, none) and distance metrics (Euclidean, inner product) for the third data set by k-nearest neighbors method resulted in the use of inner product as a similarity metric, and with no normalizations.
The auxiliary data for the third, human gene expression data was derived from the expression level of homologous mouse genes. Each class corresponded to one mouse tissue, and human gene was assigned to the class, if the homologous mouse gene was clearly expressed in that tissue. The limit was that it must belong to the fourth quartile of that gene's expression over all mouse tissues. Only those mouse tissues (21) for which there was an equivalent in human tissues were considered as classes.
Methods for visualizing similarity
Hierarchical clustering constructs a tree or dendrogram that visualizes similarity and clusteredness of data. For an example tree of gene expression data, see [7]. Data samples are located in the leaves of the tree, and similar samples occur in proximate branches. There are several variant methods for constructing the trees [3]. Here we will use the Cluster program by Eisen [16] that progressively agglomerates pairs of most similar clusters together. The program offers three variants of the clustering algorithm that differ on how the distance between clusters is defined. The first variant is centroid linkage, where the distance between clusters is defined as the distance between the means of the clusters. The second variant is complete linkage, where the distance between clusters is defined as the maximum distance between points in the clusters to be joined, and the third variant is single linkage, where the distance between clusters is defined as the minimum distance between points in the two clusters.
The tree produced by the clustering algorithm can be cut at any level to obtain disjoint clusters. Here, we are not interested in clusters per se, however, but in the visualization of similarity. The hierarchical clustering algorithms do not directly define such similarity, so we have devised two different definitions that are the best we could think of. The simple method is to order the leaves into a linear order according to how far from each other they are in the tree. The ordering is not unique; we have fixed it by using the method recommended by Eisen: in non-unique cases, use the order provided by a one-dimensional SOM.
Since it can be argued that ordering by the one-dimensional SOM is somewhat arbitrary, we additionally include an alternative that is in a way more justified. Distance between leafs is the distance measure directly induced by the dendrogram, that is, the ultrametric distance [3].
The self-organizing map (SOM) [4] is an algorithm that maps high-dimensional data nonlinearly onto a low-dimensional lattice in a topology-preserving manner. As with hierarchical clustering, the SOM can be used as both a nonlinear projection and a clustering method; clusters can be extracted from the computed SOM (see e.g. [20]).
The SOM is a discrete lattice of map nodes (marked by the dots and labels in Fig. 8). There is a model vector mi attached to each map unit i. A data sample x is projected onto the SOM display to the node having the closest model vector mw, defined in the basic SOM by
![]()
Here d is the distance measure, which in this paper is the inner product (correlation).
The SOM algorithm computes such values for the model vectors that (i) the projection becomes ordered: proximate samples on the SOM display are similarly proximate in the data space, and (ii) the projection models the data distribution; each model vector becomes the centroid of all data samples mapped to it and to its neighborhood on the map display.
There are several variants of the SOM algorithm; here we describe the original sequential one that we will later complement with new metrics. We used this 'vanilla' version of the algorithm in its basic form and without any tricks not found in basic textbooks, to avoid biasing the study in favor of SOMs.
During the iterative computation of the SOM, at step t a data sample x(t) is selected randomly, and the model vectors are updated toward the data sample according to
![]()
If inner product is used as the distance measure, the model vectors should be normalized after the adaptation step. Here, hw(x), i = α(t) exp (-d(w,i) / 2σ(t)2) is the neighborhood function, where d(w,i) is the distance of the units w and i on the SOM lattice, and α(t) and σ(t) are piecewise-linearly decreasing coefficients.
Clustering can be visualized on a SOM display using the U-matrix [[21], see Fig. 8]. In the gray-shade display, the light areas contain genes that are mutually more similar than on the dark areas. Hence, very light areas are clear clusters and dark stripes are gaps in between them. Technically, a hexagon is added in between each pair of SOM units and shaded according to the distance between their model vectors. The shade of the hexagons of the map units is the median of the neighboring hexagons.
On a SOM display, the similarity can be defined simply as the distance on the display plane. This measure does not, however, take into account the density of the model vectors that is visualized by the U-matrix. Hence, we have used distances along minimal paths on the map lattice, with weights equal to the distances between the model vectors. On light areas, such distances are shorter and on dark areas they are longer.
The SOMs of the yeast gene expression data were of the size of 20 × 25 map units (about 3 genes in a unit on the average), and were computed in two phases for a conservative number of iterations. In the first, organizing phase σ(t) decreased from 11 to 3, and α(t) from 0.2 to 0.02. In the second, fine-tuning phase σ(t) decreased from 3 to 1, and α(t) from 0.02 to zero. The best map of 4 randomly initialized SOMs was selected according to the (local) cost function [4].
Similarly, the SOMs of the mouse gene expression data were of the size of 22 × 27 map units (about 2.7 genes in a unit on the average). In the first, organizing phase σ(t) decreased from 11 to 3, and α(t) from 0.2 to 0.02. In the second, fine-tuning phase σ(t) decreased from 3 to 1, and α(t) from 0.02 to zero. The best map of 7 randomly initialized SOMs was selected according to the (local) cost function [4].
Multidimensional scaling (MDS) attempts to represent the data as points in a small-dimensional space such that all pairwise distances of data points are preserved. It can be used for constructing a non-linear projection from the high-dimensional expression space to a two-dimensional display plane.
There are several variants of multidimensional scaling that differ in the details of the cost function. We will compare two of them, Sammon's projection and non-metric multidimensional scaling (NMDS), that have favorable properties for the used trustworthiness measure. Sammon's projection [22] minimizes the mean-square error in the pairwise distances, normalized by the original distances. Hence, it emphasizes the preservation of short distances, which is important for trustworthiness. Non-metric multidimensional scaling [23] attempts to preserve the rank order of the distances; the rank order is used to measure errors in the trustworthiness measure.
Sammon's projection and non-metric multidimensional scaling do not have parameters to select, but the optimization can get caught in local minima, depending on the initialization. We computed the Sammon's projection with 10 different random initializations and selected the one with the smallest cost. Non-metric multidimensional scaling was computed only once because of the very long computational time.
Measuring trustworthiness and detecting genes for which the visualization is suspect
When visualizing similarities of data samples, the local similarities are the most salient: the first perceptions are which samples are proximate, and which proximate samples form groups. Hence, to measure how trustworthy a visualization is, we should focus on the preservation of local similarities, i.e., the proximities. To avoid biases in the comparison studies, we will use a simple non-parametric measure of whether samples within a set of closest samples on the display are in fact closest in the expression space as well.
Let us first consider some alternative measures. The most straightforward way of defining the neighborhood would be to fix a radius and include all samples within the ball with the fixed radius. The problem would be that, since the density of data varies, the amount of data within the ball would similarly vary considerably as well. Additionally, selecting a good neighborhood radius would require prior knowledge of the data density. These problems can be solved by defining the neighborhood to consist of the k nearest neighbors, where k is selected based on the number of nearby samples we are interested in analyzing.
The second decision that needs to be made is how to measure similarity preservation within the neighborhood of proximate samples. In principle, preservation of all distances could be directly measured, but we discounted this possibility because it would bias the study in favor of MDS methods that try to directly preserve all distances. We considered it enough that the samples are within the neighborhood. In any case, preservation of distances within the neighborhoods is taken into account when one varies the size of the neighborhood, which we did in the experiments.
The third decision that needs to be made is how to measure errors in trustworthiness for the samples that are visualized proximate but are in fact different. The simplest measure would be counts of erroneous data samples. This would, however, be only a rough measure and hence not very discriminative between the methods. Hence, we decided to measure the distance from the neighborhood even though it might bias the results slightly towards favoring MDS methods. We made the (arbitrary) decision to measure the distances in a rank scale.
Based on the above reasoning, we ended up in a measure of assessing the trustworthiness of visualizations. We consider a projection onto a display trustworthy if the set of k closest neighbors of a sample on the display are also close by in the original space. This is measured for all data samples.
More formally, let N be the number of data samples and r(xi, xj) be the rank of the data sample xj in the ordering according to distance from xi in the original data space. Denote by Uk(xi) the set of those data samples that are in the neighborhood of sample xi in the visualization display but not in the original data space. Our measure of trustworthiness of the visualization, M1, is defined by
![]()
where A(k) = 2/(Nk (2N - 3k - 1)) scales the values between zero and one. The worst attainable values of M1 may, at least in principle, vary with k, and were estimated in Figures 2 and 3 with random projections and with random neighborhoods.
Trustworthiness is one side of the coin; the other is that some neighborhoods of k points in the original space may not be preserved because of discontinuities in the projection. As a result of the latter kinds of errors, not all proximities existing in the original data are visible in the visualization.
The errors caused by discontinuities may be quantified as follows, analogously to the errors in trustworthiness. Let Vk(xi) be the set of those data samples that are in the neighborhood of the data sample xi in the original space but not in the visualization, and let 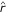 (xi, xj) be the rank of the data sample xj in the ordering according to distance from xi in the visualization display. The effects of discontinuities of the projection are quantified by how well the original neighborhoods are preserved, measured by
(xi, xj) be the rank of the data sample xj in the ordering according to distance from xi in the visualization display. The effects of discontinuities of the projection are quantified by how well the original neighborhoods are preserved, measured by
![]()
In case of ties in rank ordering, all compatible rank orders are assumed equally likely, and averages of the error measures are computed.
There exists a simple way of increasing the trustworthiness of a display: discarding the samples for which the display is the least trustworthy, and analyzing them separately. We will do this by iteratively finding the data sample that reduces most the trustworthiness, and removing it from the visualization. The process is continued until a suitable number of the most untrustworthy data samples have been removed, or a desirable level of trustworthiness has been attained. A similar method can be used to find where the visualization has broken the continuity of a neighborhood. The idea is to study which neighborhood sample pairs reduce M2 the most. Locating the sample representing the center of the neighborhood and the sample missing from the neighborhood on the visualization will reveal if separate areas on the display are in fact close by in the data space.
Learning metrics
The learning metrics principle
The learning metrics [14,15] are based on the assumption that changes in the primary data space are important if they cause changes in another (auxiliary) data space.
Formally, denote a primary data sample by x and its functional class by c. During learning, the data occurs in pairs (x, c). The squared distance measure of the data space is changed locally to measure the important differences, that is, the differences among the distributions of the functional classes p(c|x). When the differences are measured by the Kullback-Leibler divergence DKL, the distances become locally
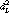 (x, x + dx) ≡ DKL (p(c|x)||p(c|x + dx)) = dxT J(x) dx, (5)
(x, x + dx) ≡ DKL (p(c|x)||p(c|x + dx)) = dxT J(x) dx, (5)
where J(x) is the Fisher information matrix with parameters x. The conditional distribution p(c|x) can be computed using the Bayes rule from a standard estimator for the joint distribution, such as Mixture Discriminant Analysis (MDA2) [24], or obtained directly from a "mixture of experts" [25]. For more details see [14]. The metric can in principle be extended to non-local distances by computing (approximate) path integrals, but for computational reasons we resort to the local approximations. The approximation has worked satisfactorily for nearest-neighbor searches in empirical tests, particularly when complemented with a kind of regularization: in practice the metric will often be singular for very high-dimensional spaces, and hence we will add to it a portion of the Euclidean distance,
(x, x + dx) ≡ dxT [λI + (1 - λ) J(x)] dx, (6)
where I is the identity matrix. The coefficient λ is selected using a validation set. The regularization makes the local approximations more feasible for non-local distances as well.
A difference in this paper, compared to the earlier works on learning metrics, is that the yeast data set lies on the surface of a hypersphere. For such data, the density estimators should also be defined on the hypersphere. Technically, instead of using Gaussian kernels, we used the so-called von Mises-Fisher kernels that are analogs of Gaussians on the hypersphere [26]. (We used 30 kernels.) The local distances (6) are still Euclidean on the hypersphere, but in practice non-local distances also need to be computed. When computing distances from x, we have projected all vectors to the tangent plane of the hypersphere at x, with the distance from x scaled to be equal to the arc length from x. Distances on the tangent plane are then computed with (6).
Self-organizing map in the new metric
In the first step of a SOM iteration, the best matching unit is sought in the new metric dL (Eq. 6; cf. also the discussion after the equation). The steepest descent update rule for learning metrics turns out [14] to be the same as in the Euclidean metric. Here, the update is applied in the tangent plane, and the results are transformed back to the hypersphere. It can be shown that the resulting update rule moves mi toward x along the shortest route on the hypersphere, such that their angle reduces by the fraction given by hwi(t).
The underlined genes in Figure 8, for which the metric had changed the most, were found as follows. We sought 20 nearest neighbors of each gene, in both the old inner product metrics and the learning metric. The two sets were compared, and the proportion of neighbors that had remained the same was computed. The 202 genes with the smallest proportion (at most 13 neighbors remained the same) were selected.
Authors' contributions
SK, EC, JN, and PT developed the overall plan. SK, JN and MO were responsible for the results with learning metrics (MO did the actual simulation). SK and JV were responsible for measuring the trustworthiness of the visualization methods (JV did the simulations). PT and EC carried out the biological analysis of the results. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgments
This work was supported by the Academy of Finland, in part by grants 50061 and 52123. We wish to thank Mr. Arto Klami and Mr. Leo Lahti for help with some of the simulations.
Contributor Information
Samuel Kaski, Email: samuel.kaski@hut.fi.
Janne Nikkilä, Email: janne.nikkila@hut.fi.
Merja Oja, Email: merja.oja@hut.fi.
Jarkko Venna, Email: jarkko.venna@hut.fi.
Petri Törönen, Email: toronen@hytti.uku.fi.
Eero Castrén, Email: Eero.Castren@uku.fi.
References
- Mao J, Jain AK. Artificial neural networks for feature extraction and multivariate data projection. IEEE Trans Neural Networks. 1995;6:296–317. doi: 10.1109/72.363467. [DOI] [PubMed] [Google Scholar]
- Goodhill GJ, Sejnowski TJ. Unifying objective function for topographic mappings. Neural Comput. 1997;9:1291–1303. [Google Scholar]
- Jain AK, Dubes RC. Algorithms for Clustering Data. New Jersey: Prentice Hall, Englewood Cliffs; 1988. [Google Scholar]
- Kohonen T. Self-Organizing Map. 3. Berlin: Springer; 2001. [Google Scholar]
- Borg I, Groenen P. Modern Multidimensional Scaling. Springer; 1997. [Google Scholar]
- Bhattacharjee A, Richards WG, Staunton J, Li C, Monti S, Vasa P, Ladd C, Beheshti J, Bueno R, Gillette M, Loda M, Weber G, Mark EJ, Lander ES, Wong W, Johnson BE, Golub TR, Sugarbaker DJ, Meyerson M. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci USA. 2001;98:13790–13795. doi: 10.1073/pnas.191502998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes TR, Marton MJ, Jones AR, Roberts CJ, Stoughton R, Armour CD, Bennett HA, Coffrey E, Dai H, He YD, Kidd MJ, King AM, Meyer MR, Slade D, Lum PY, Stepaniants SB, Shoemaker DD, Gachotte DD, Chakraburtty K, Simon J, Bard M, Friend SH. Functional discovery via a compendium of expression profiles. Cell. 2000;102:109–126. doi: 10.1016/s0092-8674(00)00015-5. [DOI] [PubMed] [Google Scholar]
- Kaski S, Nikkilä J, Törönen P, Castrén E, Wong G. Analysis and visualization of gene expression data using self-organizing maps. In Proceedings of NSIP-01, IEEE-EURASIP Workshop on Nonlinear Signal and Image Processing: June 3–6 2001; Baltimore, Maryland; Proceedings on CD-ROM. 2001.
- Tamayo P, Slonim D, Mesirov J, Zhu Q, Kitareewan S, Dmitrowsky E, Lander ES, Golub TR. Interpreting patterns of gene expression with self-organizing maps: Methods and application to hematopoietic differentiation. Proc Natl Acad Sci USA. 1999;96:2907–2912. doi: 10.1073/pnas.96.6.2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Törönen P, Kolehmainen M, Wong G, Castrén E. Analysis of gene expression data using self-organizing maps. FEBS Lett. 1999;451:142–146. doi: 10.1016/S0014-5793(99)00524-4. [DOI] [PubMed] [Google Scholar]
- Torkkola K, Gardner RM, Kaysser-Kranich T, Ma C. Self-organizing maps in mining gene expression data. Information Sciences. 2001;139:79–96. doi: 10.1016/S0020-0255(01)00158-X. [DOI] [Google Scholar]
- Su AI, Cooke MP, Ching KA, Hakak Y, Walker JR, Wiltshire T, Orth AP, Vega RG, Sapinoso LM, Moqrich A, Patapoutian A, Hampton GM, Schultz PG, Hogenesch JB. Large-scale analysis of the human and mouse transcriptomes. Proc Natl Acad Sci USA. 2002;99:4465–4470. doi: 10.1073/pnas.012025199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaski S, Sinkkonen J, Peltonen J. Bankruptcy analysis with self-organizing maps in learning metrics. IEEE Trans Neural Networks. 2001;12:936–947. doi: 10.1109/72.935102. [DOI] [PubMed] [Google Scholar]
- Sinkkonen J, Kaski S. Clustering based on conditional distributions in an auxiliary space. Neural Comput. 2002;14:217–239. doi: 10.1162/089976602753284509. [DOI] [PubMed] [Google Scholar]
- EisenLab http://rana.lbl.gov/
- Venna J, Kaski S. Neighborhood preservation in nonlinear projection methods: An experimental study. In: Dorffner G, Bischof H, Hornik K, editor. In Proceedings of the International Conference on Artificial Neural Networks – ICANN 2001; Vienna. Berlin: Springer; 2001. pp. 485–491. [Google Scholar]
- Oja M, Nikkilä J, Törönen P, Wong G, Castrén E, Kaski S. Exploratory clustering of gene expression profiles of mutated yeast strains. In: Zhang W, Shmulevich I, editor. In Computational and Statistical Approaches to Genomics. Boston, MA: Kluwer; 2002. pp. 65–78. [Google Scholar]
- Mewes HW, Frishman D, Guldener U, Mannhaupt U, Mayer K, Mokrejs M, Morgenstern B, Munsterkotter M, Rudd S, Weil B. MIPS: a database for genomes and protein sequences. Nucleic Acids Res. 2002;30:31–34. doi: 10.1093/nar/30.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vesanto J, Alhoniemi E. Clustering of self-organizing map. IEEE Trans Neural Networks. 2000;11:586–600. doi: 10.1109/72.846731. [DOI] [PubMed] [Google Scholar]
- Ultsch A. Self-organizing neural networks for visualization and classification. In: Opitz O, Lausen B, Klar R, editor. In Information and Classification. Berlin: Springer-Verlag; 1993. pp. 307–313. [Google Scholar]
- Sammon JW., Jr A nonlinear mapping for data structure analysis. IEEE Trans Comput. 1969;C-18:401–409. [Google Scholar]
- Kruskal JB. Multidimensional scaling by optimizing goodness of fit to a nonmetric hypothesis. Psychometrica. 1964;29:1–27. [Google Scholar]
- Hastie T, Tibshirani R, Buja A. Flexible discriminant and mixture models. In: Kay J, Titterington D, editor. In Neural Networks and Statistics. Oxford University Press; 1995. [Google Scholar]
- Peltonen J, Klami A, Kaski S. Learning More Accurate Metrics for Self-Organizing Maps. In: Jos R Dorronsoro, editor. In Proceedings of the International Conference on Artificial Neural Networks – ICANN 2002; Madrid. Berlin: Springer; 2002. pp. 999–1004. [Google Scholar]
- Mardia KV. Statistics of directional data. JR Stat Soc [Ser B] 1975;37:349–393. [Google Scholar]