

Abstract
Knowledge of 15N chemical shift anisotropy is prerequisite both for quantitative interpretation of nuclear spin relaxation rates in terms of local dynamics, and for the use of residual chemical shift anisotropy (RCSA) as a constraint in structure determination. Accurate measurement of the very small RCSA from the difference in 15N chemical shift under isotropic and weakly aligning liquid crystalline conditions is very sensitive to minute differences in sample conditions, such as pH or ionic strength. For this reason, chemical shifts were measured for the same solution, under static liquid crystalline alignment, and under magic angle spinning conditions where alignment relative to the magnetic field is removed. Measurements were made for 14 well-resolved G-N1 and 6 U-N3 15N nuclei in a sample of tRNAVal. Fitting these RCSA data together with 15N-1H dipole-CSA cross-correlated relaxation measurements to the recently refined structural model of tRNAVal yields the magnitude, asymmetry, and orientation of the 15N CSA tensors.
Imino 15N resonances contain a wealth of information on both the structure and dynamics of nucleic acids. In contrast to the very crowded spectral region of the (deoxy-)ribose resonances, the imino 1H-15N correlation spectrum is often remarkably well resolved, even for larger systems. Imino-imino NOEs provide important connectivity information between adjacent base pairs; 2hJNNcouplings across hydrogen bonds can identify the hydrogen bonded partner and yield information on H-bond strength;1,2 the 15N relaxation rates provide information on both the rotational diffusion tensor and the amplitude and time scale of internal dynamics;3-5 the 15N-1H residual dipolar coupling (RDC) provides information on the orientation of the N-H bond in weakly aligned systems; and the residual chemical shift anisotropy (RCSA)6-8 can provide a second independent measurement for the orientation of the nucleotide relative to the alignment tensor. For RCSA information to be independent of that contained in RDCs, it is required that the 15N-1H dipolar and 15N CSA tensors are not linearly correlated.9,10 Both the interpretation of relaxation rates in terms of dynamics and the use of RCSA as a structural restraint require accurate knowledge of the imino 15N CSA tensor. Quantum chemical computational work indicates that the CSA tensor is strongly affected by hydrogen bonding;11,12 however, the only experimental data on these CSA tensors were obtained from solid state NMR studies on free nucleosides.12,13
The present study aims to define experimentally the 15N CSA tensors for G-N1 and U-N3 nuclei in Watson-Crick base-paired nucleotides, under conditions commonly used for analysis of solution NMR data. Analogous to prior work in proteins,14 we derive the 15N CSA tensors from the RCSA observed for different nucleotides in the same molecule, tRNAVal, upon weak alignment in a dilute liquid crystalline phase of Pf1.15 With the structure of tRNAVal accurately known (PDB entry 1K4C),16 its alignment relative to the magnetic field is defined by one-bond 15N-1H residual dipolar couplings (RDCs). The chemical shift change, Δδ, between aligned and isotropic phase is given by14
| (1) |
where θij is the angle between the δii principal axis of the traceless CSA tensor and the Ajj principal axis of the diagonalized traceless molecular alignment tensor. Because Δδ values are quite small, in the parts per billion (ppb) range, exceptional care needs to be taken that this difference is not impacted by small changes in sample conditions (pH, concentration, ionic strength). For this reason, measurements were carried out on the same solution, with one aliquot measured under static, aligned conditions in a Shigemi microcell, and measurements of isotropic chemical shifts under slow magic angle spinning conditions (∼630 Hz, to avoid precipitation of Pf1 phage), using a Bruker 4-mm triple resonance MAS probehead. The temperature of the MAS sample was adjusted to be the same (28 °C) as that of the static sample by ensuring an identical difference in chemical shift between the water resonance and internal trimethylsilylpropionic acid, and verified by ensuring that the difference between static and MAS imino 1H and 15N chemical shifts is not correlated with their temperature coefficients (Supporting Information).
The superimposed imino 1H-15N HSQC spectra, recorded under aligned and isotropic conditions, reveal small but readily measured chemical shift differences (Figure 1 and Supporting Information). Repeated measurements indicate that the reproducibility of the measured Δδ(15N) values is ca 5 ppb, yielding a 3 ppb random error in their averaged values, with the entire Δδ range spanning 170 ppb. Optimization of the fit between measured and predicted RCSA values by adjusting the 15N CSA tensor is quite sensitive to uncertainty in the atomic coordinates (structural noise), and for this reason we also measured 15N relaxation rates to further restrain the CSA tensor. By fitting the ratio of the transverse relaxation rate, R2, and the CSA/N-H dipolar cross-correlated relaxation rate, Γ, this fit becomes insensitive to small imperfections in the dynamic and structural model used to generate this ratio.5 However, because the approximation of an axially symmetric CSA tensor collinear to the dipolar interaction is less appropriate for imino 15N sites than for peptide amide 15N, reasonably accurate knowledge of the diffusion tensor is required for analysis of the R2/Γ ratios, which were derived from R1, R2, and 15N-{1H} NOE values. Rates were fit to the axially symmetric diffusion model, using the recently RDC-refined structural model of tRNAVal (PDB entry 1K4C).16 As expected for these R2/Γ ratios, which are relatively insensitive to details of the dynamic model employed, they cluster in narrow ranges (1.43 ± 0.11 for U-N3 and 1.26 ± 0.08 for G-N1 at 500 MHz, 1.86 ± 0.15 and 1.55 ± 0.09 at 800 MHz), and provide strong constraints on the magnitude and orientation of the 15N CSA tensor. We note, however, that optimizing the fit between experimental and predicted values of both Δδ and R2/Γ values by iterative adjustment of the 15N CSA tensor is sensitive to the N-H bond length used to calculate the dipolar interaction. The strength of the 15N-1H dipolar interaction relates inversely to the magnitude of the RDC-derived alignment tensor and the optimized CSA tensor therefore scales with <rNH−3>. High resolution crystal structures, hydrogen bond 2hJNN couplings, as well as quantum chemical calculations point to a shorter N1…N3 hydrogen bond length for U-A than for G-C Watson Crick basepairs.2,17 Equilibrium N-H bond lengths of rNH = 1.036 and 1.050 Å, computed for such Watson-Crick base-paired U and G imino groups, respectively,2 need to be adjusted for zero-point stretching and bending motions in order to derive an effective bond length, rNHeff, which determines the observed RDCs and NMR relaxation rates.18,19 Assuming that the zero-point motion corrections to the 15N-1H dipolar interactions are similar for peptide and imino N-H groups yields rNHeff values of 1.043 (G) and 1.057 (U) Å.19 It is worth noting that if the same rNH bond lengths (1.04 Å) were used for U and G, generalized order parameters (S2) for U (0.91±0.05) fall significantly below those of G (0.97±0.02), whereas higher and very similar values are obtained for rNHeff = 1.043 Å for G (S2 = 0.99±0.03), and rNHeff = 1.057 Å for U (S2 = 0.97±0.06). Heteronuclear 15N-{1H} NOE values (0.76±0.11), very close to the theoretical limit, also point to the absence of significant root-mean-squared amplitudes of the internal dynamics for the bases in the tRNA Watson-Crick base-pairs.
Figure 1.
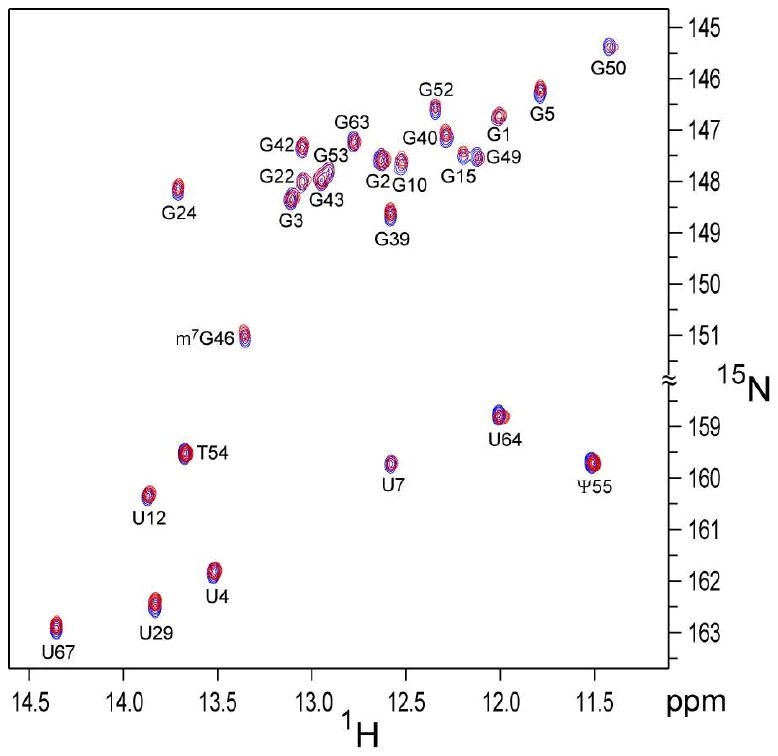
Superposition of the imino region of the 600 MHz tRNAVal HSQC spectra (0.7 mM in 90% H2O, pH 7, 80 mM NaCl, 5 mM MgCl2, 28 °C) in 10.5 mg/mL Pf1, recorded either on a static sample in a 300 μL Shigemi microcell (red) or in a 40 μL magic angle rotor, spinning at 630 Hz (blue).
Under the assumption that the 15N CSA tensor has the same value for all Watson-Crick base-paired G-N1 sites, and similarly for all U-N3, the CSA tensor values are obtained from an iterative non-linear fitting procedure that restrains the δ33 component of the CSA tensor to be orthogonal to the base plane. Excellent agreement is observed between measured and predicted Δδ values, not only when these Δδ values are included in the CSA optimization procedure (filled symbols in Figure 2), but also when evaluating the agreement in a cross-validated manner (open symbols). CSA tensor parameters summarized in Table 1 show good agreement with solid state data regarding the magnitudes of the tensors, but differ considerably in asymmetry (Supporting Info Table S3).12,13 As predicted by quantum chemical calculations, for a traceless shift tensor, hydrogen bonding strongly increases the difference between δ22 and δ33.11 For the shorter U-A hydrogen bonds, |δ33| becomes larger than |δ11|. The sign of the angle between δ22 and the G:N1-H bond is the same as previously calculated,11 but its magnitude is ca 6° smaller. The quality of the fit between observed and predicted Δδ values is limited both by structural noise, and by the assumption that all tensors are the same for a given nucleotide type. In this respect it is interesting to note that if the analysis is carried out using a high quality homology model for tRNAVal (used as the starting model for RDC refinement)20, the prediction becomes 35% worse for G and 83% worse for U. If the experimental Δδ values and the tRNAVal model are assumed to be error free, scatter seen in Figure 2 can be attributed to random variations between nucleotides, σ(δ), in the CSA tensor. Variations in H-bond length are expected to impact δ22 and δ33 in opposite directions, with a correlated small change in the orientation of these two principal axes and little effect on δ11.11 Using these assumptions, we require σ(δ33) = 8 ppm for U, 14 ppm for G, to reproduce the scatter seen in Figure 2. These σ(δ33) values correspond to upper limits for the rms variations in H-bond length of ±0.13 Å for U-A and ±0.29 Å for G-C.11
Figure 2.
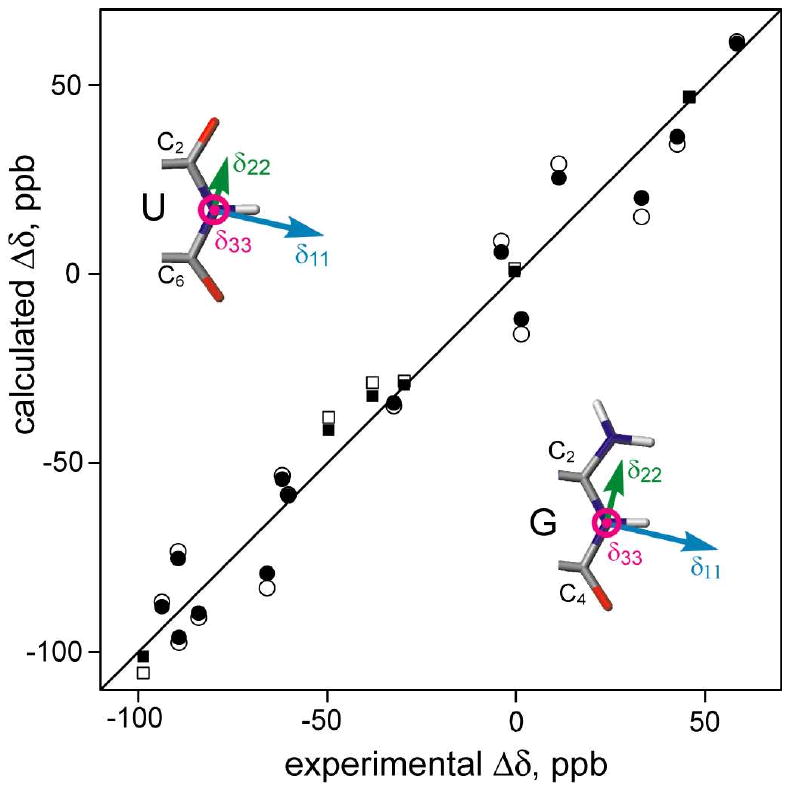
Correlations between the experimental and predicted Δδ(15N). Filled symbols correspond to Δδ values for a CSA tensor optimized with all nucleotides of a given type (circles for G:N1; squares for U:N3) included in the CSA fit. Open symbols represent jack-knifed cross-validation results, where each (predicted/observed) pair is shown for a CSA tensor obtained without using the experimental data of that nucleotide.
Table 1.
Average chemical shift tensor values for imino 15N in Watson-Crick base-paired nucleotides.a
| β,° | δ11, ppm | δ22, ppm | δ33, ppm | |
|---|---|---|---|---|
| G:N1 | 13.1±1.4 | 77.9±1.7 | -12.4±2.2 | -65.5±2.5 |
| U:N3 | 11.4±2.4 | 68.5±1.6 | 2.3±2.9 | -70.8±3.2 |
Using rNHeff =1.043 Å for G and rNHeff =1.057Å for U.
Supplementary Material
Acknowledgments
This work was supported in part by the Intramural Research Program of the NIDDK, NIH and NIH grant AI33098 (AP). We thank Werner Maas and Jochem Struppe (Bruker Instruments) for help with the MAS measurements.
Footnotes
Supporting Information Available: One table with measured Δδ values and relaxation rates; one table with temperature coefficients. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Dingley AJ, Grzesiek S. J Am Chem Soc. 1998;120:8293–8297. [Google Scholar]
- 2.Barfield M, Dingley AJ, Feigon J, Grzesiek S. J Am Chem Soc. 2001;123:4014–4022. doi: 10.1021/ja003781c. [DOI] [PubMed] [Google Scholar]
- 3.Tjandra N, Feller SE, Pastor RW, Bax A. J Am Chem Soc. 1995;117:12562–12566. [Google Scholar]
- 4.Dayie KT, Wagner G, Lefevre JF. Annu Rev Phys Chem. 1996;47:243–282. doi: 10.1146/annurev.physchem.47.1.243. [DOI] [PubMed] [Google Scholar]
- 5.Fushman D, Tjandra N, Cowburn D. J Am Chem Soc. 1998;120:10947–10952. [Google Scholar]
- 6.Sanders CR, Hare BJ, Howard KP, Prestegard JH. Prog Nucl Magn Reson Spectrosc. 1994;26:421–444. [Google Scholar]
- 7.Cornilescu G, Marquardt JL, Ottiger M, Bax A. J Am Chem Soc. 1998;120:6836–6837. [Google Scholar]
- 8.Hansen AL, Al-Hashimi HM. J Magn Reson. 2006;179:299–307. doi: 10.1016/j.jmr.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 9.Tate S, Shimahara H, Utsunomiya-Tate N. J Magn Reson. 2004;171:284–292. doi: 10.1016/j.jmr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 10.Grishaev A, Ying JF, Bax A. J Am Chem Soc. 2006;128:10010–10011. doi: 10.1021/ja0633058. [DOI] [PubMed] [Google Scholar]
- 11.Czernek J, Fiala R, Sklenar V. J Magn Reson. 2000;145:142–146. doi: 10.1006/jmre.2000.2091. [DOI] [PubMed] [Google Scholar]
- 12.Stueber D, Grant DM. J Am Chem Soc. 2002;124:10539–10551. doi: 10.1021/ja012485c. [DOI] [PubMed] [Google Scholar]
- 13.Hu JZ, Facelli JC, Alderman DW, Pugmire RJ, Grant DM. J Am Chem Soc. 1998;120:9863–9869. [Google Scholar]
- 14.Cornilescu G, Bax A. J Am Chem Soc. 2000;122:10143–10154. [Google Scholar]
- 15.Hansen MR, Mueller L, Pardi A. Nat Struct Biol. 1998;5:1065–1074. doi: 10.1038/4176. [DOI] [PubMed] [Google Scholar]
- 16.Grishaev A, Ying J, Canny MD, Pardi A, Bax A. J Biomol NMR. 2008;42:99–109. doi: 10.1007/s10858-008-9267-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dingley AJ, Masse JE, Peterson RD, Barfield M, Feigon J, Grzesiek S. J Am Chem Soc. 1999;121:6019–6027. [Google Scholar]
- 18.Case DA. J Biomol NMR. 1999;15:95–102. doi: 10.1023/a:1008349812613. [DOI] [PubMed] [Google Scholar]
- 19.Yao L, Voegeli B, Ying JF, Bax A. J Am Chem Soc. 2008;130:16518–16520. doi: 10.1021/ja805654f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grishaev A, Tugarinov V, Kay LE, Trewhella J, Bax A. J Biomol NMR. 2008;40:95–106. doi: 10.1007/s10858-007-9211-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.