

Abstract
Background
Malignant granular cell tumor (MGCT) is a rare disease entity. Forty-one well-documented MGCTs have been reported in the world literature.
Case Report
This report describes a patient who presented with a MGCT of the lung and reviews the preoperative evaluation, pathologic features and differential diagnosis of the disease. This case represents the first report of resected primary pulmonary MGCT.
Conclusions
Diagnosis of MGCT is based on histology of the primary tumor, immunohistochemistry, and exclusion of tumors that may mimic granular cell tumor.
Keywords: Lung, granular, neoplasm, tumor, surgery.
Introduction
Granular cell tumors (GCT) are uncommon, usually benign neoplasms, most frequently originating from tongue, skin, and breast [1,2]. Since the first case of GCT from the trachea and bronchus was described in the late 1930s, less than 80 cases have been reported as originating from in the tracheobronchial tree, all of which were benign. One report suggested a MGCT co-existing with small cell lung cancer; however, no histology proof was available since the tumor was not resected [3]. In this report, we describe a patient who presented with a MGCT of the lung and discuss the preoperative considerations and evaluation, differential diagnosis, and the pathologic features of this rare malignancy.
Case Report
A 32-year-old Caucasian woman who was in her usual state of health until approximately 2 months ago was admitted for her new onset of chest pain and shortness of breath. She had a longstanding history of smoking since the age of 9, roughly 2–3 packs per day. Physical examination was within normal limits; no evidence of peripheral adenopathy or cutaneous nodules was noted. A chest x-ray and CT scan revealed a solitary pulmonary mass (Fig. 1, 2). The mass was round and smooth, 2.8 cm in diameter, and centrally located within the left lower lobe.
Figure 1.
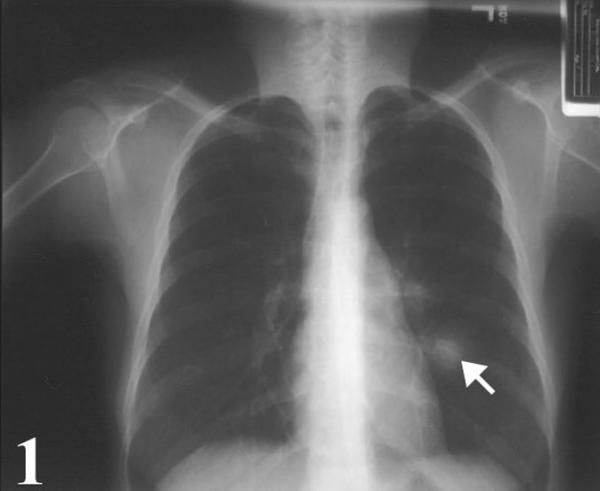
Chest radiograph showing a mass lesion (arrow) intimately associated with a segmental bronchus of the left lower lobe.
Figure 2.
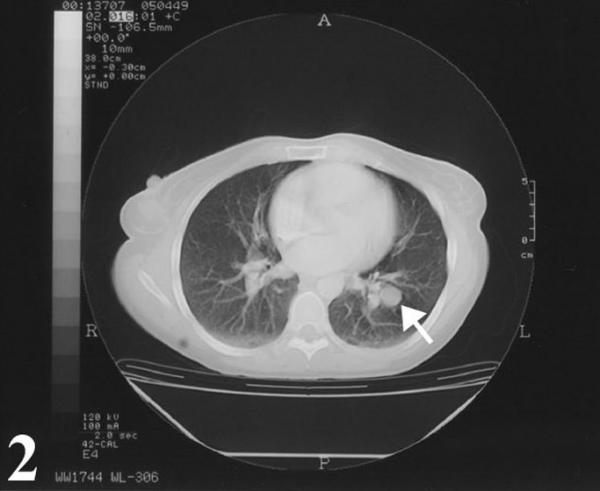
Chest computed tomogram showing localized expansion of mass, which measured 2.8 × 2.0 cm in size.
Additional work-up including abdominal and pelvic CT scans revealed no extra-pulmonary lesions. Because of her significant smoking history, clinical symptoms and imaging studies, a tentative preoperative diagnosis of lung cancer was made. A flexible bronchoscopy was then performed, which revealed no evidence of endobronchial lesions. Subsequently, a lateral thoracotomy was performed. In-situ, the centrally located mass was smooth by palpation and was in close proximity to the pulmonary hilum. There were no visible invasive or metastatic lesions in the adjacent organs or lymph nodes. Four core biopsies were taken from the mass, but no definite pathologic diagnosis of tumor was made on intraoperative frozen section. A diagnostic lobectomy was subsequently performed. The patient had an uneventful postoperative course and was discharged from hospital 4 days postoperatively.
Macroscopically, the tumor was a well-defined solitary mass, measuring 2.8 × 2.0 × 2.0 cm in size. It was located at 2.2 cm from the bronchial margin of resection and 1.5 cm from the over-lining pleural surface. The cut surface of the tumor had a whitish tan appearance and firm consistency. The mass was well-circumscribed but not encapsulated, and did not infiltrate the bronchial wall. The surrounding pulmonary parenchyma had a slightly congested appearance. Six lymph nodes were identified in the peri-bronchial region, the largest measured 1.1 cm in diameter.
Microscopically, the mass was intimately associated with a segmental bronchus and had grown circumferentially along the submucosal and muscular wall. The mass was made of sheets of granular cells. The neoplastic cells displayed moderate atypia, with a marked increase in cellularity and nuclear/cytoplasm (N:C) ratio, greater pleomorphism, nucleolar prominence, identifiable mitotic figures, and prominent spindling of tumor cells (Fig. 3). The cytoplasmic granules were PAS-positive and resistant to diastase digestion. Immunohistochemical staining for S-100 protein revealed prominent cytoplasm reaction products in the tumor cells. Staining for vimentin was weakly positive. Tumor cells exhibited increased expression of both p53 and Ki67, with more than 4% and 25% of tumor cells positive for Ki-67 and p53, respectively. The tumor cells did not stain for carcinoembryonic antigen, cytokeratins, epithelial membrane antigen, actin, desmin, myoglobin, smooth muscle actin, CD34, or HMB-45. All six lymph nodes were negative for tumor metastases. The lesion was pathologically diagnosed as a MGCT based on the morphologic findings, results of an immunohistochemical panel, increased p53 expression, and an elevated nuclear proliferative index.
Figure 3.
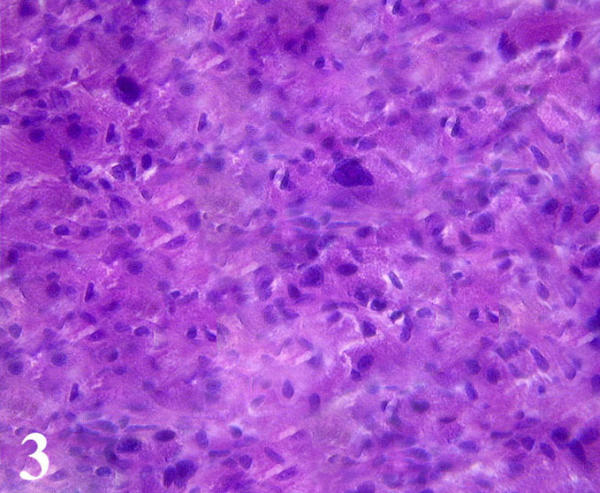
Microscopically, the tumor was made of sheets of granular cells with moderate atypia, prominent spindling, increased nuclear/cytoplasm ratio and greater pleomorphism. Note two giant tumor cells were in the central field of the photograph. (Hematoxylin and eosin; × 400)
Discussion
MGCT is a rare disease entity that was first described by Ravich et al, in 1945 [4]. Since the originally described case, forty-one additional MGCTs have been reported in various body sites, including skin, oral cavity, breast, abdominal wall, chest wall and urinary bladder [5,6]. Differentiation between benign and malignant GCTs is often difficult, since only the presence of metastasis may ultimately establish malignancy. Because MGCTs are extremely rare, attempts have been made to identify specific histologic features that would predict malignant behavior. Based on the cumulative experience of previous case reports of MGCTs, six histologic criteria are considered to be important [1,7]. These malignant features included spindling of the tumor cells, the presence of vesicular nuclei with large nucleoli, increased mitotic rate (>2 mitoses/10 high-power fields at 200× magnification), a high nuclear-to cytoplasm (N:C) ratio, pleomorphism, and necrosis [7]. Histologically, MGCT is diagnosed when three or more of the six criteria are fulfilled. In addition to the histologic features, upregulation of p53 as well as Ki67, a nuclear proliferative marker, has been recently found to correlate with an aggressive clinical course and malignant behavior. In a recent study, p53 immunostains were negative in all benign cases [7], while p53 expression was seen in greater than 10% of the cell population in 21 of 25 (79%) MGCTs. Similarly, benign GCTs showed immunoreactivity of Ki-67 in 1 % or less of the tumor cell population, while fourteen of 25(56%) malignant tumors had Ki67 immunopositivity in up to 30% of the cell population. Our case fulfilled five of the six histologic features of malignant GCT.
Increased expression of p53 and an elevated Ki-67 expression in the tumor cells further supported the diagnosis. In addition, malignant tumors that may mimic MGCT were excluded based on histologic and immunophenotypic features. These excluded neoplasms are alveolar soft part sarcoma, rhabdomyosarcoma, leiomyoma, leiomyosarcoma, angiosarcoma, malignant fibrous histocytoma, melanoma and malignant peripheral nerve sheath tumor (MPNST). Furthermore, the possibility of a metastatic malignant GCT originating from an extrapulmonary site was excluded by the thorough preoperative evaluation. Metastatic malignant GCT to the lung usually presents with bilateral nodules rather than a single solitary mass.
MGCT is more commonly seen in women with a female to male ratio being 2.4:1. Patient age at presentation ranges from 23 to 82 years with the average age at diagnosis being 48 years. The more common benign counterpart has been described in all age groups, including newborns. Therefore, age of the patient is not a particularly helpful factor when attempting to distinguish benign from malignant form of the disease. Size of MGCTs ranges from 0.5 cm to 17 cm and approximately half of these are 4 cm or larger at the time of diagnosis. The size of the primary lesion alone is not a reliable predictor of the malignant behavior because many of the MGCT were less than 4 cm. The options for treatment of GCT of the lung are based on tumor size. Small tumors (<1.0 cm) may be treated by bronchoscopic extirpation, laser therapy, or sleeve resection. Larger tumors (>1.0 cm) that usually extend beyond the tracheobronchial cartilage are usually treated by surgical resection [4]. However, no uniform management of malignant GCT is yet available, since this is the first documented pulmonary malignant GCT.
Acknowledgments
Acknowledgement
We thank Dr. Sam Wiseman for critical reviewing the manuscript.
Contributor Information
Ming Jiang, Email: jiangm@roswellpark.org.
Timothy Anderson, Email: timothy.anderson@roswellpark.org.
Chukwumere Nwogu, Email: chukwumere.nwogu@roswellpark.org.
Dongfeng Tan, Email: dongfeng.tan@roswellpark.org.
References
- Khansur T, Balducci L, Tavassoli M. Granular cell tumor: clinical spectrum of the benign and malignant entity. Cancer. 1987;60:220–222. doi: 10.1002/1097-0142(19870715)60:2<220::aid-cncr2820600217>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Deavers M, Guinee D, Koss MN, Travis WD. Granular cell tumors of the lung. Clinicopathologic study of 20 cases. Am J Surg Pathol. 1995;19:627–635. doi: 10.1097/00000478-199506000-00002. [DOI] [PubMed] [Google Scholar]
- Lauro S, Trasatti L, Bria E, Gelibter A, Larosa G, Vechione A. Malignant bronchial Abrikossoff's tumor and small cell lung cancer. Anticancer Res. 2001;21:563–565. [PubMed] [Google Scholar]
- Ravich A, Stout AP, Ravich RA. Malignant granular cell myoblastoma involving the urinary bladder. Ann Surg. 1945;121:361–372. doi: 10.1097/00000658-194503000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardines L, Cheung L, LiVolsi V, Hendrickson S, Brooks JJ. Malignant granular cell tumors: report of a case and review of the literature. Surgery. 1994;116:49–54. [PubMed] [Google Scholar]
- Matsumoto H, Kojima Y, Inoue T, Takegana S, Tsuda H, Kobayashi A, Watanabe K. A malignant granular cell tumor of the stomach: report of a case. Surg Today. 1996;26:119–122. doi: 10.1007/BF00311775. [DOI] [PubMed] [Google Scholar]
- Fanburg-Smith JC, Meis-Kindblom JM, Fante R, Kindblom LG. Malignant granular cell tumor of soft tissue. Diagnostic criteria and clinicopathologic correlation. Am J Surg Pathol. 1998;22:779–794. doi: 10.1097/00000478-199807000-00001. [DOI] [PubMed] [Google Scholar]