

Summary
TRIC channels function as monovalent cation-specific channels that mediate counter ion movements coupled with ryanodine receptor-mediated Ca2+ release from intracellular stores in muscle cells. Mammalian tissues differentially contain two TRIC channel subtypes: TRIC-A is abundantly expressed in excitable cells, whereas TRIC-B is ubiquitously expressed throughout tissues. Here, we report the physiological role of TRIC-B channels in mouse perinatal development. TRIC-B-knockout neonates were cyanotic owing to respiratory failure and died shortly after birth. In the mutant neonates, the deflated lungs exhibited severe histological defects, and alveolar type II epithelial cells displayed ultrastructural abnormalities. The metabolic conversion of glycogen into phospholipids was severely interrupted in the mutant type II cells, and surfactant phospholipids secreted into the alveolar space were insufficient in the mutant neonates. Moreover, the mutant type II cells were compromised for Ca2+ release mediated by inositol-trisphosphate receptors, despite Ca2+ overloading in intracellular stores. Our results indicate that TRIC-B channels take an active part in Ca2+ signalling to establish specialised functions in type II cells and are thus essential for perinatal lung maturation.
Keywords: Ca2+ release, IP3 receptor, Alveolar epithelial cell, Counter ion channel, Endoplasmic reticulum, TRIC-B (TMEM38B)
INTRODUCTION
Calcium ions are an essential secondary messenger in cellular signal-transduction pathways, and Ca2+ release from intracellular stores regulates important physiological functions, including cell-fate decisions and cellular maturation. Ryanodine and inositol 1,4,5-trisphosphate (IP3) receptor subtypes on the sarco/endoplasmic reticulum (SR/ER) comprise a unique family of intracellular Ca2+ release channels that mediate Ca2+ mobilisation in response to various stimuli (Meissner, 1994; Bezprozvanny and Ehrlich, 1995; MacLennan et al., 2002; Patterson et al., 2004). Release of Ca2+ from the SR/ER generates a negative potential on the luminal side, which is likely to inhibit subsequent Ca2+ release processes. Therefore, in addition to the Ca2+ uptake and storage functions of intracellular stores, counter ion movements seem to be essential for efficient Ca2+ release in order to balance the membrane potential (Meissner, 1983).
We have been conducting a survey of the molecular components that support the multiple functions of the SR/ER (e.g. Nishi et al., 1999; Takeshima et al., 2000; Cai et al., 2009) and recently identified two TRIC (trimeric intracellular cation) channel subtypes, namely TRIC-A and TRIC-B (TMEM38A and TMEM38B - Mouse Genome Informatics) (Yazawa et al., 2007). TRIC-A contains three putative transmembrane segments and assembles into a bullet-shaped homotrimer to function as a monovalent cation-selective channel. Knockout mice lacking both TRIC-A and TRIC-B channels suffer embryonic cardiac failure, and the mutant cardiac myocytes display severe dysfunction in SR Ca2+ handling. Moreover, the SR from TRIC channel-deficient skeletal muscle shows reduced K+ permeability and weakened Ca2+ release. Therefore, TRIC channels function as counter ion channels that synchronise with ryanodine receptor-mediated Ca2+ release in striated muscle cells (Yazawa et al., 2007). The role of TRIC channels expressed in non-muscle cells, however, remains to be studied. We focused on the neonatal lethality that occurs in TRIC-B-knockout mice, and this study identified an essential role for TRIC-B in perinatal lung maturation.
MATERIALS AND METHODS
TRIC-B-knockout mice
TRIC-B-knockout mice of the C57BL6J and 129 strain genetic backgrounds were generated and genotyped as described previously (Yazawa et al., 2007). Haematological assessment was carried out using a blood gas analyser (model GASTAT-601, Techno Medica, Japan), glucose tester and lactate meter (Glutest Ace and Lactate Pro, Arkray, Japan). Mouse experiments were conducted with the approval of the Animal Research Committee at Kyoto University, according to the regulations regarding animal experimentation at Kyoto University.
Immunochemical analysis
Neonatal lung tissues were homogenised in a buffer containing 0.3 M sucrose, 20 mM Tris-HCl (pH 7.4) and protease inhibitors, and centrifuged to remove tissue debris and nuclei. The post-nuclear fractions (10-50 μg protein) were run on SDS-polyacrylamide gels and the separated proteins transferred onto nylon filters for immunoblot analysis (Takeshima et al., 2000). Primary antibodies used were: anti-TRIC channel subtypes (Yazawa et al., 2007), aquaporin 5 (AQP5) (Alomone Labs, Israel), surfactant associated protein B (SP-B) (Millipore), prosurfactant protein C (proSP-C) (Millipore), ABC transporter A3 (ABCA3) (Santa Cruz), purinergic P2Y2 receptor (Alomone Labs), protease activated receptor 4 (PAR4) (Cell Signaling Technology), phospholipase C β3 (PLCβ3) (Santa Cruz), GTP-binding protein α-subunit q/11 (Gαq/11) (Santa Cruz), IP3 receptor type 1 (IP3R1) (Affinity BioReagents), IP3 receptor type 3 (IP3R3) (BD Transduction Laboratories), sarco/endoplasmic Ca2+-ATPase 2 (SERCA2; ATP2A2) (Santa Cruz), actin (Sigma) and ER-lumenal polypeptide chain binding protein (BiP) (BD Transduction Laboratories). Immunoreactions were visualised using horseradish peroxidase-conjugated secondary antibodies (DAKO, Denmark) and an Enhanced ECL Chemiluminescence Kit (GE Healthcare).
For immunohistochemistry, the neonatal lung was fixed using Ca2+/Mg2+-free PBS (PBS-) containing 4% paraformaldehyde (PFA) and embedded in Tissue-Tek OCT compound (Sakura Finetek, Japan) for preparing cryosections (∼10 μm). After endogenous peroxidase was quenched with 3% H2O2 and the nonspecific reaction was blocked with 1% bovine serum albumin and 5% normal goat serum, the cryosections were exposed to AQP5, SP-B or proSP-C antibodies. For confocal microscopy (Olympus, Japan), immunoreactions and nuclei were visualised with Alexa Fluor 488-conjugated secondary antibody (Molecular Probes) and with propidium iodide (Dojindo, Japan), respectively.
Anatomical analysis
Mice were fixed in 3% PFA, 2.5% glutaraldehyde and 0.1 M sodium cacodylate (pH 7.4), and tissues were washed, dehydrated and embedded in Epon (Takeshima et al., 1994). For light microscopy, tissue sections (∼1 μm) were stained with 0.1% Toluidine Blue solution. For electron microscopy (JEM-200CX, JEOL), ultrathin sections (∼80 nm) were stained with uranyl acetate and lead citrate. For Sudan Black B staining analysis, lung tissues were fixed with PBS-containing 4% PFA, immersed in sucrose solutions (up to 30%) and frozen in OCT compound. Tissue sections (∼10 μm) were treated with 50% ethanol and then stained with a solution containing 0.1% Sudan Black B and 50% ethanol. After rinsing with 50% ethanol, the specimens were counterstained with Kernechtrot's Nuclear Fast Red (Sigma).
Phospholipid analysis
The lung was cut into small pieces and digested in PBS-containing 0.5 mg/ml collagenase (Nitta Gelatin, Japan) at 37°C for 30 minutes. The digested tissue solution was centrifuged (700 g for 10 minutes), and the supernatant was recovered as the interstitial fraction. Total lipids in the fraction were extracted with a chloroform-methanol mixed solvent, and phospholipids were quantified by conventional colourimetric determination (Fiske and SubbaRow, 1925). For further analysis of phospholipid components (Taguchi et al., 2007), after adding 1-heptadecanoyl-sn-glycero-3-phosphocholine (17:0-LPC, Avanti Polar Lipids) as an internal standard, lipids were extracted from the interstitial fraction and subjected to liquid chromatography-electrospray ionisation mass spectrometry/tandem mass spectrometry (LC-ESIMS/MS) analysis using the 4000 Q-Trap Quadrupole Linear Ion Trap Hybrid Mass Spectrometer (Applied Biosystems) equipped with Acquity ultra-performance liquid chromatography (Waters). Lipid samples were separated using a reverse-phase C18 column, and phospholipid fractions were injected into the mass spectrometer using an autosampler.
Ca2+ imaging analysis
Lungs from newborn mice were washed with solution I (0.9% NaCl, 0.1% glucose, 30 mM HEPES, 6 mM KCl, 3 mM Na2HPO4, 3 mM NaH2PO4, 3 mM MgSO4, 1.5 mM CaCl2, 100 μg/ml streptomycin and 100 units/ml penicillin, pH 7.4), minced using a chopper and digested in CaCl2/MgSO4-free solution I supplemented with 5 mM EGTA and 10 units/ml elastase (Worthington) at 37°C for 20 minutes. The resulting cell solution was treated with ∼0.5 mg/ml DNase I (Wako, Japan) for 5 minutes, and then sequentially passed through nylon filters (160-, 44- and 15-μm mesh). Single cells obtained were incubated in mouse IgG-coated plates (37°C for 30 minutes, twice), and unattached cells were collected by centrifugation and cultured on cover glasses in Dulbecco's Modified Eagle's Medium supplemented with 10% foetal bovine serum, 100 μg/ml streptomycin and 100 units/ml penicillin. For fluorometric Ca2+ imaging, cultured alveolar cells were loaded with 5 μM Fura-2 AM (Dojindo) in a physiological salt solution (PSS: 150 mM NaCl, 4 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 5.6 mM glucose, 5 mM HEPES, pH 7.4). A cooled CCD camera mounted on a microscope equipped with a polychrometer (Meta Fluor Imaging System, Universal Imaging) was used to capture fluorescence images with alternating excitation at 340 and 380 nm and emission at greater than 510 nm.
RESULTS
Respiratory failure in TRIC-B-knockout neonates
We recently generated TRIC-B-knockout mice by gene targeting (Yazawa et al., 2007). Heterozygous mice were indistinguishable from wild-type littermates in growth, development and reproduction. By crossing the heterozygous mice, TRIC-B-knockout neonates were delivered at the expected Mendelian frequency, although they were marginally smaller in body size (data not shown). The knockout neonates wriggled their bodies and gasped for breath, and died shortly after birth (Fig. 1A). Respiratory gas exchange is normally initiated at birth, when the mice turn pink and rapidly inflate their lungs, as indicated by the appearance of white patches on the thorax. By contrast, although TRIC-B-knockout mice initiated normal respiratory efforts, they failed to inflate their lungs, remaining cyanotic and succumbing within an hour. In accordance with this observation, our haematological assessment detected insufficient plasma O2 pressure, elevated CO2 pressure and reduced pH in the mutant neonates (Table 1). Therefore, TRIC-B-knockout neonates die due to severe hypoxia and acidosis resulting from respiratory failure.
Fig. 1.

Respiratory failure and deflated lungs in TRIC-B-knockout neonates. (A) Wild-type (WT) and TRIC-B-knockout (B-/-) neonates. (B) Lung tissues from wild-type and TRIC-B-knockout neonates. The TRIC-B-knockout lungs were congested and small in terms of volume. (C) Photomicroscopy images of the lungs from wild-type (upper row) and TRIC-B-knockout (bottom row) mice. The tissue specimens were sectioned and stained with Toluidine Blue. Scale bar: 40 μm.
Table 1.
Haematological data for TRIC-B-knockout neonates
| Parameter | WT | B(−/−) |
|---|---|---|
| pH | 7.49±0.10 (4) | 6.79±0.05** (3) |
| pO2 (Torr) | 140.1±1.4 (4) | 48.4±1.8** (3) |
| pCO2 (Torr) | 15.5±3.2 (4) | 72.4±17.9** (3) |
| HCO3− (mmol/l) | 10.0±1.2 (4) | 10.6±1.4 (3) |
| Hb (g/dl) | 10.3±0.8 (4) | 10.7±1.7 (3) |
| Glucose (mg/dl) | 26.9±2.2 (15) | 28.5±3.9 (10) |
| Lactate (mmol/l) | 16.8±0.4 (15) | 16.9±0.5 (10) |
The data represent the mean±s.e.m. The numbers of neonates examined are shown in parentheses. Statistical differences between the genotypes are marked with asterisks (P<0.01 by Student's t-test).
WT, wild type; B(−/−), knockout.
Although the lungs from TRIC-B-knockout neonates showed normal wet-weight levels (data not shown), the tissues were deflated, the intra-alveolar septa remained thick and air spaces were diminished (Fig. 1B). Histological observations confirmed atelectasis neonatorum in the mutant neonates (Fig. 1C). In the wild-type lungs stained with Toluidine Blue, pulmonary alveoli were well-organised by embryonic day (E) 17.5, and there were many black dots indicative of surfactant protein aggregates in the alveolar spaces. By contrast, although the TRIC-B-knockout lungs retained the surfactant aggregates, they did not form organised alveoli even at the neonatal stage. Therefore, alveolar hypoplasia seems to be the major cause of death in the mutant neonates.
TRIC channel subtypes are differentially expressed in all tissues. Although the mature lungs of adult mice contain moderate levels of both TRIC-A and TRIC-B channels (Yazawa et al., 2007), immunoblotting clearly indicated that TRIC-B expression is dominant over TRIC-A expression in the neonatal lung (Fig. 2A). In the mutant neonates, the loss of TRIC-B channels might specifically damage a certain cell type in the lung.
Fig. 2.
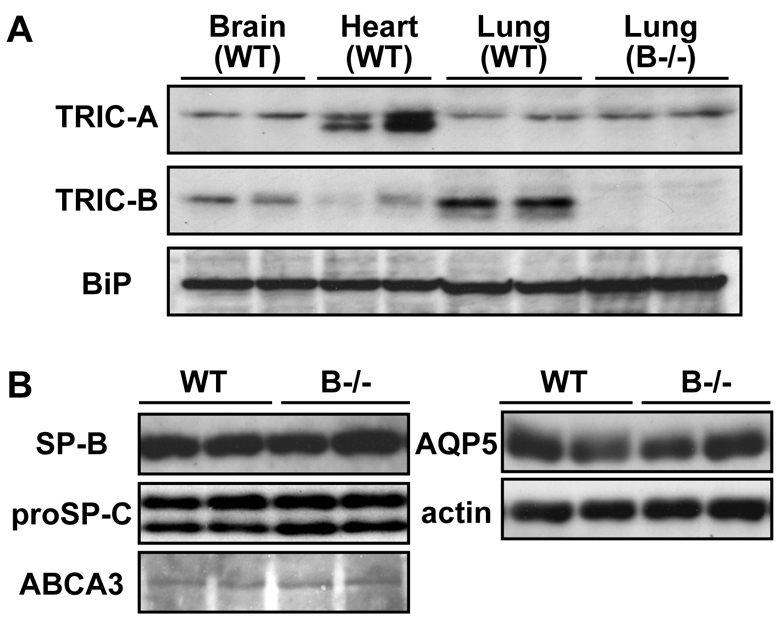
TRIC channel subtypes in neonatal tissues and alveolar epithelial marker proteins in TRIC-B-knockout mice. (A) Immunoblot analysis of TRIC channels in wild-type and TRIC-B-knockout neonates. Total tissue homogenates were examined using antibodies specific to TRIC channel subtypes and to the loading control BiP (HSPA5). (B) Immunoblot analysis of alveolar epithelial markers in neonatal lungs. Total lung homogenates were examined using antibodies to type II cell markers (mature SP-B, proSP-C and ABCA3), a type I marker (AQP5) and to the loading control actin.
Ultrastructural abnormalities of alveolar type II cells lacking TRIC-B channels
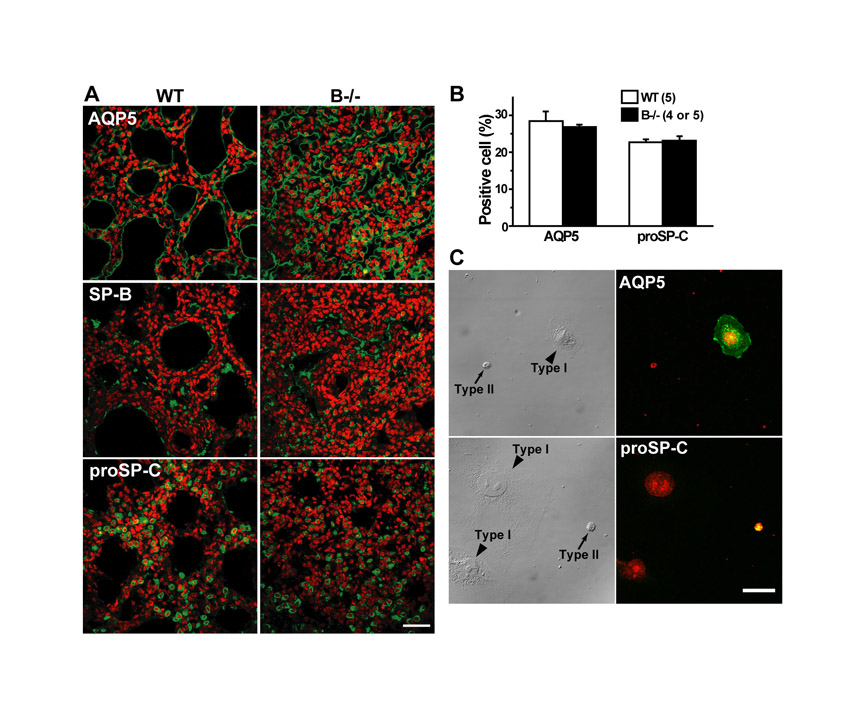
Our histological analysis also suggested the persistent accumulation of glycogen-rich cells in the TRIC-B-knockout neonatal lung (Fig. 1C). To confirm this, glycogen-rich cells were visualised by metachromasy for Toluidine Blue staining (Fig. 3A). Indeed, the proportion of glycogen-rich cells in the alveolar epithelium of the TRIC-B-knockout lung was more than twofold higher than the control value of the wild-type lung (Fig. 3B). The alveolar epithelium is composed of type I and type II cells; the type I cells are squamous and surround the alveolar spaces, whereas the type II cells are cylindrical and serve to secrete surfactants. Immature type II cells excessively preserve sugar to generate glycogen deposits in the cytoplasm, and during their morphological and functional maturation the stored glycogen is converted into phospholipids to produce lamellar bodies composed of surfactant lipids and proteins (Ridsdale and Post, 2004). Therefore, the excess of glycogen-rich cells suggests that the functional maturation of type II cells is severely interrupted in the TRIC-B-knockout neonates. However, immunoblot analysis (Fig. 2B) showed normal expression of type I (AQP5) and type II [mature SP-B (SFTPB), proSP-C (SFTPC) and ABCA3] cell markers in the TRIC-B-knockout lungs, and immunohistochemical analysis (see Fig. S1 in the supplementary material) detected a normal population of type I and type II marker-positive cells in the mutant alveoli. These observations indicate that the fundamental cell-fate decision occurs normally in the alveolar epithelium of TRIC-B-knockout mice.
Fig. 3.

Glycogen accumulation and poor lamellar bodies in TRIC-B-knockout type II cells. (A) Histological analysis of neonatal mouse lungs stained with Toluidine Blue. The metachromasy indicates an increased glycogen content in pulmonary alveoli from TRIC-B-knockout neonates. (B) Statistical analysis of glycogen-rich cells among alveolar epithelial cells in wild-type and TRIC-B-knockout lungs. Alveolar epithelial cells exhibiting a purple-red colour after Toluidine Blue staining were classified as glycogen-rich. More than 1870 alveolar epithelial cells were randomly examined for each genotype for the statistical comparison. (C) Electron microscopy of lungs from E18.5 wild-type and TRIC-B-knockout embryos. (Upper row) Pulmonary alveoli, confirming the predominance of glycogen-rich cells in the TRIC-B-knockout lungs. The control lung contained alveolar type II cells equipped with peripheral saccules (arrowheads), whereas immature type II cells with poor saccules (arrowheads) or without saccules (asterisks) were observed in the TRIC-B-knockout lung. (Lower row) Typical type II cells, poor saccule formation, accumulated glycogen deposits and insufficient lamellar body formation in the TRIC-B-knockout lung. Gly, glycogen deposit; Nu, nucleus; Lb, lamellar body. (D) Statistical analysis of lamellar body-positive cells among alveolar epithelial cells. (E) Statistical analysis of lamellar body numbers observed in individual cells. (F) Statistical analysis of lamellar body sizes. The lamellar body was morphologically assigned in electron microscopy images and the size was analysed using ImageJ (NIH). For the statistical analyses, more than 780 alveolar epithelial cells and more than 152 lamellar bodies were examined for each genotype. The data represent the mean±s.e.m., the numbers of mice examined are shown in parentheses, and statistical differences between the genotypes are marked with an asterisk (P<0.05 by Student's t-test). Scale bars: 10 μm in A; 2 μm in C.
In wild-type neonates, electron microscopy predominantly detected mature alveolar type II cells equipped with apical microvilli and containing numerous lamellar bodies. In TRIC-B-knockout neonates, however, the deflated lung predominantly contained immature type II cells, as indicated by sparse microvilli, large glycogen deposits and insubstantial lamellar bodies (Fig. 3C). Statistical evaluations confirmed that TRIC-B-knockout neonates contained fewer lamellar body-positive cells than wild-type neonates (Fig. 3D). Furthermore, both the quantity and size of lamellar bodies were obviously insufficient in TRIC-B-knockout type II cells (Fig. 3E,F). Thus, lamellar body formation is seriously impaired in the mutant type II cells.
Impaired phospholipid synthesis and secretion in alveolar type II cells lacking TRIC-B channels
Based on the poor lamellar bodies detected in TRIC-B-knockout neonates, we predicted reduced phospholipid contents in the mutant alveolar type II cells as well as in the alveolar space. To examine phospholipid deposits in intracellular lamellar bodies, we utilised Sudan Black B to differentially stain phospholipids and nuclei black and pink, respectively (Fig. 4A). The area of black-stained granules in the TRIC-B-knockout lung was significantly smaller than that in the wild-type lung (Fig. 4B). In order to assess surfactant phospholipids in the alveolar space, we prepared interstitial extracts from the neonatal lungs by a mild collagenase treatment. Total phospholipid levels in the interstitial fractions from TRIC-B-knockout lungs were significantly lower than in those from wild-type lungs (Fig. 4C). Therefore, both the biosynthesis and secretion of surfactant phospholipids are impaired in the mutant type II cells.
Fig. 4.

Impaired phospholipid synthesis and secretion in TRIC-B-knockout type II cells. (A) Photomicroscopy images of neonatal mouse lung tissues stained with Sudan Black B. Scale bar: 50 μm. (B) Quantification of black-stained granules that indicate phospholipid deposits in type II cells. (C) Colourimetric quantification of total phospholipids in interstitial fractions. (D) LC-ESIMS/MS quantification of surfactant phosphatidylcholine (PC) species in interstitial fractions. The PC contents were normalised using an internal standard of 17:0-lyso PC (see Fig. S2 in the supplementary material). The data represent the mean±s.e.m., the numbers of the lungs examined are shown in parentheses, and statistical differences between the genotypes are marked with an asterisk (P<0.05 by Student's t-test).
To further examine the secreted phospholipid species, the interstitial fractions were subjected to LC-ESIMS/MS analysis (see Fig. S2A,B in the supplementary material). To reduce the surface tension at the alveolar air-liquid interface, alveolar surfactant contains a large amount of phosphatidylcholine (PC) (∼80% of total phospholipids), and lamellar bodies contain an abundance of PC species carrying palmitate residues (Rooney, 1985). Not only the major 16:0-16:1 and 16:0-16:0 PC species (Fig. 4D), but also the minor PC species (see Fig. S2C in the supplementary material), were clearly reduced in the interstitial fractions from the TRIC-B-knockout lungs. Phosphatidylglycerol (PG) is the second major phospholipid in lung surfactant, accounting for ∼10% of the total phospholipids (Rooney, 1985). Major PG species, including 16:0-16:1 and 16:0-16:0 PG, were also decreased in the mutant lung (see Fig. S2D in the supplementary material). Moreover, we detected lower amounts of phosphatidylethanolamine, phosphatidylserine and phosphatidylinositol species in the mutant lung (see Fig. S2E in the supplementary material).
After lamellar bodies are secreted into the alveolar space, the surfactant undergoes a sequential morphological transformation to tubular myelin, small vesicles and mono- and multi-layers. The transformation processes depend upon the phospholipid and protein composition of the secreted lamellar bodies. Tubular myelin is not essential for alveolar inflation (Ikegami et al., 2001), but seems to contribute to the formation of lipid layers at the air-liquid interface (Hallman, 2004). In the electron microscopy analysis, tubular myelin structures were frequently observed in the wild-type neonatal lungs, whereas we could not detect typical tubular myelin in TRIC-B-knockout neonates (see Fig. S3 in the supplementary material). Together with the results from the LC-ESIMS/MS analysis, this ultrastructural abnormality provides further evidence of the insufficient quantity and deranged composition of the surfactant phospholipids secreted into the alveolar space of the TRIC-B-knockout lung.
Compromised Ca2+ release in alveolar type II cells lacking TRIC-B channels
Because TRIC channels support Ca2+ release from the muscle SR, and alveolar type II cells exhibit morphological and metabolic abnormalities in TRIC-B-knockout neonates, we next surveyed Ca2+ signalling defects in the mutant type II cells. When primary alveolar cells were prepared in culture, type II cells were distinguishable from type I cells by their appearance: the type I cells were squamous, whereas the type II cells were cuboidal. This assignment was further confirmed by immunostaining for the alveolar epithelial markers AQP5 and proSP-C (see Fig. S1C in the supplementary material). It is well recognised that type II cells gradually lose their specialised features in primary culture. To minimise this dedifferentiation and to standardise cellular conditions, cultured type II cells were subjected to Ca2+ imaging analysis after ∼72 hours.
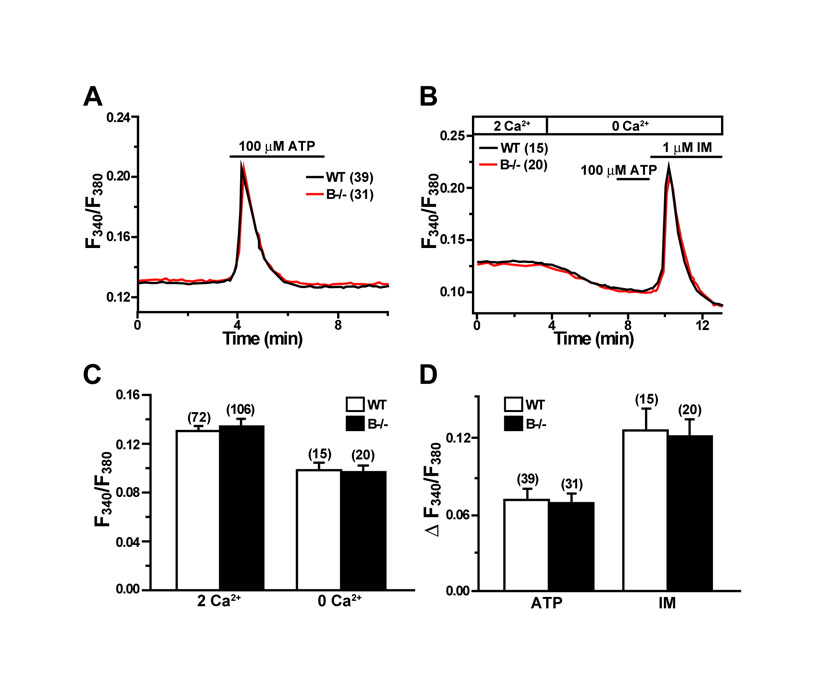
Fura-2 measurements detected three major defects in cultured TRIC-B-knockout type II cells. The first striking abnormality was that resting Ca2+ levels in the mutant cells were significantly lower than those in wild-type cells in a normal bathing solution. However, Ca2+ removal from the bathing solution reduced cytoplasmic Ca2+ levels in both the mutant and wild-type type II cells, and similar resting levels were observed in both under Ca2+-free conditions (Fig. 5A,B). Several molecular mechanisms can be proposed for the impressive fluctuation in the resting Ca2+ level seen in the TRIC-B-knockout cells (see Discussion).
Fig. 5.

Compromised Ca2+ handling in TRIC-B-knockout type II cells. (A) Impaired ATP-evoked Ca2+ transients and facilitated Ca2+ responses induced by ionomycin (IM) in TRIC-B-knockout type II cells. Fura-2 Ca2+ imaging was performed in type II cells derived from more than four neonates of each genotype, and averaged ratiometric traces are shown. (B) Statistical analysis of resting cytoplasmic Ca2+ levels in normal and Ca2+-free bathing solutions. (C) Statistical analysis of ATP-evoked Ca2+ transients and the subsequent ionomycin-induced Ca2+ responses. (D) Enhanced ionomycin-induced Ca2+ responses and normal store-operated calcium entry (SOCE) activity in TRIC-B-knockout type II cells. (E) Statistical analysis of ionomycin-induced responses and following SOCE activities. The data represent the mean±s.e.m., the numbers of cells examined are shown in parentheses, and statistical differences between the genotypes are marked with asterisks (P<0.01 by Student's t-test).
Because the ryanodine receptor agonist caffeine did not evoke detectable Ca2+ transients, it is likely that type II cells do not contain intracellular stores equipped with ryanodine receptors (data not shown). Surfactant secretion from type II cells is stimulated by purinergic P2Y2 receptor (P2RY2) activation, which results in IP3 receptor-mediated Ca2+ release (Haller et al., 1998). In agreement with the previous report, ATP application to cultured type II cells generated Ca2+ transients. The second phenotype of TRIC-B-knockout type II cells was impaired agonist-induced Ca2+ transients (Fig. 5A,C). Because the PAR4 (F2RL3) receptor is also expressed in type II cells (Ando et al., 2007), IP3 receptor-mediated Ca2+ release can be induced by a PAR4 agonist peptide (sequence AYPGKF). TRIC-B-knockout type II cells also exhibited weakened PAR4 agonist-evoked Ca2+ transients (see Fig. S4A,B in the supplementary material). It is unlikely that the compromised IP3 receptor-mediated Ca2+ release was due to defects in phosphatidylinositol turnover, as no differences were observed in the expression of signalling proteins between the TRIC-B-knockout and wild-type lungs (see Fig. S5 in the supplementary material).
The third phenotype of TRIC-B-knockout type II cells was that they contained Ca2+-overloaded intracellular stores. Application of the Ca2+ ionophore ionomycin (Fig. 5D,E) and the Ca2+ pump inhibitors thapsigargin and cyclopiazonic acid (see Fig. S4C-F in the supplementary material) evoked highly enhanced Ca2+ responses in the mutant cells. Therefore, despite the presence of Ca2+-overloaded stores, IP3 receptor-mediated Ca2+ release was weakened in the mutant cells. Indeed, TRIC-B-knockout type II cells showed poor Ca2+ transients by P2Y2 receptor activation, but facilitated Ca2+ responses by ionomycin in a continuous recording (Fig. 5A). Agonist-induced IP3 generation often results in activation of Ca2+ influx in many cell types (Putney, 2007). This Ca2+ entry involves signalling from depleted intracellular stores to cell surface Ca2+ channels, a process referred to as store-operated calcium entry (SOCE). Cultured type II cells showed typical SOCE responses, and no abnormality was detected in SOCE in TRIC-B-knockout cells (Fig. 5D and see Fig. S4 in the supplementary material).
The three atypical features in Ca2+ handling described above are specific to alveolar type II cells in TRIC-B-knockout mice, as we could not detect any abnormalities in the Ca2+ imaging data from mutant alveolar type I cells (see Fig. S6 in the supplementary material), cardiac myocytes (see Fig. S7 in the supplementary material) or embryonic fibroblasts (see Fig. S8 in the supplementary material). These observations imply that abnormal Ca2+ handling is closely linked to the morphological and biochemical defects in TRIC-B-knockout type II cells during perinatal maturation.
DISCUSSION
TRIC-A protein forms a monovalent cation-selective channel in lipid bilayer membranes and functions as a counter ion channel coupled with ryanodine receptor-mediated Ca2+ release in striated muscle cells (Yazawa et al., 2007). In our current collaborative study, recombinant TRIC-B protein displayed a similar channel-forming activity (our unpublished observations). Knockout mice lacking TRIC-A channels are fertile, whereas TRIC-B-knockout mice show neonatal lethality. Double-knockout mice lacking both TRIC subtypes are embryonic lethal, and the aggravated lethality indicates in vivo compensatory functions between the subtypes (Yazawa et al., 2007). In this study, we found weakened IP3 receptor-mediated Ca2+ release from Ca2+-overloaded intracellular stores in TRIC-B-knockout alveolar type II cells (Fig. 5). These abnormalities in stored Ca2+ handling closely resemble those observed in embryonic cardiomyocytes lacking both TRIC channel subtypes (Yazawa et al., 2007). Therefore, TRIC-B channels predominantly expressed in the neonatal lung seem to function as counter ion channels synchronised with IP3 receptor-mediated Ca2+ release and act to support efficient agonist-induced Ca2+ responses in alveolar type II cells (Fig. 6). In addition to muscle and alveolar type II cells, TRIC channel subtypes are likely to contribute to physiological Ca2+ release in various additional cell types.
Fig. 6.
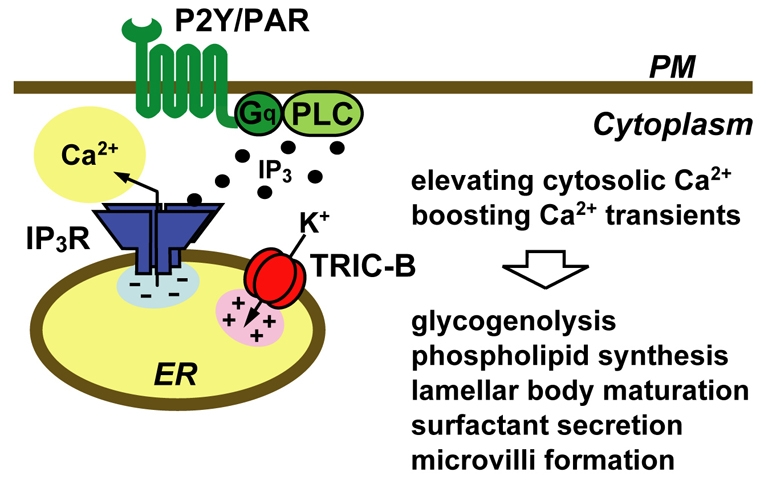
Proposed role of the TRIC-B channel in type II cells. By functioning as a counter ion channel coupled with IP3 receptor-mediated Ca2+ release in intracellular stores, TRIC-B channels seem to stimulate agonist-induced Ca2+ transients and maintain resting Ca2+ levels in type II cells. Because cellular Ca2+ signals are probably involved in the functional and morphological maturation of type II cells at the perinatal stage, impaired Ca2+ handling caused by the TRIC-B deficiency is likely to disturb the maturation processes, leading to respiration failure.
The `idling' levels of IP3 generation and IP3 receptor gating presumably constitute a Ca2+ leakage machinery to modulate Ca2+ stores and to elevate cytoplasmic Ca2+ levels under basal conditions in several cell types (Mozhayeva and Mozhayeva, 1996; Lomax et al., 2002; White and McGeown, 2003). In this study, we detected reduced resting Ca2+ levels in TRIC-B-knockout type II cells (Fig. 5). In addition to compromising agonist-induced Ca2+ release, the loss of TRIC-B channels might inhibit the proposed Ca2+ leakage machinery. Because the contents of Ca2+ stores are restricted in a balance between Ca2+ uptake and release, it is reasonable that the TRIC-B-channel deficiency inhibits Ca2+ leakage generated by basal phosphatidylinositol turnover and thus produces Ca2+-overloaded stores in type II cells. Alternatively, the loss of TRIC-B channels could interfere with basal SOCE activity, which could also elevate resting cytoplasmic Ca2+ levels. This seems rather unlikely, however, because TRIC-B-knockout type II cells retained regular SOCE responses under several store-depleted conditions (Fig. 5 and see Fig. S4 in the supplementary material).
During the functional maturation of alveolar epithelial cells, type I cells differentiate from immature type II cells in response to TGFβ/Smad signalling (Bhaskaran et al., 2007) and become AQP5 positive (Williams, 2003), whereas type II cells start to abundantly synthesise phospholipids and surfactant associated proteins A to D (SP-A to D; SFTPA to D) (Rooney et al., 1994). Based on immunochemical data with alveolar marker proteins (Fig. 2B and see Fig. S1 in the supplementary material), TRIC-B channels are not involved in cellular differentiation of alveolar epithelium. However, insufficient glycogen breakdown and phospholipid synthesis (Figs 3 and 4) probably result from compromised Ca2+ handling in TRIC-B-knockout type II cells, as sugar and lipid metabolisms are controlled by several key Ca2+-dependent enzymes, including glycogen phosphorylase, pyruvate dehydrogenase and fatty acid synthase (Davis and Kauffman, 1986; Terrand et al., 2001; Nasser et al., 2004). The combination of reduced resting Ca2+ levels and insufficient agonist-evoked Ca2+ transients presumably inactivates these Ca2+-dependent enzymes, and thereby might inhibit the conversion of glycogen into phospholipids during the perinatal maturation of TRIC-B-knockout type II cells. Moreover, abnormal Ca2+ handling is likely to inhibit the formation of lamellar bodies containing phospholipids and surfactant proteins (Fig. 3), as Ca2+-dependent fusion between juvenile lamellar bodies is required for their morphological maturation (Dietl and Haller, 2005). The impaired biosynthesis of phospholipids, together with their poor secretion (Fig. 4) and atypical structural features in the alveolar space (see Fig. S3 in the supplementary material), led us to conclude that these abnormalities combine to produce a lethal defect in lung inflation in TRIC-B-knockout neonates (Fig. 6).
Our data further raise the possibility that impaired Ca2+ handling could interrupt Ca2+-dependent kinase/phosphatase signalling in TRIC-B-knockout type II cells. Calcineurin is a Ca2+/calmodulin-dependent phosphatase that directly dephosphorylates nuclear factors of activated T cells (NFATs) in the cytoplasm to promote their translocation into the nucleus and participation in the transcriptional regulation of diverse genes. Mutant mice with a pulmonary epithelium-specific deletion of the calcineurin b1 (Ppp3r1) gene show respiratory failure immediately after birth, and in the mutant lungs the expression of SP-A to D and ABCA3 is almost absent. Therefore, the expression of the type II cell-specific proteins is transcriptionally upregulated under the control of the calcineurin-NFAT cascade during perinatal maturation (Dave et al., 2006). By contrast, normal expression levels of SP-B, proSP-C and ABCA3 were observed in the TRIC-B-knockout lungs (Fig. 2B), indicating that calcineurin-NFAT signalling is not significantly affected in the mutant type II cells. Although the roles in lung development of the Ca2+/calmodulin-dependent kinases, protein kinase C subtypes and Ca2+-dependent tyrosine kinases are largely unknown, it is interesting to speculate that insufficient signals from such Ca2+-dependent kinases might amplify or attenuate the primary defects that result from the gene ablation in TRIC-B-knockout type II cells.
Supplementary material
Supplementary material for this article is available at http://dev.biologists.org/cgi/content/full/136/14/2355/DC1
Supplementary Material
We thank Naoya Onohara for reading the manuscript. This work was supported in part by research grants from the Ministry of Education, Culture, Sports, Science and Technology, the Ministry of Health and Welfare of Japan, US National Institutes of Health, the Kurata Memorial Foundation, the Japan Foundation for Applied Enzymology and the Novartis Foundation. Deposited in PMC for release after 12 months.
References
- Ando, S., Otani, H., Yagi, Y., Kawai, K., Araki, H., Nakamura, T., Fukuhara, S. and Inagaki, C. (2007). Protease-activated receptor 4-mediated Ca2+ signaling in mouse lung alveolar epithelial cells. Life Sci. 81, 794-802. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny, I. and Ehrlich, B. E. (1995). The inositol 1,4,5-trisphosphate (InsP3) receptor. J. Membr. Biol. 145, 205-216. [DOI] [PubMed] [Google Scholar]
- Bhaskaran, M., Kolliputi, N., Wang, Y., Gou, D., Chintagari, N. R. and Liu, L. (2007). Trans-differentiation of alveolar epithelial type II cells to type I cells involves autocrine signaling by transforming growth factor beta 1 through the Smad pathway. J. Biol. Chem. 282, 3968-3976. [DOI] [PubMed] [Google Scholar]
- Cai, C., Masumiya, H., Weisleder, N., Matsuda, N., Nishi, M., Hwang, M., Ko, J.-K., Lin, P., Thornton, A., Zhao, X. et al. (2009). MG53 nucleates assembly of cell membrane repair machinery. Nat. Cell Biol. 11, 56-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave, V., Childs, T., Xu, Y., Ikegami, M., Besnard, V., Maeda, Y., Wert, S. E., Neilson, J. R., Crabtree, G. R. and Whitsett, J. A. (2006). Calcineurin/Nfat signaling is required for perinatal lung maturation and function. J. Clin. Invest. 116, 2597-2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, L. H. and Kauffman, F. C. (1986). Calcium-dependent activation of glycogen phosphorylase in rat pheochromocytoma PC12 cells by nerve growth factor. Biochem. Biophys. Res. Commun. 138, 917-924. [DOI] [PubMed] [Google Scholar]
- Dietl, P. and Haller, T. (2005). Exocytosis of lung surfactant from the secretory vesicle to the air-liquid interface. Annu. Rev. Physiol. 67, 595-621. [DOI] [PubMed] [Google Scholar]
- Fiske, C. H. and SubbaRow, Y. (1925). The colorimetric determination of phosphorus. J. Biol. Chem. 66, 375-400. [Google Scholar]
- Haller, T., Ortmayr, J., Friedrich, F., Völkl, H. and Dietl, P. (1998). Dynamics of surfactant release in alveolar type II cells. Proc. Natl. Acad. Sci. USA 95, 1579-1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallman, M. (2004). Lung surfactant, respiratory failure, and genes. New Engl. J. Med. 350, 1278-1280. [DOI] [PubMed] [Google Scholar]
- Ikegami, M., Elhalwagi, B. M., Palaniyar, N., Dienger, K., Korfhagen, T., Whitsett, J. A. and McCormack, F. X. (2001). The collagen-like region of surfactant protein A (SP-A) is required for correction of surfactant structural and functional defects in the SP-A null mouse. J. Biol. Chem. 276, 38542-38548. [DOI] [PubMed] [Google Scholar]
- Lomax, R. B., Camello, C., Van Coppenolle, F., Petersen, O. H. and Tepikin, A. V. (2002). Basal and physiological Ca2+ leak from the endoplasmic reticulum of pancreatic acinar cells. Second messenger-activated channels and translocons. J. Biol. Chem. 277, 26479-26485. [DOI] [PubMed] [Google Scholar]
- MacLennan, D. H., Abu-Abed, M. and Kang, C.-H. (2002). Structure-function relationships in Ca2+ cycling proteins. J. Mol. Cell. Cardiol. 34, 897-918. [DOI] [PubMed] [Google Scholar]
- Meissner, G. (1983). Monovalent ion and calcium ion fluxes in sarcoplasmic reticulum. Mol. Cell. Biochem. 55, 65-82. [DOI] [PubMed] [Google Scholar]
- Meissner, G. (1994). Ryanodine receptor/Ca2+ release channels and their regulation by endogenous effectors. Annu. Rev. Physiol. 56, 485-508. [DOI] [PubMed] [Google Scholar]
- Mozhayeva, M. G. and Mozhayeva, G. N. (1996). Evidence for the existence of inositol (1,4,5)-trisphosphate- and ryanodine-sensitive pools in bovine endothelial cells. Ca2+ releases in cells with different basal level of intracellular Ca2+. Pflugers Arch. 432, 614-622. [DOI] [PubMed] [Google Scholar]
- Nasser, J. A., Hashim, S. A. and Lachance, P. A. (2004). Calcium and magnesium ATPase activities in women with varying BMIs. Obes. Res. 12, 1844-1850. [DOI] [PubMed] [Google Scholar]
- Nishi, M., Komazaki, S., Kurebayashi, N., Ogawa, Y., Noda, T., Iino, M. and Takeshima, H. (1999). Abnormal features in skeletal muscle from mice lacking mitsugumin29. J. Cell Biol. 147, 1473-1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson, R. L., Boehning, D. and Snyder, S. H. (2004). Inositol 1,4,5-trisphosphate receptors as signal integrators. Annu. Rev. Biochem. 73, 437-465. [DOI] [PubMed] [Google Scholar]
- Putney, J. W., Jr (2007). Recent breakthroughs in the molecular mechanism of capacitative calcium entry (with thoughts on how we got here). Cell Calcium 42, 103-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridsdale, R. and Post, M. (2004). Surfactant lipid synthesis and lamellar body formation in glycogen-laden type II cells. Am. J. Physiol. Lung Cell Mol. Physiol. 287, L743-L751. [DOI] [PubMed] [Google Scholar]
- Rooney, S. A. (1985). The surfactant system and lung phospholipid biochemistry. Am. Rev. Respir. Dis. 131, 439-460. [DOI] [PubMed] [Google Scholar]
- Rooney, S. A., Young, S. L. and Mendelson, C. R. (1994). Molecular and cellular processing of lung surfactant. FASEB J. 8, 957-967. [DOI] [PubMed] [Google Scholar]
- Taguchi, R., Nishijima, M. and Shimizu, T. (2007). Basic analytical systems for lipidomics by mass spectrometry in Japan. Methods Enzymol. 432, 185-211. [DOI] [PubMed] [Google Scholar]
- Takeshima, H., Iino, M., Takekura, H., Nishi, M., Kuno, J., Minowa, O., Takano, H. and Noda, T. (1994). Excitation-contraction uncoupling and muscular degeneration in mice lacking functional skeletal muscle ryanodine-receptor gene. Nature 369, 556-559. [DOI] [PubMed] [Google Scholar]
- Takeshima, H., Komazaki, S., Nishi, M., Iino, M. and Kangawa, K. (2000). Junctophilins: a novel family of junctional membrane complex proteins. Mol. Cell 6, 11-22. [DOI] [PubMed] [Google Scholar]
- Terrand, J., Papageorgiou, I., Rosenblatt-Velin, N. and Lerch, R. (2001). Calcium-mediated activation of pyruvate dehydrogenase in severely injured postischemic myocardium. Am. J. Physiol. Heart Circ. Physiol. 281, H722-H730. [DOI] [PubMed] [Google Scholar]
- White, C. and McGeown, J. G. (2003). Inositol 1,4,5-trisphosphate receptors modulate Ca2+ sparks and Ca2+ store content in vas deferens myocytes. Am. J. Physiol. Cell Physiol. 285, C195-C204. [DOI] [PubMed] [Google Scholar]
- Williams, M. C. (2003). Alveolar type I cells: molecular phenotype and development. Annu. Rev. Physiol. 65, 669-695. [DOI] [PubMed] [Google Scholar]
- Yazawa, M., Ferrante, C., Feng, J., Mio, K., Ogura, T., Zhang, M., Lin, P.-H., Pan, Z., Komazaki, S., Kato, K. et al. (2007). TRIC channels are essential for Ca2+ handling in intracellular stores. Nature 448, 78-82. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}