

Abstract

Primary allylic selenosulfates (seleno Bunte salts) and selenocyanates transfer the allylic selenide moiety to thiols giving primary allylic selenosulfides, which undergo rearrangement in the presence of PPh3 with the loss of selenium to give allylically rearranged allyl alkyl sulfides. This rearrangement may be conducted with prenyl-type selenosulfides to give isoprenyl alkyl sulfides. Alkyl secondary and tertiary allylic disulfides, formed by sulfide transfer from allylic heteroaryl disulfides to thiols, undergo desulfurative allylic rearrangement on treatment with PPh3 in methanolic acetonitrile at room temperature. With nerolidyl alkyl disulfides this rearrangement provides an electrophile-free method for the introduction of the farnesyl chain onto thiols. Both rearrangements are compatible with the full range of functionality found in the proteinogenic amino acids and it is demonstrated that the desulfurative rearrangement functions in aqueous media, enabling the derivatization of unprotected peptides. It is also demonstrated that the allylic disulfide rearrangement can be inducted in the absence of phosphine at room temperature by treatment with piperidine, or simply by refluxing in methanol. Under these latter conditions the reaction is also applicable to allyl aryl disulfides, providing allylically rearranged allyl aryl sulfides in good yields.
Introduction
The development of methods for the functionalization of biopolymers, especially peptides and proteins, under the mildest possible conditions, dubbed ligation, is a current frontier in organic chemistry.1–16 High chemoselectivity, stability, and compatibility with protic solvents, and even aqueous media, are requirements for such ligations.
Thiols, and in particular cysteine residues, have proven popular targets for chemoselective ligation.1,2,17 Native chemical ligation (NCL) for the synthesis of peptides and proteins, and its enzyme-promoted biochemical equivalent, expressed protein ligation,18–21 is one such reaction that takes advantage of the sulfhydryl group and which uses it to great advantage in the highly chemoselective formation of amide bonds in aqueous solution.10,21–31 Another thiol-based method, the selective formation of mixed disulfides, is both one of the oldest and most enduring of ligation methods.1,2,32–38 The mildness of the disulfide ligation and its established chemoselectivity for the cysteine thiol in the presence of all the proteinogenic amino acids stands in stark contrast to the various other methods for cysteine functionalization, most of which involve the capture of the cysteine thiol by electrophilic species, and which consequently have obvious potential chemoselectivity issues.1,2,39 The practicality of the disulfide ligation, with its direct applicability to cysteine-containing peptides, also contrasts with the various ingenious indirect methods that have been developed for the preparation of S-functionalized cysteine derivatives,17 including, for example, the Michael addition of thiols to dehydroalanine units,40 the alkylation of thiolates with peptide-based β-halo-alanine units,41–43 and other electrophiles,44,45 the opening of peptide-based aziridines by thiolates,46,47 and the synthesis of peptides with previously functionalized cysteine building blocks,48–50 each of which requires the synthesis of modified peptides. The many advantages of the disulfide ligation are offset, however, by its impermanence, which results from the lability of the disulfide bond in the presence of thiols and other reducing agents.
Consideration of the practical advantages of the disulfide ligation, and the disadvantages of its impermanence, led us to investigate methods for rendering it permanent. The result of our search was the dechalocogenative allylic seleneosulfide and disulfide ligations, whose scope we describe in full in this article.51,52
Results and Discussion
Background
The phosphine-promoted desulfurative allylic rearrangement of diallyl disulfides to give allyl sulfides53–57 proceeds by way of a [2,3]-sigmatropic rearrangement via a diallyl thiosulfoxide intermediate58,59 and, thus, is closely related to the well known Evans-Mislow rearrangement of allylic sulfoxides.60 This dechalcogenative rearrangement, which may also be induced to operate in the reverse direction on treatment of allyl sulfides with elemental sulfur,61 and which is potentially important in the chemistry of essential oils derived from garlic,62 appeared to us to hold promise as a means of rendering disulfides permanent if it could be caused to function at ambient temperatures in protic solvents. The seminal work of Höfle and Baldwin revealed that the reaction is rapid in benzene at room temperature provided that the thiosulfoxide is removed from the equilibrium. Thus, the diallyl disulfides 1 and 2 rearrange via the thiosulfoxides 3 and 4 to the thermodynamically more stable disulfides 5 and 6 via two sequential sigmatropic rearrangements with half-lives of 79 and 14 mins, respectively, at 24 °C (Scheme 1).58
Scheme 1.

Rearrangement of Diallyl Disulfides58
In the absence of a thiophile, simple alkyl allyl sulfides 7 and 8 were found by Höfle and Baldwin to be stable at room temperature and could be purified by vacuum distillation, whereas 10 and 11 were reported to lose sulfur spontaneously at room temperature, albeit without mention of a timescale for this process.58 A comparable dependence of rearrangement rate on substituent pattern was observed by Moore and Trego in their early work on the reaction.53 Pseudo first order rate constants were measured by Höfle and Baldwin for the reaction of the alkyl allyl disulfides 7–11 with PPh3 in benzene at 60 °C leading to the conclusion that increased bulk around the thiosulfoxide reduces its concentration in the equilibrium and so retards reduction.58 While this remains possible, we consider it more likely that the equilibrium is shifted more toward the thiosulfoxide when a more highly substituted alkene is formed at the expense of a less substituted one, and when a more highly substituted C-S bond is replaced by a less substituted one (Scheme 2).58
Scheme 2.

Pseudo-first Order Rate Constants for Phosphine-mediated Rearrangement inBenzene58
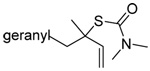
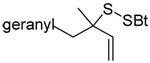
A single example of the corresponding deselenative rearrangement of a diallyl diselenide, that of di(geranyl) diselenide to geranyl linalyl selenide, was reported to proceed with a half life of approximately 2.5 h at 25 °C on exposure to excess PPh3 in chloroform, thereby indicating the selenium version of this reaction to be considerably faster than the original sulfur protocol.63 Guillemin and coworkers subsequently were able to obtain crude preparations (~80% pure) of diallyl diselenide, dicrotyl diselenide, and diprenyl diselenide, characterize them spectroscopically, and study their reactions with tributylstannane, indicating such diselenides to have at least moderate stability at room temperature.64
Preliminary Studies
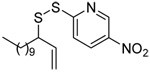
We initiated our investigations with a brief survey of methods of mixed disulfide formation, of solvents, and of phosphines for the allylic disulfide rearrangement. As shown in Table 1, allylic pyridyl sulfides,1,2,65 allylic thiosulfonates,38 and allylic Bunte salts (S-allyl thiosulfates)66,67 all proved adept at the transfer of allylic sulfides to a range of simple thiols. The phosphine-promoted rearrangement, however, was not so simple. Thus, it was necessary to heat the alkyl geranyl disulfide 18 with PPh3 in toluene at reflux in order to achieve efficient conversion to the rearranged linalyl sulfide 19 (Table 1, entry 1). It is important to note that, consistent with our aim of developing a practical process suitable for application to biologically relevant systems, our reactions were carried out with a minimal excess of phosphine, unlike the work of Höfle and Baldwin.58 The difference in the amount of phosphine presumably accounts for the differences in reaction times and temperatures observed. Under the same conditions the phenyl geranyl disulfide 20, however, gave the unrearranged phenyl geranyl sulfide 21 (Table 1, entry 2), thereby drawing attention to the relatively fine dividing line between the desired dechalcogenative allylic rearrangement and the better known nucleophilic removal of a sulfur atom from a disulfide.68 As is evident from the comparison of entries 1 and 2 in Table 1, the replacement of an alkylthiyl moiety by an arylthiyl group is sufficient to tip the balance in favor of nucleophilic attack by the phosphine on the native disulfide. Entry 3 of Table 1 illustrates how the replacement of PPh3 by the more nucleophilic hexaethylphosphoramine enabled the reaction temperature to be reduced to the ambient but, unfortunately, with continued formation of the simple desulfurization product. Finally, Table 1, entry 4, ruled out any future use of hexaethylphosphoramine, at least in the context of amino acid and peptide-based systems, owing to both transesterification and racemization evidenced by incorporation of deuterium from the solvent at the α-center.
Table 1.
Preliminary Survey of Disulfides and Phosphines.
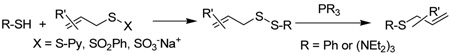 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Thiol | Donor | Disulfide (% yield) | Solvent | Temp (°C) | PR3 | Product (% yield) |
| 1 | Me(CH2)7SH | PhMe | 110 | PPh3 | |||
| 17 | 18 (72) | 19 (71) | |||||
| 2 | PhSH | PhMe | 110 | PPh3 | |||
| 17 | 20 (95) | 21 (69) | |||||
| 3 | PhSH | 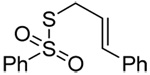 |
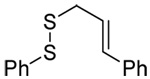 |
C6D6 | rt | P(NEt2)3 | 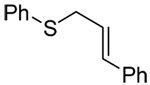 |
| 22 | 23 (73) | 24 (80) | |||||
| 4 | 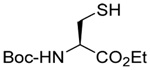 |
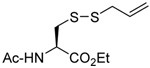 |
CD3OD | rt | P(NEt2)3 | 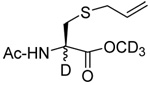 |
|
| 25 | 26 | 27 (77) | 28 (73) | ||||
Allylic Selenosulfide Rearrangement
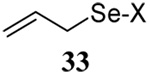
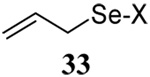
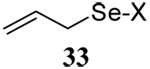
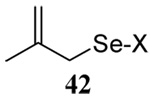
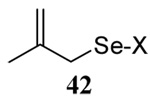
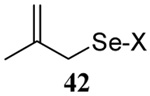
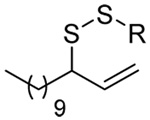
The somewhat inauspicious results reported in Table 1 led us to consider the possibility of the deselenative rearrangement of Se-allylic selenosulfides, which should be more facile than the desulfurative rearrangement of the allylic disulfides.69 The clean formation of simple mixed alkyl selenosulfides can be realized at room temperature by the reaction of Se-alkyl seleno-Bunte salts with thiols.70,71 Accordingly, a number of Se-allyl seleno Bunte salts were prepared by the reaction of potassium seleno sulfate, prepared in situ from potassium sulfite and selenium powder, with allyl halides. These compounds were typically orange crystalline solids that, while not indefinitely stable, could be handled in air at room temperature. Reaction with a range of aliphatic thiols then resulted in the formation of a series of selenosulfides, that could be readily detected by thin layer chromatography and/or NMR spectroscopy, but which were allowed to undergo the subsequent deselenative rearrangement, either with or without the addition of phosphine as required (Scheme 3, Table 2).
Scheme 3.

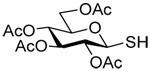
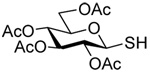
Formation and Rearrangement of Se-Allylic Selenosulfides
Table 2.
Formation and Rearrangement of Selenosulfides
| Entry | Thiol | Donor | Donor (% yield) |
Product a,b | Rearrangement yield (equiv of PPh3) |
||
|---|---|---|---|---|---|---|---|
| X = SO3−K+ | X = CN | from X = SO3−K+ | from X = CN | ||||
| 1 | Me(CH2)15SH | 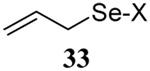 |
56 | 84 | 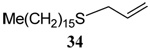 |
74% (0) | 70% (0) |
| 2 | 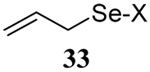 |
56 | 84 |  |
70% (0) | 70% (0) | |
| 3 | 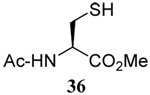 |
 |
56 | 84 | 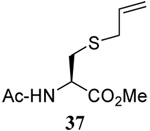 |
61% (2.5) | 80% (0) |
| 4 | 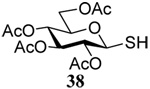 |
 |
56 | 84 | 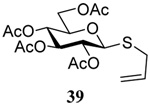 |
50% (2.5)c | 55 (2.5)d |
| 5 |  |
 |
56 | 84 | 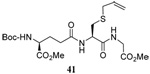 |
58% (2.5) | 75% (0) |
| 6 | 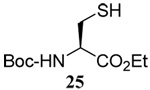 |
 |
58 | 73 | 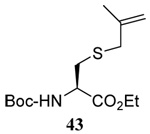 |
62% (2.5) | 68% (0) |
| 7 |  |
 |
58 | 73 | 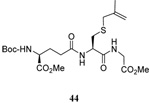 |
60% (2.5) | 71% (0) |
| 8 | 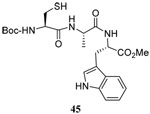 |
 |
58 | 73 | 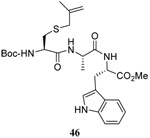 |
60% (2.5) | 65% (2.5) |
| 9 | Me(CH2)7SH | 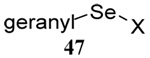 |
40 | 80 |  |
62% (2.5) | 70% (2.5) |
| 10 | 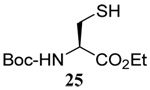 |
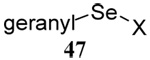 |
40 | 80 | 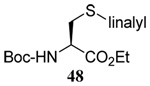 |
46% (2.5) | 66% (2.5) |
| 11 |  |
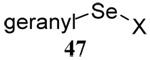 |
40 | 80 | 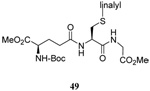 |
43% (2.5) | 55% (2.5) |
| 12 |  |
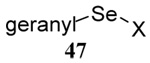 |
40 | 80 | 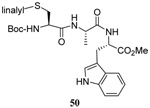 |
51% (2.5) | 50% (2.5) |
| 13 | 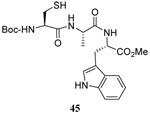 |
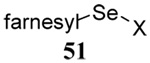 |
35 | 85 | 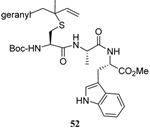 |
47% (2.5) | 40% (2.5) |
| 14 | 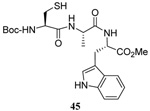 |
 |
28 | 75 |  |
31% (2.5) | 65% (2.5) |
Unless otherwise stated all reactions were conducted in MeOH
at room temperature
T = 65 °C
CDCl3 was used as a solvent at T = 65 °C.
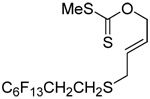
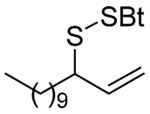
We have also prepared the Se-allyl selenosulfides by the reaction of allyl selenocyanates64 with thiols, a process that also takes place readily at room temperature (Scheme 3), and which had been reported for the formation of simple diaryl selenosulfides.72 The formation of allyl selenosulfides from the reaction of allylselenols with disulfides was not investigated because of anticipated difficulties in the preparation and handling of the allylic selenols.64 Overall, it is clear from Table 2 that allyl selenocyanates may generally be prepared in higher yields than the corresponding Se-allyl seleno Bunte salts, but that in most cases both selenenylation systems perform the transfer of the Se-allyl group to the thiol comparably well, as evidenced by the yield of the subsequent rearrangement products.
It is of some interest to note that the primary allylic selenocyanates prepared and employed in this study were readily handled at room temperature and showed no tendency to undergo rapid rearrangement. This observation parallels that of Riague and Guillemin, who had earlier prepared several of the same allylic selenocyanates and reported their purification by vacuum distillation,73 but it stands in contrast to the reported chemistry of simple allylic thiocyanates which are reported to undergo [2,3]-sigmatropic rearrangement to the corresponding allylic isothiocyanates in a matter of hours at room temperature.74,75 Based on the work of Sharpless with 2-methyl-3-selenocyanato-1-heptene, secondary and presumably tertiary selenocyanates can be expected to undergo a [1,3]-sigmatropic rearrangement to their primary regioisomers.63
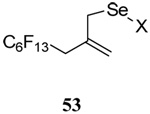
The transfer of a simple allyl or methallyl group did not necessitate the addition of phosphine and proceeded spontaneously over a 2 h period at room temperature. In the case of geranyl and farnesyl Se-Bunte salts and selenocyanates, proceeding with allylic rearrangement to the linalyl and nerolidyl sulfides, respectively, it was necessary to add PPh3 to provoke the loss of selenium. The more difficult rearrangement of the geranyl and farnesyl systems corresponds to the pattern observed originally with the allylic disulfides (Scheme 2), with the prenyl system 9 undergoing the phosphine-promoted rearrangement least rapidly. This is clearly the consequence of the formation of the less stable isoprenyl substitution pattern. The anomeric thiol 38 rearranged slowly over a period of several days at room temperature in the presence of PPh3, and much more rapidly at 65 °C. The fluorous substituted methallyl system 53, unlike the simple methallyl transfer reactions, necessitated the addition of PPh3 in order to proceed at room temperature. These latter two observations are understood in terms of the mechanism of Scheme 3, with the formation of the intermediate selenosulfoxide disfavored by either an electron-withdrawing substituent on the allyl group or by the involvement of the sulfur atom in an exo-anomeric type interaction that serves to remove electron density from sulfur. When the reaction takes place with the formation of a new stereogenic center this was typically obtained as an almost equimolar mixture of two diastereomers and, accordingly, no attempt was made to distinguish the isomers.
The range of examples presented in Table 2, which were all conducted at room temperature with the exception of the carbohydrate-based example, displays the broad functional group compatibility of the method. A series of further experiments in which the reaction of cysteine derivative 25 with the Se-methallyl seleno Bunte salt 42 and PPh3 was conducted in methanol at room temperature in the presence of equimolar amounts of N-benzyloxycarbonyl tyrosine benzyl ester, N-benzyloxycarbonyl methionine, Nα-benzyloxycarbonyl lysine methyl ester, N-benzyloxycarbonyl arginine methyl ester, and N-benzyloxycarbonyl aspartic acid α-methyl ester, with no loss of yield and with recovery of the spectator amino acid, further confirmed the applicability of the reaction in the presence of most standard functional groups.
Unfortunately, all attempts at the preparation of tertiary allylic seleno Bunte salts or the equivalent selenocyanates, such as would be necessary for the introduction of a simple geranyl or farnesyl group to a sulfide, failed. This result, which is not surprising in view of the instability of secondary allylic selenocyanates and even phenylselenides noted by Sharpless,63 forced us to return to the allylic disulfides.
Allylic Disulfide Rearrangement
The early work on the allylic disulfide rearrangement (Scheme 2)53,58 indicated the considerable effect of substituents in the allylic moiety on reaction rate with the secondary and tertiary allylic disulfides undergoing rearrangement several orders of magnitude more rapidly than their primary counterparts in hot benzene, thereby providing grounds for hope that the rearrangement could be induced to function as required at room temperature with the correct substituent pattern. In addition, consideration of the mechanism of the allylic disulfide rearrangement leads to the hypothesis that the reaction should be facilitated in polar solvents capable of stabilizing the dipolar thiosulfoxide intermediate.76,77 Indeed, the early workers in the field noted a 6–10 fold increase in the rate of desulfurative rearrangement of 1,3-dimethylbut-2-enyl t-butyl disulfide with PPh3 on going from benzene to ethanol/benzene (9/2) at 80 °C,53 and Höfle and Baldwin commented on the instability of methanolic solutions of 10 and 11 with respect to rearrangement, as compared to the neat samples.58
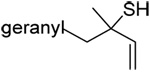
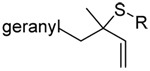
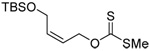
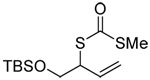
A series of secondary and tertiary allylic thiols were prepared by the [3,3]-sigmatropic rearrangement of a variety of primary allylic thiocarbonyl derivatives (Scheme 4, Table 3).78–83 While this chemistry was straightforward, caution was necessary in the conversion of the rearrangement products to the thiols as these were found to undergo a known80 1,3-shift to the isomeric primary allylic thiols under basic conditions. The optimum conditions for this cleavage involved the reduction of the thiol carbamates with lithium aluminum hydride,80 and the cleavage of the dithiocarbonates with ethanolamine. The thiols were then allowed to react with either 2,2’-dipyridyl disulfide, 2,2’-di(5-nitropyridyl) disulfide, or 2,2’-di(1,3-benzothiazolyl) disulfide1,2,32–34,65,84–87 for conversion to the corresponding allyl heteroaryl disulfides and ultimate transfer to the target thiols (Scheme 4, Table 3). It is noteworthy that sulfenylating agents derived from hindered allylic thiols have drawn little attention so far, with only a limited number of allylic benzothiazolyl disulfides being described in the literature.88,89 In addition to these allylic heteroaryl disulfides, we also prepared a single example 75 of an S-allyl Se-phenyl selenosulfide, by reaction of the thiol with N-phenylselenophthalimide, as it had been reported that the selenosulfides were more effective at sulfenyl transfer than the corresponding disulfides.37
Scheme 4.

Preparation of Secondary and Tertiary Allylic Thiols and Heteroaryl Disulfides
Table 3.
Preparation of Allylic Thiols and the Corresponding Allyl Heteroaryl Disulfides
| Allylic Xanthates (% yield) | Rearranged Thiocarbamate (% yield) | Thiol (% yield) | Disulfide (% yield) |
|---|---|---|---|
 |
 |
 |
|
| 61 (80) | 62 (99) | 63 (93) | R = 2-Py 64 (96) R = 4-NO2-2-Py 65 (86) R = Bt 66 (81) |
 |
 |
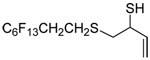 |
|
| 67 (92) | 68 (99) | 69 (−) | 70 (15) |
 |
 |
 |
|
| 71 (80) | 72 (83) | 73 (87) | R = SBt 74 (83) R = SePh 75 (56) |
 |
 |
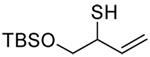 |
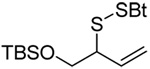 |
| 76 (87) | 77 (94) | 78 (−) | 79 (45 from 77) |
As anticipated these allylic heteroaryl disulfides enabled smooth sulfenyl transfer to a selection of primary thiols in either benzene or methanol/acetonitrile mixtures at room temperature (Table 4). Addition of either PPh3 or (4-dimethylaminophenyl)diphenylphosphine then provoked the desired desulfurative allylic rearrangement. For those reactions that were carried out in benzene, the sulfenyl transfer was affected at room temperature, after which the phosphine was added and the reaction mixture heated to reflux to bring about the rearrangement. On the other hand, as expected from mechanistic considerations, when reactions were conducted in methanol and acetonitrile no heating was required for the entire sequence. With the more highly substituted allyl heteroaryl disulfides sulfenyl transfer was relatively slow, but was accelerated very significantly by the addition of triethylamine. Interestingly, sulfenyl transfer from the selenosulfide 75 was also slow, but was accelerated in the presence of triethylamine, and we conclude that there is no practical advantage to the use of the selenosulfide transfer protocol.90 No obvious consequence in terms of reactivity was observed when the sulfenyl transfer group was changed from 5-nitro-2-pyridyl, to 2-pyridyl, to 2-benzothiazolyl, but the differing polarities of the corresponding heteroaryl sulfides formed as byproducts can be used to advantage in the case of otherwise difficult purifications. Similarly, no significant difference in the rate of the desulfurative rearrangement was observed on changing from PPh3 to (4-dimethylaminophenyl)diphenylphosphine 84, but the latter did afford a practical advantage in terms of purification of the final products.
Table 4.
Formation and Rearrangement of Secondary and Tertiary Allylic Disulfide
| Thiol | Sulfenating agent | Solvent | PR3 | T (°C) | Product (% yield) |
|---|---|---|---|---|---|
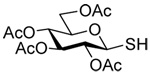 |
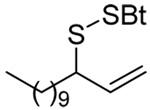 |
C6H6 | PPh3 | 80 | |
| 38 | 66 | 80 (73) ds: 12/1 | |||
 |
C6H6 | PPh3 | 80 |  |
|
| 38 | 70 | 81 (75) ds: 7.9/1 | |||
 |
 |
C6H6 | PPh3 | 80 | |
| 38 | 74 | 82 (66) ds: 1.8/1 | |||
 |
 |
MeOH:MeCN (1:1) | Ph2P-(4-C6H4NMe2) | rt | 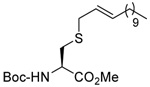 |
| 83 | 66 | 84 | 85 (85) | ||
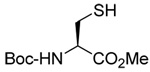 |
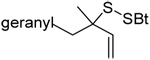 |
MeOH:MeCN (1:1) | PPh3 | rt | |
| 83 | 74 | 86(84) ds: 1.87/1 | |||
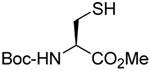 |
MeOH:MeCN (1:1) | PPh3 | rt | ||
| 83 | 75 | 86 (80) mixture of isomers | |||
 |
MeOH:MeCN (1:1) | Ph2P-(4-C6H4NMe2) | rt | 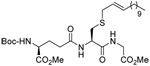 |
|
| 40 | 65 | 84 | 87 (70) | ||
| MeOH:MeCN (1:1) | PPh3 | rt | 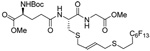 |
||
| 40 | 70 | 88 (75) | |||
 |
MeOH:MeCN (1:1) | PPh3 | rt | 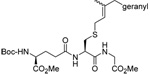 |
|
| 40 | 74 | 89 (70) ds: 1.7/1 | |||
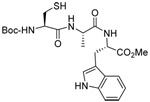 |
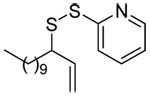 |
MeOH:MeCN (1:1) | PPh3 | rt | 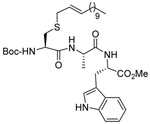 |
| 45 | 64 | 90 (80) | |||
 |
MeOH:MeCN (1:1) | PPh3 | rt | 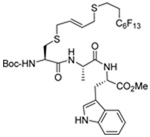 |
|
| 45 | 70 | 91 (79) | |||
 |
 |
MeOH:MeCN (1:1) | PPh3 | rt | 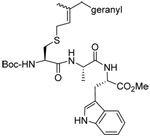 |
| 45 | 74 | 92 (79) ds: 1.6/1 |
The desulfurative rearrangement of the secondary allylic disulfides takes place with high E-selectivity as anticipated on the basis of related [2,3]-sigmatropic rearrangements such as the Evans-Mislow rearrangement60 or the [2,3]-Wittig rearrangement.91 With the tertiary allylic disulfides E/Z mixtures of the rearranged products are formed that slightly favor the E-isomer. Interestingly, in the case of the nerolidyl selenosulfide 75 the product 86 was obtained as a more complex mixture of isomers, presumably due to isomerization of the central alkene in the resulting farnesyl chain by the selenide byproducts in combination with light.92 When the reaction of the cysteine derivative 83 with the sulfenyl transfer agent 66 and PPh3 was carried out in a mixture of deuteriomethanol and deuterioacetonitrile no incorporation of deuterium at the amino acid α-center was observed, indicating that the reaction conditions do not provoke racemization.
The examples presented in Table 4 attest to the broad functional group compatibility of the formation and desulfurative rearrangement of secondary and tertiary allylic disulfides and draw attention to the complementary nature of the process with the deselenative allylic selenosulfide protocol, with all classes of primary, secondary, and tertiary allylic sulfides accessible by one of the two reaction sequences at room temperature and without the need for electrophilic reagents.
Peptide Functionalization in Aqueous Media
The applicability of the desulfurative allylic rearrangement to unprotected peptides in aqueous media was first probed with glutathione 93 and sulfenylating agent 64. In the event, reaction of disulfide 64 with glutathione 93 in a 2/1/1 mixture of Tris Buffer/MeCN/THF, with a substrate concentration of 0.02 M at room temperature, followed by addition of PPh3 smoothly yielded the lipidated glutathione 94 in 70% yield (Scheme 5).
Scheme 5.

Functionalization of Glutathione
The chemoselectivity of the rearrangement was further explored with the commercial decapeptide fibronectin fragment 95.93,94 This decapeptide was selected as a proving ground due to the dense array of the more challenging amino acid side chains that it presents. As with glutathione 93, sulfenylation of the cysteine moiety of the decapeptide 95, followed by phosphine-promoted rearrangement, was accomplished at room temperature in aqueous buffer, with the aid of organic co-solvents, and resulted in the isolation of the lipidated decapeptide 96 in 70% yield (Scheme 6). The location of the tridecene chain on the cysteine group in 96 was established by MS/MS experiments.
Scheme 6.

Functionalization of a Fibronectin Fragment
As a final demonstration of the protocol in aqueous media we prepared an octapeptide 101 in which the cysteine residue was located four residues from a C-terminal methionine unit, so as to mimic the Cys-AA1-AA2-Met sequence targeted by the farnesyl transferase enzymes.95–97 The other amino acids in this synthetic peptide were selected so as to provide a measure of steric hindrance to the cysteine moiety. Two tetrapeptides were assembled by routinely98,99 and 97, which was destined to become the N-terminal fragment of the target, was saponified, converted to the ethylthio ester 98 with HATU and ethanethiol,100,101 and finally released from the carbamate protecting group (Scheme 7). This sequence resulted in an inseparable mixture of two diastereomers 99 at the C-terminal alanine, a problem known to plague thioester synthesis for NCL.102 Coupling of thioester 99 with tetrapeptide 100 under conditions of NCL afforded the target octapeptide 101 in 51% yield as a 1.3/1 mixture of two diastereomers, which was separated by preparative RP-HPLC. The major diastereoisomer, presumed to be the all l-octapeptide, was then allowed to react with the nerolidyl benzothiazolyl disulfide 74 under buffered aqueous conditions at room temperature, followed by addition of PPh3 leading, overall, to the farnesylated octapeptide 102 in 66% isolated yield (Scheme 7). This farnesylation reaction occurred with the formation of E/Z-isomers mixtures in approximately equal ratio in line with the model experiments in organic solution (Table 4).
Scheme 7.

Synthesis of an Octapeptide by Native Chemical Ligation and Subsequent Farnesylation
The examples of Scheme 5–Scheme 7 are distinct from other recent functionalizations of sulfur in cysteine containing peptides17 as they require neither the use of electrophilic reagents,1,2,39 nor the incorporation of modified amino acid residues into the peptide backbone prior to functionalization,40–47 nor the synthesis of peptides with previously functionalized amino acid building blocks.48–50 These factors combine to render this chemistry both highly chemoselective, and highly efficient in terms of steps required on the actual peptide target. In contrast to other methods involving the addition of nucleophilic thiols to dehydroalanine groups inserted into peptides,40 the products are obtained as single diastereomers with the sole exception being the formation of E/Z-mixtures when the allylic sulfide contains a trisubstituted alkene. We anticipate that the use of a water-soluble phosphine should be possible in the above experiments, with the proviso that it not be sufficiently nucelophilic and/or basic to lead to the type of problems outlined for hexaethylphosphoramine encountered in our preliminary survey of conditions.
Rearrangement in the Absence of Phosphines
Finally, we turn to the question of the requirement for a phosphine to promote the desulfurative rearrangement of the allylic disulfides. Several factors indicated that conditions may be found under which the reaction would function in the absence of the phosphine. For example, the attempted purification of disulfide 103 by chromatography over silica gel resulted in the isolation of 13% of the allylically rearranged thioether 85 along with the disulfide itself (Scheme 8).52 Secondly, on standing in deuteriochloroform that had not been treated to remove acidic impurities the same disulfide 103 underwent partial rearrangement to 85 with loss of sulfur.52 More tangentially related are the reported use of morpholine or piperidine to drive the related allylic sulfoxide/sulfenate rearrangement,60 and the uptake of molecular sulfur by allylic sulfides leading to rearranged allylic disulfides.56
Scheme 8.

Rearrangement in the Absence of Phosphines
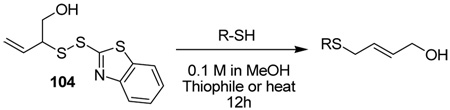
The apparent acid-catalyzed rearrangement of 103 to 85 with loss of sulfur could not be developed into a satisfactory general method. However, after some investigation, we have found that the rearrangement can be achieved by stirring with two equivalents of piperidine at room temperature in methanol, or simply by heating to reflux in methanol with no added thiophile for a period of several hours. Examples of rearrangements so-conducted are presented in Table 5 using a sulfenyl transfer reagent 104 derived by removal of the silyl group from sulfenyl donor 79. Appropriate control experiments conducted with PPh3 at room temperature are also reported in Table 5.
Table 5.
Desulfurative Rearrangement of Allylic Disulfides in the Absence of Phosphine
 | |||||
|---|---|---|---|---|---|
| Entry | Thiol | Product | Method |
||
| PPh3 % yield, E/Z | Piperidine % yield, E/Z | Heat % yield E/Z | |||
| 1 |  |
45 E only |
62 2.6:1 |
78 E only |
|
| 105 | |||||
| 2 |  |
- | - | 67 E only |
|
| 83 | 106 | ||||
| 3 | 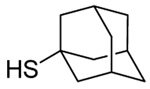 |
- | - | 68 E only |
|
| 107 | |||||
| 4 | Me(CH2)7SH | - | - | 75 E only |
|
| 108 | |||||
| 5 | 4-Me-C6H4-SH | 18 E only |
66 2.5:1 |
66 E only |
|
| 109 | |||||
| 6 | PhSH | - | - | 63 E only |
|
| 110 | |||||
| 7 | 4-Cl-C6H4-SH | - | - | 62 E only |
|
| 111 | |||||
| 8 | 4-NO2-C6H4SH | - | - | 20 E only |
|
| 112 | |||||
| 9 | 2-PySH | - | - | 45 E only |
|
| 113 | |||||
| 10 | 2-HO-C6H4SH | - | - | 57 E only |
|
| 114 | |||||
It is of some interest that both the piperidine and the refluxing methanol conditions enable the desulfurative rearrangement of allyl aryl disulfides for the first time, thereby opening up new avenues for the synthesis of allyl aryl sulfides and extending the overall scope of the reaction. The rearrangements of the allyl aryl disulfides presented in Table 5, and conducted in the absence of phosphine but with the aid of either piperidine or hot methanol, are to be contrasted with the attempted promotion of such a rearrangement set out in Table 1, when the excision of sulfur was observed without the allylic rearrangement. We also note the application of the overall process to a tertiary thiol and draw attention to the absence of racemization in the case of the cysteine derivative as determined in the usual manner by the use of deuteriomethanol as solvent.
Conclusions
The deselenative allylic rearrangement of primary allyl selenosulfides and the desulfurative rearrangement of secondary and tertiary allylic are powerful and complementary techniques for the synthesis of primary, secondary, and tertiary allylic sulfides at room temperature. The preparation of the selenosulfides and disulfides requires no electrophilic reagent, leading to highly chemoselective methods for the permanent modification of thiols. The chemoselectivity of the two reactions enables their application to unprotected peptides in aqueous media in the presence of all types of standard amino acid side chains, thereby providing a powerful means for the direct and permanent modification of cysteine containing peptides.
Supplementary Material
Full experimental protocols, and copies of spectra for all new compounds.
Acknowledgement
We thank the NIH (AI 56575 and GM 62160) for partial support of this work, and the Ministère des Affaires Étrangères, France, for the award of a Lavoisier Fellowship to FB. DC thanks Professor Yasuhiro Kajihara, Yokoyama City University, for a helpful discussion regarding native chemical ligation.
References
- 1.Hermanson GT. Bioconjugate Techniques. San Diego: Academic Press; 1996. [Google Scholar]
- 2.Lundblad RL. Chemical Reagents for Protein Modification. 3 ed. Boca Raton: CRC Press; 2005. [Google Scholar]
- 3.van Swieten PF, Leeuwenburgh MF, Kessler BM, Overkleeft HS. Org. Biomol. Chem. 2005;3:20–27. doi: 10.1039/b412558d. [DOI] [PubMed] [Google Scholar]
- 4.Peri F, Nicotra F. Chem Comm. 2004:623–627. doi: 10.1039/b308907j. [DOI] [PubMed] [Google Scholar]
- 5.Pratt MR, Bertozzi CR. Chem. Soc. Rev. 2005;34:58–68. doi: 10.1039/b400593g. [DOI] [PubMed] [Google Scholar]
- 6.Prescher JA, Dube DH, Bertozzi CR. Nature. 2004;430:873–877. doi: 10.1038/nature02791. [DOI] [PubMed] [Google Scholar]
- 7.Hang HC, Bertozzi CR. Acc. Chem. Res. 2001;34:727–736. doi: 10.1021/ar9901570. [DOI] [PubMed] [Google Scholar]
- 8.Nilsson BL, Hondal RJ, Soellner MB, Raines RT. J. Am. Chem. Soc. 2003;125:5268–5269. doi: 10.1021/ja029752e. [DOI] [PubMed] [Google Scholar]
- 9.Bode JW. Curr. Op. Drug Disc. Dev. 2006;9:765–775. [PubMed] [Google Scholar]
- 10.Nilsson BL, Soellner MB, Raines RT. Ann. Rev. Biophys. Biomol. Structure. 2005;34:91–118. doi: 10.1146/annurev.biophys.34.040204.144700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kalia J, Raines RT. ChemBioChem. 2006;7:1376–1383. doi: 10.1002/cbic.200600150. [DOI] [PubMed] [Google Scholar]
- 12.Lin H, Walsh CT. J. Am. Chem. Soc. 2004;126:13998–14003. doi: 10.1021/ja045147v. [DOI] [PubMed] [Google Scholar]
- 13.Langenhan JM, Thorson JS. Current Organic Synthesis. 2005;2:59–81. [Google Scholar]
- 14.Agard NJ, Baskin JM, Prescher JA, Lo A, Bertozzi CR. ACS Chem. Biol. 2006;1:644–648. doi: 10.1021/cb6003228. [DOI] [PubMed] [Google Scholar]
- 15.Köhn M, Breinbauer R. Angew. Chem. Int. Ed. 2004;43:3106–3116. doi: 10.1002/anie.200401744. [DOI] [PubMed] [Google Scholar]
- 16.Kolb HC, Finn MG, Sharpless KB. Angew. Chem. Int. Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 17.Pachamuthu K, Schmidt RR. Chem. Rev. 2006;106:160–187. doi: 10.1021/cr040660c. [DOI] [PubMed] [Google Scholar]
- 18.Muir TW, Sondhi D, Cole PA. Proc. Natl. Acad. Sci. USA. 1998;95:6705. doi: 10.1073/pnas.95.12.6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Noren CJ, Wang JM, Perler FB. Angew. Chem. Int. Ed. 2000;39:450. [PubMed] [Google Scholar]
- 20.Muralidharan V, Muir TW. Nat. Methods. 2006;3:429. doi: 10.1038/nmeth886. [DOI] [PubMed] [Google Scholar]
- 21.Coltart DM. Tetrahedron. 2000;56:3449–3491. [Google Scholar]
- 22.Dawson PE, Muir TW, Clark-Lewis I, Kent SBH. Science. 1994;266:776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 23.Dawson PE, Kent SBH. Ann. Rev. Biochem. 2000;69:923–960. doi: 10.1146/annurev.biochem.69.1.923. [DOI] [PubMed] [Google Scholar]
- 24.Yeo DSY, Srinivasan R, Chen GYJ, Yao SQ. Chem. Eur. J. 2004;10:4664–4672. doi: 10.1002/chem.200400414. [DOI] [PubMed] [Google Scholar]
- 25.Johnson ECB, Kent SBH. J. Am. Chem. Soc. 2006;128:6640–6646. doi: 10.1021/ja058344i. [DOI] [PubMed] [Google Scholar]
- 26.Brik A, Ficht S, Yang Y-Y, Bennett CS, Wong C-H. J. Am. Chem. Soc. 2006;128:15026–15033. doi: 10.1021/ja065601q. [DOI] [PubMed] [Google Scholar]
- 27.Liu L, Hong Z-Y, Wong C-H. ChemBioChem. 2006;7:429–432. doi: 10.1002/cbic.200500437. [DOI] [PubMed] [Google Scholar]
- 28.Hondal RJ, Nilsson BL, Raines RT. J. Am. Chem. Soc. 2001;123:5140–5141. doi: 10.1021/ja005885t. [DOI] [PubMed] [Google Scholar]
- 29.Clive DLJ, Hisaindee S, Coltart DM. J. Org. Chem. 2003;68:9247–9254. doi: 10.1021/jo030192r. [DOI] [PubMed] [Google Scholar]
- 30.Tchertchian S, Hartley O, Botti P. J. Org. Chem. 2004;69:9208–9214. doi: 10.1021/jo049471k. [DOI] [PubMed] [Google Scholar]
- 31.Wu B, Chen J, Warren JD, Chen G, Hua Z, Danishefsky SJ. Angew. Chem. Int. Ed. 2006;45:4116–4125. doi: 10.1002/anie.200600538. [DOI] [PubMed] [Google Scholar]
- 32.Kenyon GL, Bruice TW. Methods Enzym. 1977;47:407–430. doi: 10.1016/0076-6879(77)47042-3. [DOI] [PubMed] [Google Scholar]
- 33.Wynn R, Richards FM. Methods Enzym. 1995;251:351–356. doi: 10.1016/0076-6879(95)51138-5. [DOI] [PubMed] [Google Scholar]
- 34.Faulstich H, Heintz D. Methods Enzym. 1995;251:357–366. doi: 10.1016/0076-6879(95)51139-3. [DOI] [PubMed] [Google Scholar]
- 35.Macindoe WM, van Oijen AH, Boons G-J. Chem. Commun. 1998:847–848. [Google Scholar]
- 36.Grayson EJ, Ward SJ, Hall AJ, Rendle PM, Gamblin DP, Batsanov AS, Davis BG. J. Org. Chem. 2005;70:9740–9754. doi: 10.1021/jo051374j. [DOI] [PubMed] [Google Scholar]
- 37.Gamblin DP, Garnier P, van Kasteren S, Oldham NJ, Fairbank AJ, Davis BG. Angew. Chem. Int. Ed. 2004;43:828–833. doi: 10.1002/anie.200352975. [DOI] [PubMed] [Google Scholar]
- 38.Gamblin DP, Garnier SJ, Ward NJ, Oldham AJ, Fairbanks AJ, Davis BG. Org. Biomol. Chem. 2003;1:3642–3644. doi: 10.1039/b306990g. [DOI] [PubMed] [Google Scholar]
- 39.Ludolph B, Eisele F, Waldmann H. J. Am. Chem. Soc. 2002;124:5954–5955. doi: 10.1021/ja025768t. [DOI] [PubMed] [Google Scholar]
- 40.Zhu Y, van der Donk WA. Org. Lett. 2001;3:1189–1192. doi: 10.1021/ol015648a. [DOI] [PubMed] [Google Scholar]
- 41.Thayer DA, Yu HN, Galan MC, Wong C-H. Angew. Chem. Int. Ed. 2005;44:4596–4599. doi: 10.1002/anie.200500090. [DOI] [PubMed] [Google Scholar]
- 42.Jobron L, Hummel G. Org. Lett. 2000;2:2265–2267. doi: 10.1021/ol006019o. [DOI] [PubMed] [Google Scholar]
- 43.Pachamuthu K, Zhu X, Schmidt RR. J. Org. Chem. 2005;70:3720–3723. doi: 10.1021/jo0482357. [DOI] [PubMed] [Google Scholar]
- 44.Cohen SB, Halcomb RL. Org. Lett. 2001;3:405–407. doi: 10.1021/ol006908b. [DOI] [PubMed] [Google Scholar]
- 45.Cohen SB, Halcomb RL. J. Am. Chem. Soc. 2002;124:2534–2543. doi: 10.1021/ja011932l. [DOI] [PubMed] [Google Scholar]
- 46.Galonic DP, van der Donk W, Gin DY. J. Am. Chem. Soc. 2004;126:12712–12713. doi: 10.1021/ja046793x. [DOI] [PubMed] [Google Scholar]
- 47.Galonic DP, Ide ND, van der Donk WA, Gin DY. J. Am. Chem. Soc. 2005;127:7359–7369. doi: 10.1021/ja050304r. [DOI] [PubMed] [Google Scholar]
- 48.Palomo JM, Lumbierres M, Waldmann H. Angew. Chem. Int. Ed. 2006;45:477–481. doi: 10.1002/anie.200503298. [DOI] [PubMed] [Google Scholar]
- 49.Durek T, Alexandrov K, Goody RS, Hildebrand A, Heinemann I, Waldmann H. J. Am. Chem. Soc. 2004;126:16368–16378. doi: 10.1021/ja046164n. [DOI] [PubMed] [Google Scholar]
- 50.Kragol G, Lumbierres M, Palomo JM, Waldmann H. Angew. Chem. Int. Ed. 2004;43:5839–5842. doi: 10.1002/anie.200461150. [DOI] [PubMed] [Google Scholar]
- 51.Crich D, Krishnamurthy V, Hutton TK. J. Am. Chem. Soc. 2006;128:2544–2545. doi: 10.1021/ja057521c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Crich D, Brebion F, Krishnamurthy V. Org. Lett. 2006;8:3593–3596. doi: 10.1021/ol061381+. [DOI] [PubMed] [Google Scholar]
- 53.Moore CG, Trego GR. Tetrahedron. 1962;18:205–218. [Google Scholar]
- 54.Evans MB, Higgins GMC, Moore CG, Porter M, Saville B, Smith JF, Trego BR, Watson AA. Chem. & Ind. 1960:897. [Google Scholar]
- 55.Blackburn GM, Ollis WD. J. Chem. Soc., Chem. Commun. 1968:1261–1262. [Google Scholar]
- 56.Barnard D, Houseman TH, Porter M, Tidd BK. J. Chem. Soc., Chem. Commun. 1969:371–372. [Google Scholar]
- 57.Pilgram K, Phillips DD, Korte F. J. Org. Chem. 1964;29:1844–1847. [Google Scholar]
- 58.Höfle G, Baldwin JE. J. Am. Chem. Soc. 1971;93:6307–6308. [Google Scholar]
- 59.Kutney GW, Turnbull K. Chem. Rev. 1982;82:333–357. [Google Scholar]
- 60.Evans DA, Andrews GC. Acc. Chem. Res. 1974;7:147–155. [Google Scholar]
- 61.Baechler RD, Hummel JP, Mislow K. J. Am. Chem. Soc. 1973;95:4442–4444. [Google Scholar]
- 62.Block E, Iyer R, Grisoni S, Saha C, Belman S, Lossing FP. J. Am. Chem. Soc. 1988;110:7813–7827. [Google Scholar]
- 63.Sharpless KB, Lauer KB. J. Org. Chem. 1972;37:3973–3974. [Google Scholar]
- 64.Riague EH, Guillemin J-C. Organometallics. 2002;21:68–73. [Google Scholar]
- 65.Stoner EJ, Negrón G, Gaviño R. In: Handbook of Reagents for Organic Synthesis: Reagents for Glycoside, Nucleotide, and Peptide Synthesis. Crich D, editor. Chichester: Wiley; 2005. pp. 330–338. [Google Scholar]
- 66.Klayman DL, White JD, Sweeney TR. J. Org. Chem. 1964;29:3737–3738. [Google Scholar]
- 67.Milligan B, Swan JM. J. Chem. Soc. 1963:6008–6012. [Google Scholar]
- 68.Harpp DN, Gleason JG. J. Am. Chem. Soc. 1971;93:2437–2445. [Google Scholar]
- 69.The use of the deselenative allylic diselenide rearrangement was excluded from consideration on the grounds that selenocysteine is too uncommon in Nature to form the basis for a broad general methodology.
- 70.Patarasakulchai N, Southwell-Keely PT. Phosphorus and Sulfur. 1985;22:277–282. [Google Scholar]
- 71.Klayman DL, Günther WHH, editors. Organic selenium compounds: their chemistry and biology. New York: Wiley Interscience; 1973. [Google Scholar]
- 72.Clark ER, Al-Turaihi MAS. J. Organomet. Chem. 1977;134:181–187. [Google Scholar]
- 73.Guillemin JC, Bouayad A, Vijaykumar D. ChemComm. 2000;13:1163–1164. [Google Scholar]
- 74.Emerson DW, Booth JK. J. Org. Chem. 1965;30:2480–2481. [Google Scholar]
- 75.Carbohydrate-based allylic thiocyanates, however, require higher temperatures for rearrangement.Ferrier RJ, Vethaviyaser N. J. Chem. Soc. (C) 1971:1907–1913.
- 76.We assign the dipolar structure to the thiosulfoxides, a class of compounds, which have not been isolated or characterized spectroscopically, by reasonable analogy with the sulfoxides. Thiosulfinates (ROS(S)OR) have been isolated and studied crystallographically, spectroscopically, and computationally, with the evidence pointing to the polar structure.77
- 77.Zysman-Colman E, Nevins N, Eghbali N, Snyder JP, Harpp DN. J. Am. Chem. Soc. 2005;128:291–304. doi: 10.1021/ja0559395. [DOI] [PubMed] [Google Scholar]
- 78.Garmaise Dl, Uchiyama A, McKay AF. J. Org. Chem. 1962;27:4509–4512. [Google Scholar]
- 79.Ferrier RJ, Vethaviyasar N. J. Chem. Soc., Chem. Commun. 1970:1385–1387. [Google Scholar]
- 80.Hackler RE, Balko TW. J. Org. Chem. 1973;38:2106–2110. doi: 10.1021/jo00951a038. [DOI] [PubMed] [Google Scholar]
- 81.Ueno Y, Sano H, Okawara M. Tetrahedron Lett. 1980;21:1767–1770. [Google Scholar]
- 82.Eto M, Tajiri O, Nakagawa H, Harano K. Tetrahedron. 1998;54:8009–8014. [Google Scholar]
- 83.Nakai T, Ari-Izumi A. Tetrahedron Lett. 1976;17:2335–2338. [Google Scholar]
- 84.Talgoy MM, Bell AW, Duckworth HW. Can. J. Biochem. 1979;57:822–833. doi: 10.1139/o79-102. [DOI] [PubMed] [Google Scholar]
- 85.Kimura T, Matsueda R, Nakagawa Y, Kaiser ET. Anal. Biochem. 1982;122:274–282. doi: 10.1016/0003-2697(82)90281-0. [DOI] [PubMed] [Google Scholar]
- 86.Brzezinska E, Ternay AL. J. Org. Chem. 1994;59:8239–8244. [Google Scholar]
- 87.Rabanal F, DeGrado WF, Dutton PL. Tetrahedron Lett. 1996;37:1347–1350. [Google Scholar]
- 88.Watson AA. J. Chem. Soc. (C) 1964:2100–2107. [Google Scholar]
- 89.Morrison NJ. J. Chem. Soc., Perkin Trans. 1. 1984:101–106. [Google Scholar]
- 90.The accelerated reactions reported in the literature37 for sulfenyl transfer from selenosulfides, as compared to disulfides, also appear to be the result of the inclusion of triethylamine.
- 91.Nakai T, Mikami K. In: Org. React. Paquette LA, editor. Vol. 46. New York: John Wiley & Sons; 1994. pp. 105–209. [Google Scholar]
- 92.Barrett AGM, Barton DHR, Johnson G, Nagubandi S. Synthesis. 1978:741. [Google Scholar]
- 93.Boucaut J-C, Darribère T, Poole TJ, Aoyama H, Yamada KM, Thierry JP. J. Cell. Biol. 1984;99:1822–1830. doi: 10.1083/jcb.99.5.1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yamada KM, Kennedy DW. J. Cell. Biol. 1984;99:29–36. doi: 10.1083/jcb.99.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Casey PJ. Science. 1995;266:221. doi: 10.1126/science.7716512. [DOI] [PubMed] [Google Scholar]
- 96.Kale TA, Hsieh S-aJ, W RM, Distefano MD. Curr. Top. Med. Chem. 2003;3:1043–1074. doi: 10.2174/1568026033452087. [DOI] [PubMed] [Google Scholar]
- 97.Brunsveld L, Kuhlmann J, Alexandrov K, Wittinghofer A, Goody RS, Waldmann H. Angew. Chem. Int. Ed. 2006;45:6622–6646. doi: 10.1002/anie.200600855. [DOI] [PubMed] [Google Scholar]
- 98.Bodanszky M, Bodanszky A. The Practice of Peptide Synthesis. 2nd ed. Heidelberg: Springer-Verlag; 1994. [Google Scholar]
- 99.Goodman M, Mahwah AF, Moroder L, Toniolo C, editors. Houben-Weyl. Methods in Organic Chemistry. Synthesis of Peptides and Peptidomimetics. Vol. E22a–e. Stuttgart: Thieme; 2002–2003. [Google Scholar]
- 100.Schnolzer M, Alewood P, Jones A, Alewood D, Kent SBH. Int. J. Peptide Protein Res. 1992;40:180–193. doi: 10.1111/j.1399-3011.1992.tb00291.x. [DOI] [PubMed] [Google Scholar]
- 101.Clippingdale AB, Barrow CJ, Wade JD. J. Pept. Sci. 2000;6:225. doi: 10.1002/(SICI)1099-1387(200005)6:5<225::AID-PSC244>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 102.Kajihara Y, Yoshihara A, Hirano K, Yamamoto N. Carbohydr. Res. 2006;341:1333–1340. doi: 10.1016/j.carres.2006.04.037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Full experimental protocols, and copies of spectra for all new compounds.