

Abstract
Evidence supports a pathogenic role for free radical injury to brain regions in Alzheimer's disease (AD); however, clinical trial results are only mildly encouraging. Examining brains from The Adult Changes in Thought study offers a unique perspective. Selectively increased free radical damage to cerebral cortex was associated with AD, microvascular brain injury (μ-VBI), and current smoking, but not with antioxidant supplement usage. Our results support suppression of free radical injury to brain as a therapeutic target for AD and μ-VBI; however, future clinical trials should consider other antioxidants or doses than those identified in our study.
Evidence from transgenic mouse models, autopsies and epidemiologic investigations supports a pathogenic role for increased free radical damage to diseased regions of brain in Alzheimer's disease (AD) (reviewed in Sonnen and colleagues' article 1); however, results from clinical trials of vitamin E supplements are only mildly encouraging. One demonstrated mild slowing of progression of moderate AD in the 2,000 IU α-tocopherol treated group.2 Another observed no benefit from treatment with 2,000 IU of synthetic vitamin E for three years in patients with amnestic MCI.3 The third model4 treated women with 600 IU of α-tocopherol every other day for four years and observed no difference in decline of global cognition between treated and placebo groups; however, there was a trend for partial protection of a composite verbal memory measure with α-tocopherol treatment. None of these clinical trials used vitamin C in combination with vitamin E. Humans cannot synthesize vitamin C as mice can, confounding comparison of clinical trials with investigations in transgenic mice. Moreover, vitamin C recycles α-tocopherol and thereby enhances it biological activity.5 Indeed, we have observed greater antioxidant effect in the human cerebrospinal fluid with combined α-tocopherol and vitamin C supplementation than with α-tocopherol alone,6 and the Cache County study observed an apparent protection from incident AD with vitamin C and α-tocopherol supplementation, but not with either alone.7 These data support clinical trials with combinations of antioxidants, especially vitamins E plus C.
Subjects and Methods
Here we examine brains from the Adult Changes in Thought (ACT) study, a prospective population-based longitudinal study of brain aging and incident dementia among 3,392 total participants 65 years or older and cognitively normal when enrolled and followed with biennial evaluations.8 Consumption of vitamin E, vitamin C, or both by ACT participants was not associated with reduced risk of developing dementia over 5.5 years of follow-up.9 These results present an opportunity to determine whether this apparent lack of therapeutic effect is associated with a measurable pharmacologic effect in neuropathologically characterized samples. If consumption of antioxidants did not suppress free radical damage to brain in ACT participants, then investigators should consider other doses or other antioxidants in clinical trials. Alternatively, suppressed free radical damage to brain by antioxidants in the ACT autopsy cohort would suggest that despite the desired pharmacologic effect there was no significant therapeutic effect, a result that would undermine the rationale for clinical trials of antioxidants to prevent AD.
The University of Washington institutional review board approved this project. Diagnoses were assigned at consensus diagnostic conferences using the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, criteria for dementia 10 and the criteria of the National Institute of Neurological and Communicative Disorders and Stroke and Alzheimer's Disease and Related Disorders Association for AD.11 Participants with new-onset dementia underwent at least one annual follow-up examination for verification of dementia status.
Categorization of supplement use was performed as previously described using data collected during the last evaluation prior to death.9 In brief, participants were asked at baseline and during biennial follow-up interviews whether they had taken vitamin C, vitamin E, or multivitamins for at least 1 week during the previous month. Exposures were categorized into seven mutually exclusive categories: nonuse, vitamin C alone, vitamin E alone, multivitamins alone, vitamin C plus multivitamins, vitamin E plus multivitamins, and vitamins E plus C with or without multivitamins. Body mass index (BMI) was calculated from measured weight and height. Participants were asked whether they had smoked more than 100 cigarettes in their lifetime and whether they smoked currently.9
Characteristics of the initial 228 autopsies from the ACT cohort are published.12 Seventy-one autopsies had brain regions flash frozen within 8 hours of death; all were used here. Of these, 44 (23 female) individuals did not have dementia at last evaluation and had average age (± standard deviation) of 87 ± 6 years, last cognitive assessment screening instrument score of 91 ± 6, and median of 308 days between last evaluation and death. Corresponding values for the 25 (17 female) demented patients were (± standard deviation) of 88 ± 5 years, cognitive assessment screening instrument score of 62 ± 18, and median of 450 days between last evaluation and death. Neuropathologic assessments followed published criteria for AD, Lewy body disorder (LBD), and microvascular brain injury (μ-VBI).12 F2-isoprostanes (IsoPs), measures of free radical damage to tissue and F4-neuroprostanes (NeuroPs), measures of free radical damage to neuronal membranes were quantified in duplicate as described previously.13 Statistical analyses used GraphPad Prism (San Diego, CA) software.
Results
There are potential limitations to the quantification of molecules in postmortem human brain related to antecedent illness, conditions of agonal state, and changes that occur in the time between death and tissue harvest. We addressed these concerns for the isoprostanoids by comparing values between participants in ACT who had died and donated their brains for investigation with healthy adult female rhesus macaques (9 to 19 years of age; n=6) who had been culled from an aging colony and whose necropsies showed age-related changes. We compared our results from female macaque parietal cortex with parietal cortex from woman in ACT who had Braak stage none to III, no μ-VBI, and were not current smokers (n=11). The isoprostanoids (Figure) were not significantly different in woman compared to female rhesus macaques (t test P > 0.05 for each). These data validate our measurements in humans as reflective of the in vivo condition.
Figure.
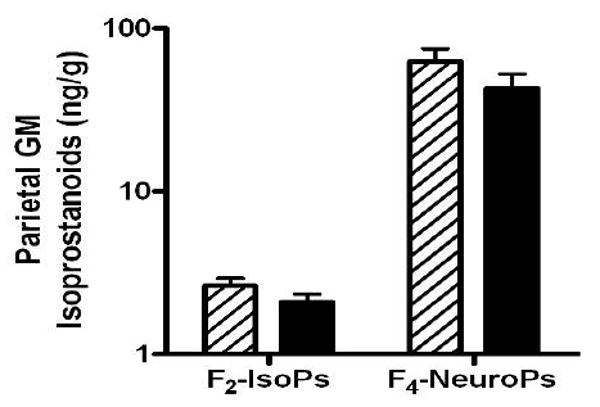
Data are isoprostanoid concentrations in parietal lobe gray matter (GM) from adult female rhesus macaques (hatched bars; aged 9 to 19 years, n = 6) and women in ACT who had Braak stage none to III (black bars), no microvascular brain injury (μ-VBI), and were not current smokers (n = 11). Two-way ANOVA had P < 0.0001 for the different isoprostanoids but P > 0.05 for comparisons between species or interaction. F2-IsoP = F2-isoprostane; F4-NeuroP = F4-neuroprostane.
We separated ACT cases based on neuropathologic features and smoking status into relatively distinct groups, as well as a group with comorbid AD plus μ-VBI. Because gross infarcts were not significantly associated with dementia in the ACT cohort once μ-VBI is included12, we did not use gross infarcts to categorize individuals. Five cases with neocortical LBD were excluded. Neither average age nor average number of years of education was significantly different among these groups (not shown). We observed significantly increased cerebral cortical F4-NeuroPs, but not F2-IsoPs in AD and current smokers, and increased levels of both isoprostanoids in individuals with μ-VBI and especially in mixed AD plus μ-VBI (Table). There was no association of isoprostanoid concentration with BMI (most recent or greatest). Isoprostanoid data from each Group were stratified into those who did not take any supplements versus those who took supplements or vitamins E plus C, and then compared by t tests. Group 4 from Table had sample sizes of 3 individuals for supplement users and nonusers; all other Groups had between 5 and 15 individuals. No significant difference in isoprostanoid concentrations was detected for any of these comparisons, including Group 1 from the Table that had minimal neuropathologic evidence of disease.
Table.
Quantitation of oxidative damage in cerebral cortex (CBR) and cerebellar cortex (CBL) in persons with Alzheimer's disease (AD), microvascular brain injury (μ-VBI), and current smokers.
| Group | N | Age (SD) | Years Education (SD) | Pathologic Classification | ng/g (mean ± SEM) | ||||
|---|---|---|---|---|---|---|---|---|---|
| AD | μ-VBI | Current Smoker | Brain Region | F2-IsoPs | F4-NeuroPs | ||||
| 1 | 22 | 86.2 (5.7) | 13.4 (3.6) | No/low | No | No | CBR | 2.2 ± 0.1 | 63.1 ± 8.2 |
| CBL | 2.2 ± 0.2 | 54.2 ± 9.5 | |||||||
| 2 | 8 | 88.0 (6.7) | 13.2 (2.2) | High | No | No | CBR | 2.7 ± 0.4 | 125.6 ± 24.8 |
| CBL | 2.0 ± 0.3 | 60.0 ± 13.1 | |||||||
| ANOVA | --/-- | */** | |||||||
| 3 | 18 | 89.0 (5.0) | 13.6 (3.6) | No/low | Yes | No | CBR | 3.0 ± 0.2 | 117.1 ± 12.7 |
| CBL | 1.9 + 0.2 | 59.9 + 10.3 | |||||||
| ANOVA | */** | */** | |||||||
| 4 | 6 | 85.8 (5.6) | 14.4 (2.1) | No/low | No | Yes | CBR | 2.7 ± 0.3 | 127.4 ± 32.3 |
| CBL | 2.7 ± 0.6 | 82.1 ± 30.2 | |||||||
| ANOVA | --/-- | **/** | |||||||
| 5 | 12 | 88.4 (4.9) | 13.3 (2.2) | High | Yes | No | CBR | 3.7 ± 0.5 | 176.2 ± 24.6 |
| CBL | 2.2 ± 0.3 | 74.7 ± 8.9 | |||||||
| ANOVA | */* | ***/*** | |||||||
Groups were defined by neuropathological features and current smoking status at last evaluation. AD was stratified as “No/low” if Braak stage was 0 to III and “High” if Braak stage was IV to VI. μ-VBI and current smoking were grouped as present or absent. Five individuals with neocortical Lewy body disease were excluded. All isoprostanoid data are an average of repeated measurements. Two-way analysis of variance (ANOVA) compared Group 1 versus other Group as the first dimension and brain region as the second dimension. Results are presented for Group/Brain region (*P < 0.05, **P < 0.01, ***P < 0.001). There was significant interaction between Group and brain region for F2-IsoPs and F4-NeuroPs in Groups 3 and 5 (P < 0.05). When appropriate, Bonferroni-corrected posttests (PT) were performed for isoprostanoid concentrations in CBR and CBL: for F2-IsoPs P < 0.01 for CBR and P > 0.05 for CBL in Groups 3 and 5; for F4-NeuroPs P < 0.01 for CBR and P > 0.05 for CBL in Groups 2, 3, 4, and 5.
Discussion
We observed relatively selectively increased free radical damage to cerebral cortical neurons in AD cases and current smokers, and more generally increased free radical damage to cerebral cortex with μ-VBI. AD and larger vessel-mediated ischemic injury cause free radical-mediated injury to brain1. Smoking causes systemic oxidative stress with increased plasma F2-IsoPs14. Our data are the first to show that smoking or μ-VBI is associated with increased free racial injury to cerebral cortex. Greatest free radical damage was observed in patients with comorbid AD and μ-VBI, raising the possibility that AD and μ-VBI may be partially additive. For the first time, we determined that typical antioxidant supplement use was not associated with suppressed basal or increased levels of free radical damage to brain from AD, μ-VBI, or smoking. In the full ACT cohort, the same estimates of antioxidant supplements were not associated with reduced risk for dementia over 5.5 years of follow-up.9 Although our analysis of autopsy samples is limited by relatively small number of observations in some groups, we did not observe any suppression of oxidative damage with antioxidant supplement use. This lack of therapeutic effect combined with no apparent pharmacologic effect in our autopsy study suggests that future clinical trials for AD or μ-VBI should consider dietary sources rather than supplements15, other antioxidants, combinations, or other doses that retain safety than those used by ACT participants.9
Acknowledgments
The authors wish to thank Dr. Arlene M. Sonnen for assistance with manuscript preparation. There are no conflicts of interest. This work was supported by AG23801, AG05136, AG006781, AG 000258, and the Nancy and Buster Alvord Endowment.
References
- 1.Sonnen JA, Breitner JC, Lovell MA, et al. Free radical-mediated damage to brain in Alzheimer's disease and its transgenic mouse models. Free Radic Biol Med. 2008;45:219–230. doi: 10.1016/j.freeradbiomed.2008.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sano M, Ernesto C, Thomas RG, et al. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer's disease. The Alzheimer's Disease Cooperative Study. N Engl J Med. 1997;336:1216–1222. doi: 10.1056/NEJM199704243361704. [DOI] [PubMed] [Google Scholar]
- 3.Petersen RC, Thomas RG, Grundman M, et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. N Engl J Med. 2005;352:2379–2388. doi: 10.1056/NEJMoa050151. [DOI] [PubMed] [Google Scholar]
- 4.Kang JH, Cook N, Manson J, et al. A randomized trial of vitamin E supplementation and cognitive function in women. Arch Intern Med. 2006;166:2462–2468. doi: 10.1001/archinte.166.22.2462. [DOI] [PubMed] [Google Scholar]
- 5.Yu BP. Cellular defenses against damage from reactive oxygen species. Physiol Rev. 1994;74:139–162. doi: 10.1152/physrev.1994.74.1.139. [DOI] [PubMed] [Google Scholar]
- 6.Quinn JF, Montine KS, Moore M, et al. Suppression of longitudinal increase in CSFF2-isoprostanes in Alzheimer's disease. Journal of Alzheimers Disease. 2004;6:93–97. doi: 10.3233/jad-2004-6110. [DOI] [PubMed] [Google Scholar]
- 7.Zandi PP, Anthony JC, Khachaturian AS, et al. Reduced risk of Alzheimer disease in users of antioxidant vitamin supplements: the Cache County Study. Arch Neurol. 2004;61:82–88. doi: 10.1001/archneur.61.1.82. [DOI] [PubMed] [Google Scholar]
- 8.Kukull WA, Higdon R, Bowen JD, et al. Dementia and Alzheimer disease incidence: a prospective cohort study. Arch Neurol. 2002;59:1737–1746. doi: 10.1001/archneur.59.11.1737. [DOI] [PubMed] [Google Scholar]
- 9.Gray SL, Anderson ML, Crane PK, et al. Antioxidant vitamin supplement use and risk of dementia or Alzheimer's disease in older adults. J Am Geriatr Soc. 2008;56:291–295. doi: 10.1111/j.1532-5415.2007.01531.x. [DOI] [PubMed] [Google Scholar]
- 10.Association AP. Diagnostic and statistical manual of mental disorders. 4th. Washington, DC: American Psychiatric Association; 1994. [Google Scholar]
- 11.McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 12.Sonnen JA, Larson EB, Crane PK, et al. Pathological correlates of dementia in a longitudinal, population-based sample of aging. Ann Neurol. 2007;62:406–413. doi: 10.1002/ana.21208. [DOI] [PubMed] [Google Scholar]
- 13.Milatovic D, VanRollins M, Li K, et al. Suppression of murine cerebral F2-isoprostanes and F4-neuroprostanes from excitotoxicity and innate immune response in vivo by alpha- or gamma-tocopherol. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;827:88–93. doi: 10.1016/j.jchromb.2005.03.037. [DOI] [PubMed] [Google Scholar]
- 14.Morrow JD, Frei B, Longmire AW, et al. Increase in circulating products of lipid peroxidation (F2-isoprostanes) in smokers. Smoking as a cause of oxidative damage. N Engl J Med. 1995;332:1198–1203. doi: 10.1056/NEJM199505043321804. [DOI] [PubMed] [Google Scholar]
- 15.Bjelakovic G, Nikolova D, Gluud LL, et al. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. Jama. 2007;297:842–857. doi: 10.1001/jama.297.8.842. [DOI] [PubMed] [Google Scholar]