

Preface
NF-κB transcription factors play a key role in many physiological processes, such as innate and adaptive immune responses, cell proliferation, cell death, and inflammation. It has become clear that aberrant regulation of NF-κB and the signaling pathways that control its activity are involved in cancer development and progression, as well as in resistance to chemo- and radiotherapy. This review discusses recent cancer-genetics and cancer-genome evidence for the involvement of NF-κB in human cancer, and discusses the therapeutic potential and benefit of targeting NF-κB in cancer and the possible complications and pitfalls of such an approach.
Keywords: Animals; Anticarcinogenic Agents; therapeutic use; Drug Resistance, Neoplasm; Humans; NF-kappa B; antagonists & inhibitors; metabolism; Neoplasms; drug therapy; metabolism; pathology; Signal Transduction; drug effects
Keywords: NF-kappaB, Signal transduction, cancer, B-cell lymphoma
Introduction
Cancer is a leading cause of death in industrialized countries1. Although mortality rates have declined in recent years due to earlier detection and more options in treatment, most cancers remain incurable. It is important to stress that cancer is not one but many different diseases, each with distinct characteristics and therapeutic requirements and options. The progressive sequence of mutations and epigenetic alterations of cancer genes promotes the malignant transformation of cancer progenitor cells by disrupting key processes involved in normal growth control and tissue homeostasis2 (Box 1). In addition, genomic instability is a common, if not universal, feature of advanced tumors. Thus, one major challenge for cancer researchers is to distinguish cancer-causing mutations from irrelevant alterations linked to complex cancer genotypes. In addition to such changes that are intrinsic to the malignant cell, tumor development and progression are also dependent on the microenvironment surrounding the malignant cell, which in many cases is inflammatory in nature3. The critical role played by the inflammatory microenvironment in the natural history and progression of most cancers offers new opportunities to therapeutic intervention based on targeting the inflammatory components of the tumor rather than the malignant cell, which is quick to mutate and acquire drug resistance4, 5.
Box 1. The six hallmarks of cancer cancera.
Self-sufficiency in proliferative growth signals
Insensitivity to growth inhibitory signals
Evasion of apoptosis
Acquisition of limitless replicative potential
Induction of angiogenesis
Induction of invasion and metastasis
The current armamentarium of medical oncology includes many active anticancer agents that are applied across tumor types. None of these broadly active anticancer drugs is an ideal panacea. Most drugs have a small therapeutic index and barely discriminate between malignant and normal cells, a problem that is further amplified by the almost inevitable onset of resistance and subsequent relapse. In recent years, the focus has shifted to new molecular therapeutics that target specific signaling pathways driving inappropriate cell growth and survival, thus offering the promise of greater specificity coupled with reduced systemic toxicity. Such strategies rely on new approaches for studying cancer genetics and cancer genomes, such as gene-expression profiling using microarrays, DNA sequencing, and genome scanning for gene copy-number alterations6–13. The successful development of a rationally designed, molecularly targeted therapy for the treatment of a specific cancer is best illustrated by the clinical launches of antibody-based therapies for the treatment of ErbB-2-/Her2 positive breast cancer, such as Herceptin14, 15, and the use of Gleevec (STI571/imatinib) as a kinase inhibitor for the treatment of Bcr-Abl-positive chronic myeloid leukemias16.
In the validation and selection of molecular targets and pathways for therapeutic intervention in cancer, the frequency with which a particular target or a pathway undergoes mutation or deregulation is a valuable indicator of its importance in the malignancy process and of the potential use of a drug that acts on that target or pathway. An important signaling pathway often altered in human cancers is the IKK/NF-κB signaling module. Aberrant NF-κB regulation has been observed in many cancers, including both solid and hematopoietic malignancies (Fig. 1), and NF-κB can affect all six hallmarks of cancer through the transcriptional activation of genes associated with cell proliferation, angiogenesis, metastasis, tumor promotion, inflammation and suppression of apoptosis17–22 (Fig. 2). Various mouse models of cancer in which IKK/NF-κB activation has been blocked by genetic means have highlighted the key role of NF-κB as a critical promoter of inflammation-linked cancers23–26. One of the most documented functions of NF-κB is its ability to promote cell survival through induction of target genes whose products inhibit the apoptotic machinery in both normal and malignant cells20, 27. NF-κB can also prevent programmed necrosis by inducing genes encoding antioxidant proteins27. Since tumor cells often use NF-κB to achieve resistance to anticancer drugs and radiations, inhibition of NF-κB activation appears as a promising option to improve the efficacy of conventional anti-cancer therapies28. However, only recently has cancer genomics proved useful to demonstrate the key role of NF-κB in carcinogenesis and identify critical steps that drive its activation. One difficulty that may have slowed down the realization that NF-κB is frequently altered in cancer is that in most cases NF-κB is activated in malignant cells not due to intrinsic mutations but in response to inflammatory stimuli originating from the microenvironment5. In other cases, which will be described below, the oncogenic mutations reside in genes that encode upstream regulators of NF-κB activity.
Figure 1. Human ca cancers that have been linked to constitutive NF-κB activation.
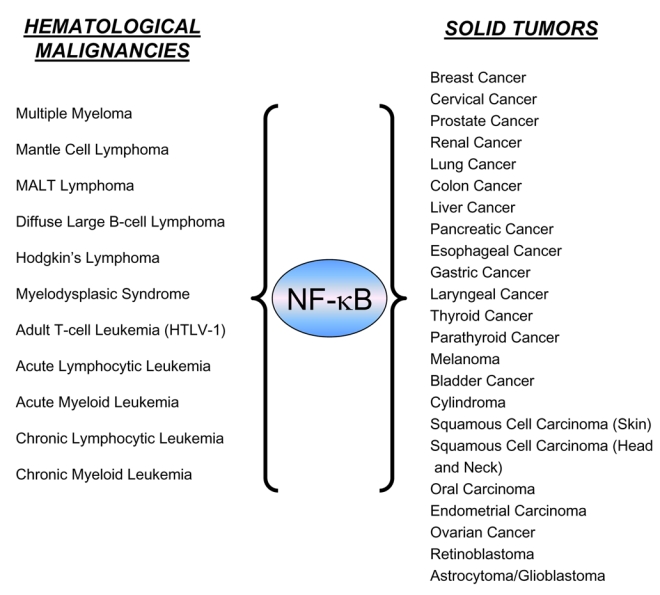
Various hematological malignancies and solid tumors that exhibit constitutive NF-κB activation (adapted from ref. 17).
Figure 2. NF-κB target genes involved in cancer development and progression.
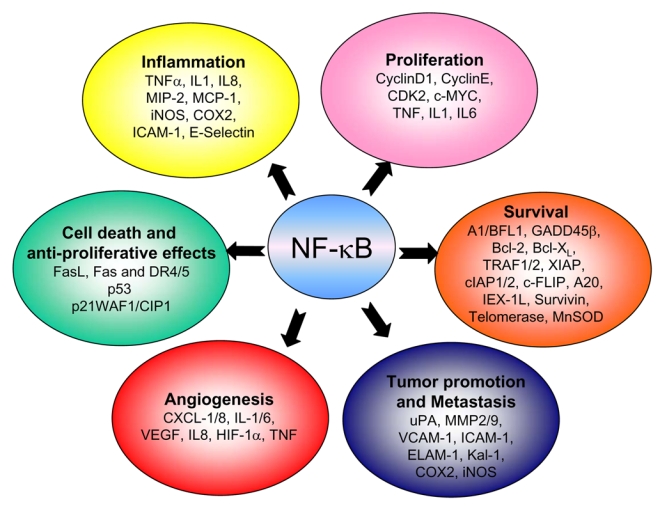
NF-κB activation affects all six hallmarks of cancer through the transcription of genes involved in cell proliferation, angiogenesis, metastasis, inflammation and suppression of apoptosis as identified in both cell lines and tissue samples (adapted from r ref. 17).
NF-κB transcription factors and their signaling pathways
In mammals, the NF-κB family is composed of five members, RelA (p65), RelB, cRel (Rel), NF-κB1 (p50 and its precursor p105) and NF-κB2 (p52 and its precursor p100)29. These proteins form homo- and heterodimeric complexes, the activity of which is regulated by two major pathways. The first one, known as the classical or canonical NF-κB activation pathway, mainly applies to RelA:p50 dimers, which, under non-stimulated conditions, are sequestered in the cytoplasm through interactions with inhibitory proteins of the IκB family. Following stimulation with a broad range of stimuli such as TNF-α or IL-1, viruses, genotoxic agents and ionizing radiation, the IκB molecules are phosphorylated by the IκB kinase complex (IKK) at specific serine residues, leading to their ubiquitination and degradation by the proteasome pathway. RelA:p50 dimers are subsequently released and free to translocate to the nucleus where they activate transcription of various target genes30. This pathway plays a major role in the control of innate immunity and inflammation31, 32. The second pathway, the so-called alternative or non non-canonical NF-κB signaling pathway, is stimulated by a more restricted set of cytokines that all belong to the TNF superfamily (e.g. BAFF, CD40L, LTβ). This pathway involves the upstream kinase NF-κB-inducing kinase (NIK) which activates IKKα, thereby leading to the phosphorylation and proteasome-dependent processing of p100, the main RelB inhibitor, thus resulting in RelB:p52 and RelB:p50 nuclear translocation and DNA binding33–37. Most importantly, all studies point out to a crucial role for the alternative pathway in controlling the development, organization and function of secondary lymphoid organs and B-cell maturation and survival32, 38.
The activation of the canonical as well as the alternative NF-κB pathways relies on the inducible phosphorylation of IκB inhibitory proteins (IκBα for the classical pathway and p100 for the alternative pathway) by the IKK complex and its subunits30. IKK is composed of two catalytic subunits, IKKα and IKKβ, and a regulatory subunit, NEMO/IKKγ. Disruption of genes encoding individual subunits has demonstrated that IKKβ and NEMO/IKKγ are required for activation of the classical NF-κB pathway by inflammatory signals, a pathway in which IKKα does not play an essential role. In contrast, RelB:p50 and RelB:p52 activation is absolutely dependent on IKKα, but not on IKKβ or NEMO/IKKγ33, 39.
Multiple myeloma and genetic abnormalities in NF-κB pathways
The most dramatic and broad example for genetic alterations that lead to activation of NF-κB in cancer was recently identified through the application of oncogenomics in multiple myeloma (MM). MM is a B cell neoplasia that accounts for 1% of all cancers and more than 10% of total hematological malignancies. MM is mainly characterized by unrestrained accumulation of antibody-secreting plasma B cells in the bone marrow, attributed to loss of apoptosis control and cell-cycle deregulation40–42. The bone marrow microenvironment has also been shown to contribute to the pathogenesis and progression of MM43. Despite advances in chemotherapy and stem cell transplantation, which have improved survival rates, MM remains an incurable disease. Thus, new treatment approaches are needed to improve the outcome of MM therapy and provide patients with longer disease-free survival. Of all the different signaling pathways constitutively activated in primary MM cells, NF-κB has recently emerged as one of the most important drivers of the tumor-promoting machinery.
Biochemical evidence
Constitutive nuclear NF-κB activity has been described in many human MM cell lines and primary myeloma cells44–46. Furthermore, MM cells have been shown to be sensitive to growth inhibition and induction of apoptosis upon treatment with various inhibitors of NF-κB signaling, including proteasome inhibitors, inhibitors of IκB phosphorylation, and IKK inhibitors47–51. Inhibition of NF-κB correlates with a decrease in expression of known anti- apoptotic NF-κB target genes, including Bcl-xL, XIAP, cIAP1, and cIAP2, as well as proliferative genes, such as CyclinD1 and IL-646, 49, 51, 52. Activation of NF-κB in bone marrow stromal cells (BMSCs) also contributes to MM pathogenesis: adherence of MM cells to BMSCs induces NF-κB-dependent cytokine transcription and secretion (e.g. IL-6, TNF-α and BAFF) by BMSCs, that in turn promote MM cell growth and survival survival53, 54, through paracrine mechanisms. NF-κB activation has also important roles in some clinical manifestations of MM, including formation of osteolytic lesions that occur in about 70% of MM patients, mostly through RANK/RANKL-mediated NF-κB activation in osteoclasts50. Osteoclasts in turn secrete soluble factors (e.g. MMPs, BMP, IGF-1) that contribute to bone metastases and MM cell proliferation and survival55–57.
Clinical evidence
Bortezomib (Velcade®, Millenium Pharmaceuticals, Inc, formerly PS-341) is a reversible 26S proteasome inhibitor that has been recently approved by the Federal Drug Administration (FDA) and the European Regulatory Agency (EMEA) for the treatment of MM. Several phase III trials have shown considerable clinical efficacy in combined therapy as well as a single agent with manageable side effects58, 59. Although bortezomib affects other signaling pathways, its efficacy may in part be due to inhibition of NF-κB activity60–62. In addition, gene expression profiling using microarrays allowed the identification of a subgroup of MM with a NF-κB signature that characterizes myeloma cells more sensitive to bortezomib63. Thalidomide (Thalomid®, Celgen Corp.), lenalidomide (Revlimid®, Celgene Corp), and arsenic trioxide (Trisenox, Cephalon, Inc.) are also active against MM64, 65. Although they can inhibit NF-κB, it is not clear whether their clinical activity is actually due to NF-κB inhibition.
Genetic evidence
Beyond these observations, the most striking evidence validating NF-κB as a critical player in the pathogenesis of MM came from two very recent studies that have led to identification of mutations in genes encoding regulators of NF-κB signaling pathways66, 67. Two laboratories used multi multi-pronged genomic and gene expression profiling approaches to identify NF-κB activating mutations in one fifth of several hundred of MM cell lines and patient samples. Gain-of-function mutations were identified in genes encoding positive regulators of NF-κB signaling, such as NIK, NF-κB1 (encoding p105/p50), NF-κB2 (encoding p100/p52), and three receptors of the TNFR superfamily, CD40, LTβR, and TACI (Fig. 3). Loss-of-function mutations occurred in genes encoding negative regulators of NF-κB activity, including TRAF2 (a negative regulator of alternative NF-κB but positive regulator of classical NF-κB), TRAF3, cIAP1/2, and CYLD (Fig. 3). Most of these genetic alterations result in activation of both the classical and alternative NF-κB pathways as evaluated by p100 processing, p52 nuclear localization, IκBα phosphorylation, increased RelA, p50, RelB and p52 DNA-binding activity, and increased NF-κB target gene expression66, 67.
Figure 3. NF-κB gain B gain- and loss loss-of of-function mutations in multiple myeloma.
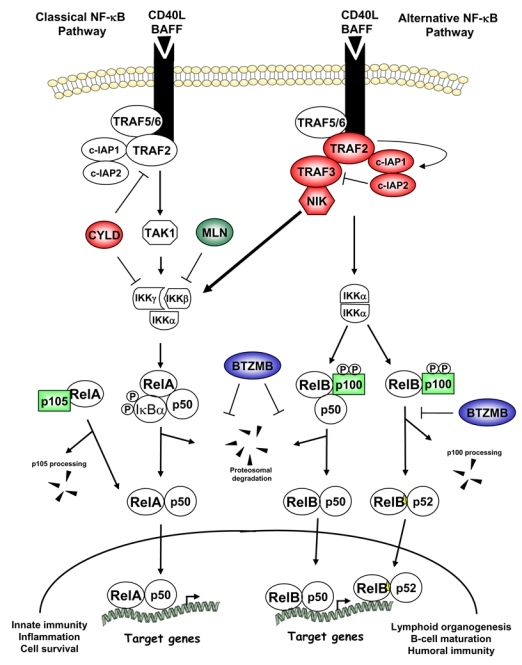
The classical and alternative NF-κB signaling pathways are represented downstream of CD40 and BAFF-R. Arrows indicate activating steps, and bars indicate inhibitory steps. Negative regulators subject to loss-of-function mutations are in red, and positive regulators affected by gain-of-function mutations are in green. Either type of mutations lead to NF-κB constitutive activation through NIK stabilization. The proteasome inhibitor bortezomib (BTZ) blocks both classical and alternative NF-κB signaling, whereas the IKKβ-specific inhibitor MLN120B (MLN) only blocks the classical NF-κB pathway. See text for further details.
NIK stabilization and NF-κB activation
One major consequence of the detected mutations in MM cells is the stabilization of NIK. The high constitutive amounts of NIK protein in MM were shown to activate both classical and alternative NF-κB signaling, and to be required for MM cell growth and survival66, 67. Normally, NIK is rapidly degraded in non-stimulated cells and this degradation is required to keep NIK activity at a low basal level68–70. Recent work indicates that NIK degradation in resting cells depends on its association with TRAF3, which serves as an adaptor that recruits NIK to an ubiquitin ligase complex composed of TRAF2 and cIAP1/271. Within this complex cIAP1/2 are directly responsible for the degradative K48-linked polyubiquitination of NIK72, 73. Receptor (CD40, LTβR, BAFF-R) engagement results in recruitment of the TRAF3: TRAF2: cIAP1/2 complex to the receptor, leading to rapid activation of TRAF2, which in turn ubiquitinates cIAP1/2 via a non-degradative K63-linkage and thereby enhance their ubiquitin ligase activity, which is now directed towards TRAF3 leading to its K48-linked ubiquitination and eventual degradation degradation71. Once TRAF3 is degraded, NIK can no longer interact with the TRAF2: cIAP1/2 complex, resulting in the stabilization of newly synthesized NIK and its eventual autophosphorylation and activation. NIK overexpression due to amplification or translocation of the NIK locus, or enhanced stability due to amino acid substitutions that disrupt NIK’s ability to bind TRAF3 occur in MM66, 67. In addition, many MM cells carry TRAF3 inactivating mutations and present accumulation of NIK66, 67. Importantly, 90% of patients with low TRAF3 amounts respond to bortezomib, with a significant increase in survival67. Collectively, these results suggest that stabilization of NIK, and subsequent NF-κB activation, is associated with bortezomib sensitivity and increased survival. Other mutations that lead to NIK stabilization include deletions of the linked cIAP1 and cIAP2 loci, and loss of TRAF266, 67. The phenotypic consequences of all of these mutations appear similar.
Based on our current knowledge it was not surprising to find that NIK stabilization results in constitutive activation of the alternative NF-κB pathway, and increased B cell survival. Analysis of aly mice, which express a non-functional NIK mutant, revealed an autonomous B cell defect, and inability to activate the alternative NF-κB pathway, a defect that is also exhibited in NIK-deficient cells74–77. Moreover, induction of the alternative NF-κB signaling pathway, especially in response to BAFF and CD40L, is crucial for normal B cell survival36, 37, 78, 79, and TRAF3 and TRAF2 both behave as negative regulators of B cell survival downstream of BAFF-R, in part via the suppression of NF-κB2/p100 processing80. Finally, it has been reported that increased expression of NIK in primary B cells, especially a non-TRAF3-binding mutant, leads to expansion of the B-cell compartment and extended B cell survival (M. Schmidt-Supprian, personal communication). Altogether, these data suggest that in response to BAFF and CD40L, the activation of the alternative NF-κB pathway via NIK stabilization is crucial for B cell survival. However, recent work by Staudt and colleagues revealed that inhibition of IKKα, the physiological target for NIK, had no effect on MM cell survival, whereas inhibition of IKKβ by the specific inhibitor MLN120B did66. The control of classical NF-κB signaling by NIK overexpression seems puzzling. Based on the characterization of aly mice as well as of NIK-knockout mice, it was suggested that NIK does not participate in the activation of the classical NF-κB pathway downstream of TNFR in normal cells 77. Nonetheless, NIK overexpression is known to activate classical NF-κB81, and thus NIK accumulation in cancer cells results in aberrant participation of NIK in activation of the classical NF-κB pathway. It should also be noted that NIK is required for the activation of both the classical and alternative NF-κB pathways in response to CD40L and BAFF in Burkitt lymphoma-derived lymphoblastoid cells82.
In sum, these observations lead to the conclusion that NIK overexpression is an important marker and driver of malignant plasma B cells and that NIK-induced IKKβ activation is required for uncontrolled B cell growth and therefore an attractive target for therapeutic intervention.
IAP antagonists and NF-κB constitutive activation
NIK overexpression was detected in MM cells that have intact TRAF3 and instead posses genetic abnormalities that prevent the expression of cIAP1 and cIAP266, 67. These observations provided the first clue that cIAP1 and cIAP2 play a negative role in the control of NIK stability. As discussed above, it is now clear that cIAP1/2 are the ubiquitin ligases that target NIK for degradation, and thus accordingly IAP antagonists can induce NIK stabilization and le lead to strong induction of classical and alternative NF-κB signaling (see below) 72, 73. However, the role of cIAP1/2 in NF-κB regulation and function is more complex. NF-κB suppresses apoptosis by inducing expression of a number of genes whose products inhibit apoptosis, including cIAP1/283. cIAP1/2 belong to the IAP family that counts eight members in human: cIAP1 and cIAP2, XIAP (X-linked IAP), NAIP (neuronal IAP), ML-IAP (melanoma IAP), Ts-IAP (testis-specific IAP), BRUCE (BIR-containing ubiquitin conjugating enzyme), and survivin84, 85. By virtue of their interaction with TRAF2, cIAP1 and cIAP2 are recruited to TNFR1 and TNFR2 signaling complexes, where they negatively modulate caspase-8 activity83, 86–88. cIAP1 and cIAP2 are also RING domain-containing ubiquitin ligases capable of promoting ubiquitination and proteosomal degradation of themselves and several of their binding partners, such as TRAF284, 89, 90 and TRAF391. cIAP1- or cIAP2-deficient mice did not reveal any significant developmental abnormalities or malfunctions in the apoptotic processes92, 93, suggesting that either these proteins are not important during normal development or that the two are fully redundant. In contrast, overexpression of cIAP1 and cIAP2 has been implicated in tumor cell survival 85, and genetic amplification of cIAP1 was suggested to promote tumorigenesis and sustain tumor growth in a mouse model of liver cancer94.
The anti-apoptotic activity of IAPs is counteracted by the natural mammalian IAP antagonist SMAC/DIABLO (second mitochondrial activator of caspases/direct IAP binding protein with low pI), which is released from mitochondria into the cytoplasm upon apoptosis induction95, 96. A number of IAP antagonists that mimic the interaction of SMAC with IAPs have been developed and are collectively called Smac mimics. These compounds promote apoptosis of various cancer cells, including MM cells, either as single agents or in synergy with other proapoptotic stimuli such as TNF-α97–99. Their efficacy was also demonstrated in a humans MM xenograft mouse model98. This activity is perplexing as several recent reports established that IAP antagonists also induced the activation of both the classical and alternative NF-κB pathways72, 73, a complex twist that might limit their use as cancer therapeutics. IAP antagonists induce IκBα phosphorylation, p100 processing, and expression of several NF-κB-dependent genes, including TNF-α, IL-8 and MCP-172, 73. Importantly, whereas blockade of TNF-α signaling suppressed IAP antagonist-induced cell death, it had no effect on NF-κB activation induced by the same molecule72. It was shown that IAP antagonists induced NIK stabilization through inhibition of cIAP1- and cIAP2-dependent-NIK ubiquitination and proteosomal degradation. It was also demonstrated that TRAF2 is a critical scaffolding link between cIAP1 and NIK. Cell stimulation with known inducers of the alternative NF-κB pathway, such as TWEAK, triggered degradation of cIAP1/2 and concomitant stabilization of NIK and p100 processing72. However, using CD40L and BAFF no ligand-induced cIAP1/2 degradation could be detected under conditions where cIAP1/2-induced TRAF3 degradation and NIK stabilization were readily observed71. Altogether, these data confirm that cIAPs are the E3 ligases responsible for NIK degradation, and are thus critical regulators of alternative NF-κB signaling. Since NIK accumulation seems to be equally important for activation of the classical NF-κB pathway in cancer cells (see above), it would be interesting to examine whether cIAP antagonists also induce NIK-dependent act activation of the classical NF-κB pathway in vivo, something that would compromise their therapeutic potential. The dual role of IAP antagonists in inducing apoptosis while activating NF-κB is presented in figure 4. It appears that the pro-apoptosic activity of these compounds is limited to a small number of unique cancer cell lines73, and under many other cases these compounds would actually, activate NF-κB signaling upon NIK stabilization and at the same time can also prevent JNK activation activation91. Thus the net effect may be inhibition of apoptosis rather than induction of apoptosis.
Figure 4. cIAPs and IAP antagonists in the regulation of NF-κB and cell death pathways.
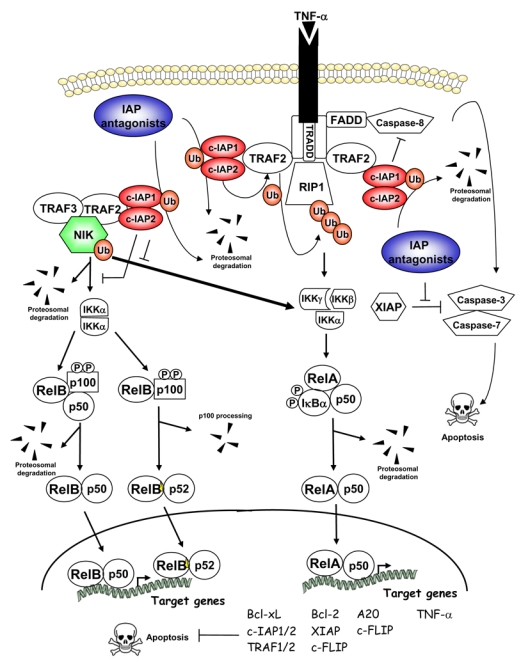
Arrows indicate activating steps, and bars indicate inhibitory steps. IAP antagonists induce cIAP1 and cIAP2 proteosomal degradation, thus leading to activation of the classical and alternative NF-κB signaling through stabilization of NIK. IAP antagonists also induce TNF-α-dependent cell death. See text for further details.
What lessons can we learn from these recent data? First, the recent findings regarding the full biochemical effects of cIAP1/2 inhibition allows a better understanding of the complex mechanisms controlling NIK stability and other aspects of TNFR signaling. We propose the following model: in absence of stimuli, TRAF3 associates with cytoplasmic NIK, and subsequently targets it to E3 complexes containing TRAF2, cIAP1 and cIAP2 which mark NIK for proteosomal degradation. Upon receptor stimulation, these regulators will be sequestered at the cytoplasmic portion of the receptor, resulting in TRAF2 activation, enhanced cIAP1/2 activity and TRAF3 degradation, thus leading to NIK stabilization. Any mutation affecting the binding of these regulators to each other and to NIK would result in NIK stabilization. Smac mimics or cIAP antagonists have the same effect. Overexpression of NIK in turn leads to constitutive activation of the classical and alternative NF-κB pathways and an increase in B cell growth and survival. The remarkable activity of bortezomib in MM patients, especially in those with TRAF3 inactivation, supports the key role of NF-κB in MM pathogenesis. Second, since NIK stabilization appears to be a new marker for malignant B cells, we believe that the specific targeting of NIK may be particularly effective in the treatment of MM and potentially other B-cell malignancies. IKKβ inhibitors should also be effective due to its increased activity in cells where NIK is persistently activated. Third, it is questionable whether IAP antagonists will find use as cancer therapeutics, unless they can selectively induce apoptosis in a broad spectrum of cancer cells despite their proven ability to stabilize NIK and thereby induce NF-κB activation. Only further preclinical studies and clinical trials will define the true potential of Smac mimics as anticancer drugs.
Oncogenomics and NF-κB inhibitors in cancer therapy
The discovery of genetic alterations that lead to constitutive NF NF-κB activition in one fifth of MM patients strikes a strong rationale for the use of NF-κB inhibitors as single-agent therapeutics in this malignancy. It also supports the idea that genomic technology should become an integral part of prospective clinical trials, helping identify which molecularly defined subsets of patients will preferentially respond to NF-κB inhibition. Indeed, a major challenge for cancer researchers and clinicians is how to identify those patients that are most likely to benefit from IKKβ or NF-κB inhibition. Beyond MM, cancer genetics and genomics approaches have been successful in identifying a subgroup of diffuse large B-cell lymphoma ell based on the NF-κB signature of gene expression profiles, and preclinical studies have validated NF-κB as a therapeutic target in this lymphoma subtype100. In addition, carboxy-terminal truncated forms of p100 have been detected in several lymphomas and leukemias, including cutaneous T cell lymphoma and B cell chronic lymphocytic leukemia101. In all cases, these mutations result in nuclear p52 accumulation, thus indicating that these types of cancer should also benefit from NF-κB inhibition. The question, however, is which of the two NF-κB signaling pathways should be targeted in these malignancies: the classical or the alternative? The NF-κB gene expression signature of patients should provide a better prediction for the choice of inhibition, and also allow the development of diagnostic and prognostic biomarkers of the response to NF-κB inhibition. Based on MM studies, NIK is an attractive candidate for inhibition, but not in those cases where processing of truncated p100 variants is NIK independent. Altogether, coupling biomarkers with pathway-targeted therapeutics should ultimately allow treatment of cancer patients to be tailored to their unique genetic abnormalities (Box 2).
Box 2. Translating basic research into new cancer therapeutics.
The fundamental understanding of the cancer genome and cancer signaling pathways leads to:
The identification of new molecular targets driving the molecular pathology and progression of human cancer and the development of drugs acting on these targets.
The identification of new and specific markers of the molecular pathology and the development of new diagnostic, pronostic and pharmacogenomics biomarkers.
Collectively, it enables the targeting of individualized treatments to patients who are most likely to benefit from a given therapy.
Much effort has been made by the pharmaceutical industry to develop IKKβ and other NF-κB inhibitors inhibitors102. However, IKKα inhibitors are yet to be described. An ideal IKK/NF-κB inhibitor to be used in molecularly molecularly-targeted therapy should prevent NF-κB activation without any effects on other signaling pathways, and be more active in malignant cells than in normal cells. One should also consider that excessive and prolonged NF-κB inhibition can be detrimental due to its important role in innate immunity. Thus, although highly efficient NF-κB inhibition is desired during administration of conventional anticancer drugs, NF-κB inhibition should be transient and highly reversible to avoid long long-term immunosuppression. In addition, by selectively targeting specific NF-κB signaling components involved in a particular disease, one may expect to minimize systemic toxicity and avoid broad suppression of innate immunity. Finally, the dose and schedule for delivering the compound needs to be carefully evaluated as a single agent, and its efficacy has to be determinated in combination with other anticancer agents. Several reports have documented the efficacy of IKK/NF-κB inhibitors in triggering apoptosis in cancer cells as single agents, and in combination either with death-inducing cytokines or chemotherapeutic drugs, an and since this area of research is moving at a rapid pace, there is hope that it will be learned how to use such compounds in the clinic. Yet one major pitfall that will have to be addressed before anti-IKK or NF-κB therapies can become successful is the surprising but pronounced ability of NF-κB activation inhibitors to enhance the production of IL-1β and related cytokines due to excessive inflammasome activation during bacterial infections infections103. This adverse effect is due to the ability of NF-κB to promote expression of genes whose products keep IL-1β processing and secretion in check. Perhaps the only way to avoid such complications would be to use IKK and NF-κB inhibitors for only a short duration in combination with broad spectrum antibiotics under well controlled hospital conditions.
Acknowledgments
Our apologies to colleagues whose important contributions are not cited due to space constraints. Many thanks to H. Authier and A. Moor Moore for invaluable technical assistance. This work was supported by grants from the NIH and AACR (M.K.), and Agence Nationale pour la Recherche, Association pour la Recherche sur le Cancer, Belgian InterUniversity Attraction Pole, Ministère de la Recherche/Cancéropole IdF, et Université Paris Descartes (V.B.).
Footnotes
Information taken from ref. 2
Contributor Information
Véronique Baud, IC, Institut Cochin CNRS : UMR8104, INSERM : U567, Université Paris Descartes - Paris V, FR.
Michael Karin, Laboratory of gene regulation and signal transduction University of California, San Diego, US.
References
- 1.World Health Organization. Global action against cancer. Geneva, Switzerland: 2005. [Google Scholar]
- 2.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 3.Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7:211–7. doi: 10.1016/j.ccr.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 4.Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest. 2007;117:1175–83. doi: 10.1172/JCI31537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–59. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 6.Nicolau M, Tibshirani R, Borresen-Dale AL, Jeffrey SS. Disease-specific genomic analysis: identifying the signature of pathologic biology. Bioinformatics. 2007;23:957–65. doi: 10.1093/bioinformatics/btm033. [DOI] [PubMed] [Google Scholar]
- 7.Segal E, Friedman N, Kaminski N, Regev A, Koller D. From signatures to models: understanding cancer using microarrays. Nat Genet. 2005;37 (Suppl):S38–45. doi: 10.1038/ng1561. [DOI] [PubMed] [Google Scholar]
- 8.Lucito R, et al. Representational oligonucleotide microarray analysis: a high-resolution method to detect genome copy number variation. Genome Res. 2003;13:2291–305. doi: 10.1101/gr.1349003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weir BA, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893–8. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang H, Han H, Mousses S, Von Hoff DD. Targeting loss-of-function mutations in tumor-suppressor genes as a strategy for development of cancer therapeutic agents. Semin Oncol. 2006;33:513–20. doi: 10.1053/j.seminoncol.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 11.Wood LD, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–13. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 12.Chanock SJ, et al. Somatic sequence alterations in twenty twenty-one genes selected by expression profile analysis of breast carcinoma carcinomas. Breast Cancer Res. 2007;9:R5. doi: 10.1186/bcr1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yosef N, et al. A supervised approach for identifying discriminating genotype patterns and its application to breast cancer data. Bioinformatics. 2007;23:e91–8. doi: 10.1093/bioinformatics/btl298. [DOI] [PubMed] [Google Scholar]
- 14.Slamon DJ, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–92. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 15.Izumi Y, Xu L, di Tomaso E, Fukumura D, Jain RK. Tumour biology: herceptin acts as an anti-angiogenic cocktail. Nature. 2002;416:279–80. doi: 10.1038/416279b. [DOI] [PubMed] [Google Scholar]
- 16.Druker BJ. STI571 (Gleevec) as a paradigm for cancer therapy. Trends Mol Med. 2002;8:S14–8. doi: 10.1016/s1471-4914(02)02305-5. [DOI] [PubMed] [Google Scholar]
- 17.Basseres DS, Baldwin AS. Nuclear factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progression. Oncogene. 2006;25:6817–30. doi: 10.1038/sj.onc.1209942. [DOI] [PubMed] [Google Scholar]
- 18.Jost PJ, Ruland J. Aberrant NF-kappaB signaling in lymphoma: mechanisms, consequences, and therapeutic implications. Blood. 2007;109:2700–7. doi: 10.1182/blood-2006-07-025809. [DOI] [PubMed] [Google Scholar]
- 19.Cilloni D, Martinelli G, Messa F, Baccarani M, Saglio G. Nuclear factor kB as a target for new drug development in myeloid malignancies. Haematologica. 2007;92:1224–9. doi: 10.3324/haematol.11199. [DOI] [PubMed] [Google Scholar]
- 20.Dutta J, Fan Y, Gupta N, Fan G, Gelinas C. Current insights into the regulation of programmed cell death by NF NF-kappaB. Oncogene. 2006;25:6800–16. doi: 10.1038/sj.onc.1209938. [DOI] [PubMed] [Google Scholar]
- 21.Luo JL, Kamata H, Karin M. The anti-death machinery in IKK/NF-kappaB signaling. J Clin Immunol. 2005;25:541–50. doi: 10.1007/s10875-005-8217-6. [DOI] [PubMed] [Google Scholar]
- 22.Burstein E, Duckett CS. Dying for NF-kappaB? Control of cell death by transcriptional regulation of the apoptotic machinery. Curr Opin Cell Biol. 2003;15:732–7. doi: 10.1016/j.ceb.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 23.Greten FR, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis colitis-associated cancer. Cell. 2004;118:285–96. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 24.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–6. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 25.Lawrence T, Bebien M, Liu GY, Nizet V, Karin M. IKKalpha limits macrophage NF-kappaB activation and contributes to the resolution of inflammation. Nature. 2005;434:1138–43. doi: 10.1038/nature03491. [DOI] [PubMed] [Google Scholar]
- 26.Pikarsky E, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–6. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 27.Luo JL, Kamata H, Karin M. IKK/NF-kappaB signaling: balancing life and death--a new approach to cancer therapy. J Clin Invest. 2005;115:2625–32. doi: 10.1172/JCI26322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakanishi C, Toi M. Nuclear factor factor-kappaB inhibitors as sensitizers to anticancer drugs. Nat Rev Cancer. 2005;5:297–309. doi: 10.1038/nrc1588. [DOI] [PubMed] [Google Scholar]
- 29.Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–60. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 30.Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109 (Suppl):S81–96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- 31.Baud V, Karin M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001;11:372–7. doi: 10.1016/s0962-8924(01)02064-5. [DOI] [PubMed] [Google Scholar]
- 32.Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–8. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 33.Derudder E, et al. RelB/p50 dimers are differentially regulated by tumor necrosis factor-alpha and lymphotoxin-beta receptor activation. J Biol Chem. 2003;278:23278–23284. doi: 10.1074/jbc.M300106200. [DOI] [PubMed] [Google Scholar]
- 34.Dejardin E, et al. The lymphotoxin-beta receptor induces different patterns of gene expression via two NF-kappaB pathways. Immunity. 2002;17:525–35. doi: 10.1016/s1074-7613(02)00423-5. [DOI] [PubMed] [Google Scholar]
- 35.Xiao G, Harhaj EW, Sun S. NF-kappaB-Inducing Kinase Regulates the Processing of NF-kappaB2 p100. Mol Cell. 2001;7:401–409. doi: 10.1016/s1097-2765(01)00187-3. [DOI] [PubMed] [Google Scholar]
- 36.Coope HJ, et al. CD40 regulates the processing of NF-kappaB2 p100 to p52. Embo J. 2002;21:5375–85. doi: 10.1093/emboj/cdf542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Claudio E, Brown K, Park S, Wang H, Siebenlist U. BAFF-induced NEMO-independent processing of NF-kappa B2 in maturing B cells. Nat Immunol. 2002;3:958–65. doi: 10.1038/ni842. [DOI] [PubMed] [Google Scholar]
- 38.Dejardin E. The alternative NF-kappaB pathway from biochemistry to biology: pitfalls and promises for future drug development. Biochem Pharmacol. 2006;72:1161–79. doi: 10.1016/j.bcp.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 39.Pomerantz JL, Baltimore D. Two pathways to NF-kappaB. Mol Cell. 2002;10:693–5. doi: 10.1016/s1097-2765(02)00697-4. [DOI] [PubMed] [Google Scholar]
- 40.Kyle RA, Rajkumar SV. Multiple myeloma. N Engl J Med. 2004;351:1860–73. doi: 10.1056/NEJMra041875. [DOI] [PubMed] [Google Scholar]
- 41.Engelhardt M, Mertelsmann R. 160 years of multiple myeloma: progress and challenges. Eur J Cancer. 2006;42:1507–9. doi: 10.1016/j.ejca.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 42.Denz U, Haas PS, Wasch R, Einsele H, Engelhardt M. State of the art therapy in multiple myeloma and future perspectives. Eur J Cancer. 2006;42:1591–600. doi: 10.1016/j.ejca.2005.11.040. [DOI] [PubMed] [Google Scholar]
- 43.Podar K, Richardson PG, Hideshima T, Chauhan D, Anderson KC. The malignant clone and the bone-marrow environment. Best Pract Res Clin Haematol. 2007;20:597–612. doi: 10.1016/j.beha.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 44.Ni H, et al. Analysis of expression of nuclear factor kappa B (NF-kappa B) in multiple myeloma: downregulation of NF-kappa B induces apoptosis. Br J Haematol. 2001;115:279–86. doi: 10.1046/j.1365-2141.2001.03102.x. [DOI] [PubMed] [Google Scholar]
- 45.Hideshima T, et al. NF-kappa B as a therapeutic target in multiple myeloma. J Biol Chem. 2002;277:16639–47. doi: 10.1074/jbc.M200360200. [DOI] [PubMed] [Google Scholar]
- 46.Bharti AC, Donato N, Singh S, Aggarwal BB. Curcumin (diferuloylmethane), down-regulates the constitutive activation of nuclear factor-kappa B and IkappaBalpha kinase in human multiple myeloma cells, leading to suppression of proliferation and induction of apoptosis. Blood. 2003;101:1053–62. doi: 10.1182/blood-2002-05-1320. [DOI] [PubMed] [Google Scholar]
- 47.Hideshima T, et al. MLN120B, a novel IkappaB kinase beta inhibitor, blocks multiple myeloma cell growth in vitro and in vivo. Clin Cancer Res. 2006;12:5887–94. doi: 10.1158/1078-0432.CCR-05-2501. [DOI] [PubMed] [Google Scholar]
- 48.Jourdan M, et al. Targeting NF-kappaB pathway with an IKK2 inhibitor induces inhibition of multiple myeloma cell growth. Br J Haematol. 2007;138:160–8. doi: 10.1111/j.1365-2141.2007.06629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sanda T, et al. Growth inhibition of multiple myeloma cells by a novel IkappaB kinase inhibitor. Clin Cancer Res. 2005;11:1974–82. doi: 10.1158/1078-0432.CCR-04-1936. [DOI] [PubMed] [Google Scholar]
- 50.Feng R, et al. SDX-308, a nonsteroidal anti-inflammatory agent, inhibits NF-kappaB activity, resulting in strong inhibition of osteoclast formation/activity and multiple myeloma cell growth. Blood. 2007;109:2130–8. doi: 10.1182/blood-2006-07-027458. [DOI] [PubMed] [Google Scholar]
- 51.Dai Y, et al. Interruption of the NF-kappaB pathway by Bay 11–7082 promotes UCN-01-mediated mitochondrial dysfunction and apoptosis in human multiple myeloma cells. Blood. 2004;103:2761–70. doi: 10.1182/blood-2003-09-3037. [DOI] [PubMed] [Google Scholar]
- 52.Mitsiades N, et al. Biologic sequelae of nuclear factor-kappaB blockade in multiple myeloma: therapeutic applications. Blood. 2002;99:4079–86. doi: 10.1182/blood.v99.11.4079. [DOI] [PubMed] [Google Scholar]
- 53.Chauhan D, et al. Multiple myeloma cell adhesion-induced interleukin-6 expression in bone marrow stromal cells involves activation of NF-kappa B. Blood. 1996;87:1104–12. [PubMed] [Google Scholar]
- 54.Landowski TH, Olashaw NE, Agrawal D, Dalton WS. Cell adhesion-mediated drug resistance (CAM-DR) is associated with activation of NF-kappa B (RelB/p50) in myeloma cells. Oncogene. 2003;22:2417–21. doi: 10.1038/sj.onc.1206315. [DOI] [PubMed] [Google Scholar]
- 55.Vande Broek I, et al. Bone marrow endothelial cells increase the invasiveness of human multiple myeloma cells through upregulation of MMP MMP-9: evidence for a role of hepatocyte growth factor. Leukemia. 2004;18:976–82. doi: 10.1038/sj.leu.2403331. [DOI] [PubMed] [Google Scholar]
- 56.Hecht M, von Metzler I, Sack K, Kaiser M, Sezer O. Interactions of myeloma cells with osteoclasts promote tumour expansion and bone degradation through activation of a complex signalling network and upregulation of cathepsin K, matrix metalloproteinases (MMPs) and urokinase plasminogen activator (uPA) Exp Cell Res. 2008;314:1082–93. doi: 10.1016/j.yexcr.2007.10.021. [DOI] [PubMed] [Google Scholar]
- 57.Hideshima T, Podar K, Chauhan D, Anderson KC. Cytokines and signal transduction. Best Pract Res Clin Haematol. 2005;18:509–24. doi: 10.1016/j.beha.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 58.Niesvizky R, et al. The relationship between quality of response and clinical benefit for patients treated on the bortezomib arm of the international, randomized, phase 3 APEX trial in relapsed multiple myeloma. Br J Haematol. 2008 doi: 10.1111/j.1365-2141.2008.07303.x. [DOI] [PubMed] [Google Scholar]
- 59.Richardson PG, Mitsiades C, Schlossman R, Munshi N, Anderson K. New drugs for myeloma. Oncologist. 2007;12:664–89. doi: 10.1634/theoncologist.12-6-664. [DOI] [PubMed] [Google Scholar]
- 60.Hideshima T, Chauhan D, Schlossman R, Richardson P, Anderson KC. The role of tumor necrosis factor alpha in the pathophysiology of human multiple myeloma: therapeutic applications. Oncogene. 2001;20:4519–27. doi: 10.1038/sj.onc.1204623. [DOI] [PubMed] [Google Scholar]
- 61.Hideshima T, et al. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001;61:3071–6. [PubMed] [Google Scholar]
- 62.Mitsiades N, et al. The proteasome inhibitor PS-341 potentiates sensitivity of multiple myeloma cells to conventional chemotherapeutic agents: therapeutic applications. Blood. 2003;101:2377–80. doi: 10.1182/blood-2002-06-1768. [DOI] [PubMed] [Google Scholar]
- 63.Mulligan G, et al. Gene expression profiling and correlation with outcome in clinical trials of the proteasome inhibitor bortezomib. Blood. 2007;109:3177–88. doi: 10.1182/blood-2006-09-044974. [DOI] [PubMed] [Google Scholar]
- 64.Hussein MA, et al. Phase 2 study of arsenic trioxide in patients with relapsed or refractory multiple myeloma. Br J Haematol. 2004;125:470–6. doi: 10.1111/j.1365-2141.2004.04941.x. [DOI] [PubMed] [Google Scholar]
- 65.Keifer JA, Guttridge DC, Ashburner BP, Baldwin AS., Jr Inhibition of NF-kappa B activity by thalidomide through suppression of IkappaB kinase activity. J Biol Chem. 2001;276:22382–7. doi: 10.1074/jbc.M100938200. [DOI] [PubMed] [Google Scholar]
- 66.Annunziata CM, et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12:115–30. doi: 10.1016/j.ccr.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Keats JJ, et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell. 2007;12:131–44. doi: 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liao G, Zhang M, Harhaj EW, Sun SC. Regulation of the NF-kappaB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J Biol Chem. 2004;279:26243–50. doi: 10.1074/jbc.M403286200. [DOI] [PubMed] [Google Scholar]
- 69.He JQ, et al. Rescue of TRAF3-null mice by p100 NF-kappa B deficiency. J Exp Med. 2006;203:2413–8. doi: 10.1084/jem.20061166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.He JQ, Saha SK, Kang JR, Zarnegar B, Cheng G. Specificity of TRAF3 in its negative regulation of the noncanonical NF-kappa B pathway. J Biol Chem. 2007;282:3688–94. doi: 10.1074/jbc.M610271200. [DOI] [PubMed] [Google Scholar]
- 71.Vallabhapurapu S, et al. Nat Immunol. 2008. TRAF2 and TRAF3 carry out non-redundant and complementary functions in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Varfolomeev E, et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell. 2007;131:669–81. doi: 10.1016/j.cell.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 73.Vince JE, et al. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell. 2007;131:682–93. doi: 10.1016/j.cell.2007.10.037. [DOI] [PubMed] [Google Scholar]
- 74.Shinkura R, et al. Alymphoplasia is caused by a point mutation in the mouse gene encoding Nf-kappa b-inducing kinase. Nat Genet. 1999;22:74–7. doi: 10.1038/8780. [DOI] [PubMed] [Google Scholar]
- 75.Karrer U, Althage A, Odermatt B, Hengartner H, Zinkernagel RM. Immunodeficiency of alymphoplasia mice (aly/aly) in vivo: structural defect of secondary lymphoid organs and functional B cell defect. Eur J Immunol. 2000;30:2799–807. doi: 10.1002/1521-4141(200010)30:10<2799::AID-IMMU2799>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 76.Yamada T, et al. Abnormal immune function of hemopoietic cells from alymphoplasia (aly) mice, a natural strain with mutant NF-kappa B-inducing kinase. J Immunol. 2000;165:804–12. doi: 10.4049/jimmunol.165.2.804. [DOI] [PubMed] [Google Scholar]
- 77.Yin L, et al. Defective lymphotoxin-beta receptor-induced NF-kappaB transcriptional activity in NIK-deficient mice. Science. 2001;291:2162–5. doi: 10.1126/science.1058453. [DOI] [PubMed] [Google Scholar]
- 78.Sasaki Y, Casola S, Kutok JL, Rajewsky K, Schmidt-Supprian M. TNF family member B cell-activating factor (BAFF) receptor-dependent and -independent roles for BAFF in B cell physiology. J Immunol. 2004;173:2245–52. doi: 10.4049/jimmunol.173.4.2245. [DOI] [PubMed] [Google Scholar]
- 79.Shulga-Morskaya S, et al. B cell-activating factor belonging to the TNF family acts through separate receptors to support B cell survival and T cell-independent antibody formation. J Immunol. 2004;173:2331–41. doi: 10.4049/jimmunol.173.4.2331. [DOI] [PubMed] [Google Scholar]
- 80.Gardam S, Sierro F, Basten A, Mackay F, Brink R. TRAF2 and TRAF3 signal adapters act cooperatively to control the maturation and survival signals delivered to B cells by the BAFF receptor. Immunity. 2008;28:391–401. doi: 10.1016/j.immuni.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 81.Malinin NL, Boldin MP, Kovalenko AV, Wallach D. MAP3K-related kinase involved in NF-kappaB induction by TNF, CD95 and IL-1. Nature. 1997;385:540–4. doi: 10.1038/385540a0. [DOI] [PubMed] [Google Scholar]
- 82.Ramakrishnan P, Wang W, Wallach D. Receptor-specific signaling for both the alternative and the canonical NF-kappaB activation pathways by NF-kappaB-inducing kinase. Immunity. 2004;21:477–89. doi: 10.1016/j.immuni.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 83.Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS., Jr NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–3. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- 84.Vaux DL, Silke J. IAPs, RINGs and ubiquitylation. Nat Rev Mol Cell Biol. 2005;6:287–97. doi: 10.1038/nrm1621. [DOI] [PubMed] [Google Scholar]
- 85.Hunter AM, LaCasse EC, Korneluk RG. The inhibitors of apoptosis (IAPs) as cancer targets. Apoptosis. 2007;12:1543–68. doi: 10.1007/s10495-007-0087-3. [DOI] [PubMed] [Google Scholar]
- 86.Shu HB, Takeuchi M, Goeddel DV. The tumor necrosis factor receptor 2 signal transducers TRAF2 and c-IAP1 are components of the tumor necrosis factor receptor 1 signaling complex. Proc Natl Acad Sci U S A. 1996;93:13973–8. doi: 10.1073/pnas.93.24.13973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rothe M, Pan MG, Henzel WJ, Ayres TM, Goeddel DV. The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell. 1995;83:1243–52. doi: 10.1016/0092-8674(95)90149-3. [DOI] [PubMed] [Google Scholar]
- 88.Rothe M, Wong SC, Henzel WJ, Goeddel DV. A novel family of putative, signal transducers associated with the cytoplasmic domain of the 75 kDa tumor necrosis factor receptor. Cell. 1994;78:681–92. doi: 10.1016/0092-8674(94)90532-0. [DOI] [PubMed] [Google Scholar]
- 89.Yang Y, Fang S, Jensen JP, Weissman AM, Ashwell JD. Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science. 2000;288:874–7. doi: 10.1126/science.288.5467.874. [DOI] [PubMed] [Google Scholar]
- 90.Li X, Yang Y, Ashwell JD. TNF-RII and c-IAP1 mediate ubiquitination and degradation of TRAF2. Nature. 2002;416:345–7. doi: 10.1038/416345a. [DOI] [PubMed] [Google Scholar]
- 91.Matsuzawa A, et al. Essential cytoplasmic translocation of a cytokine receptor-assembled signaling complex. Science. 2008;321:663–8. doi: 10.1126/science.1157340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Conte D, et al. Inhibitor of apoptosis protein cIAP2 is essential for lipopolysaccharide-induced macrophage survival. Mol Cell Biol. 2006;26:699–708. doi: 10.1128/MCB.26.2.699-708.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Conze DB, et al. Posttranscriptional downregulation of c-IAP2 by the ubiquitin protein ligase c-IAP1 in vivo. Mol Cell Biol. 2005;25:3348–56. doi: 10.1128/MCB.25.8.3348-3356.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zender L, et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell. 2006;125:1253–67. doi: 10.1016/j.cell.2006.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 96.Verhagen AM, et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 97.Schimmer AD, et al. Small-molecule antagonists of apoptosis suppressor XIAP is exhibit broad antitumor activity. Cancer Cell. 2004;5:25–35. doi: 10.1016/s1535-6108(03)00332-5. [DOI] [PubMed] [Google Scholar]
- 98.Chauhan D, et al. Targeting mitochondrial factor Smac/DIABLO as therapy for multiple myeloma (MM) Blood. 2007;109:1220–7. doi: 10.1182/blood-2006-04-015149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li L, et al. A small molecule Smac mimic potentiates TRAIL- and TNFalpha-mediated cell death. Science. 2004;305:1471–4. doi: 10.1126/science.1098231. [DOI] [PubMed] [Google Scholar]
- 100.Davis RE, Staudt LM. Molecular diagnosis of lymphoid malignancies by gene expression profiling. Curr Opin Hematol. 2002;9:333–8. doi: 10.1097/00062752-200207000-00011. [DOI] [PubMed] [Google Scholar]
- 101.Keutgens A, Robert I, Viatour P, Chariot A. Deregulated NF-kappaB activity in haematological malignancies. Biochem Pharmacol. 2006;72:1069–80. doi: 10.1016/j.bcp.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 102.Karin M, Yamamoto Y, Wang QM. The IKK NF-kappa B system: a treasure trove for drug development. Nat Rev Drug Discov. 2004;3:17–26. doi: 10.1038/nrd1279. [DOI] [PubMed] [Google Scholar]
- 103.Greten FR, et al. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell. 2007;130:918–31. doi: 10.1016/j.cell.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]