

Abstract
Natural products containing carbon-phosphorus bonds (phosphonic and phosphinic acids) have found widespread use in medicine and agriculture. Recent years have seen a renewed interest in the biochemistry and biology of these compounds with the cloning of the biosynthetic gene clusters for several family members. This review discusses the commonalities and differences in the molecular logic that lies behind the biosynthesis of these compounds. The current knowledge regarding the metabolic pathways and enzymes involved in the production of a number of natural products, including the approved antibiotic fosfomycin, the widely used herbicide phosphinothricin, and the clinical candidate for treatment of malaria FR900098, is presented. Many of the enzymes involved in the biosynthesis of these compounds catalyze chemically and biologically unprecedented transformations and a wealth of new biochemistry has been revealed through their study. These studies have also suggested new strategies for natural product discovery.
Keywords: antibiotic, fosfomycin, phosphinothricin, fosmidomycin, FR900098, phosphonates, bialaphos
Introduction
Phosphonic and phosphinic acids represent an underexploited group of bioactive compounds with great promise for the treatment of human disease. These molecules, generically termed C-P compounds, are similar to phosphate esters and anhydrides, but contain carbon-phosphorus bonds (C-P in phosphonates and C-P-C in phosphinates) in place of one or more oxygen-phosphorus bonds. They are nearly isosteric to the labile phosphate esters, but are quite stable and can withstand harsh chemical treatments, such as boiling in strong acid or base (1). These unique structural and chemical features lend useful biological properties to many phosphonic and phosphinic acids, a feature that has not gone unnoticed. The past few decades have seen extensive use of C-P compounds in medicine and agriculture and thousands of papers describe their use (2). Prominent man-made examples include glyphosate, one of the most widely applied herbicides in the world, and alendronate, a commonly prescribed treatment for osteoporosis. Naturally produced phosphonates were first identified in 1959 opening a new chapter in the biology of phosphorus (3).
While humans have only recently begun to appreciate the biological potential of C-P compounds, Nature has long recognized their value as exemplified by the biosynthesis of a wide variety of phosphonic and phosphinic acids by living organisms. These include phosphonylated macromolecules such as lipids, exopolysaccharides and glycoproteins and a fascinating array of small bioactive molecules, which are the primary focus of this review. In some organisms these compounds are the dominant phosphorus-containing molecules in the cell. For example, eggs of the freshwater snail Helisoma contain 95% of their phosphorus in the form of 2-aminoethylphosphonate-modified phosphonoglycans (4), whereas the sea amenaea Tealia possesses up to 50% of its phosphorus in a variety of phosphonolipids, phosphonoglycans and phosphonoglycoproteins (5). Other organisms, such as the protist Tetrahymena, have lower overall levels of phosphonate, but still synthesize as much as 30% of their membrane lipids in the form of phosphonolipids (6). The prevalence of C-P compounds in nature is perhaps best exemplified by the recent discovery that as much as 20-30% of the available phosphorus in the world's oceans is comprised of phosphonic acids (7). Given that phosphorus is often a limiting nutrient, this fact indicates that C-P compounds play an important, perhaps critical role, in the global environment.
A particularly important group of phosphonate natural products includes more than a dozen natural products with potent bioactive properties, Figure 1. These include antibiotics such as fosfomycin, dehydrophos and plumbemycin; herbicides such as phosphinothricin tripeptide (PTT) and phosphonothrixin; antimalarial compounds such as FR900098 and fosmidomycin; and the antihypertensive peptides K4 and K-26. Members of the actinobacteria produce the majority of these compounds, but many other microbes including Bacillus sp., Pseudomonas sp. and some filamentous fungi also synthesize C-P compounds (8).
Figure 1.
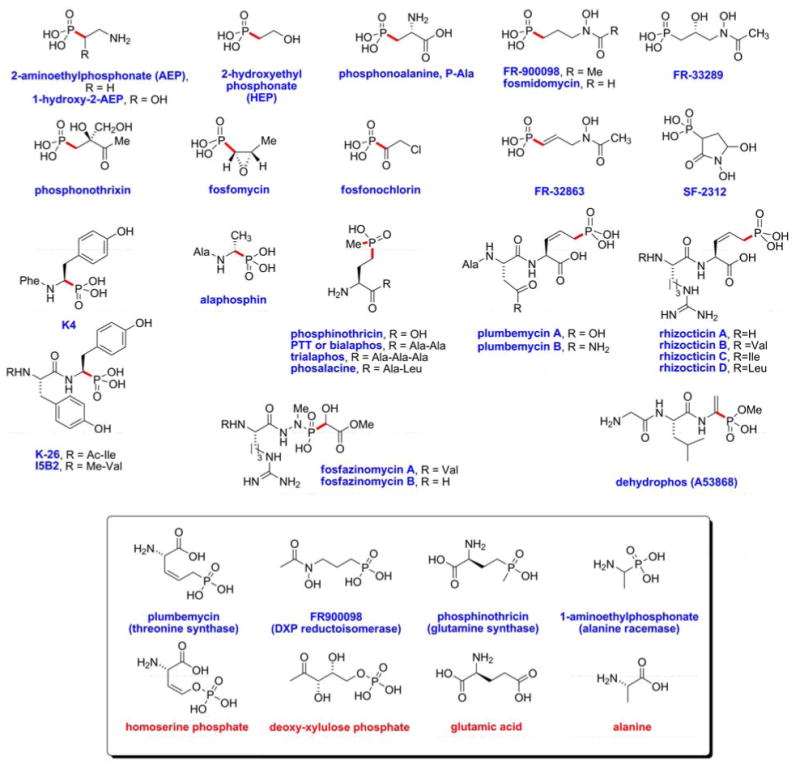
Structures of naturally occurring small molecule phosphonates and phosphonates. The P-C bonds are highlighted in red. The inset shows for a representative sampling the resemblance between the phosphonate/phosphonate and the substrate of the enzyme they target.
The bioactivity of both manmade and naturally produced C-P compounds stems from their structural similarity to analogous phosphate-esters and carboxylic acids, competing with these analogs for binding to enzyme active sites (Figure 1, inset). Because the affinity of C-P compounds is often high, many phosphonates and phosphinates act as potent competitive inhibitors. Moreover, some C-P compounds such as fosfomycin have functional groups that react with the enzyme causing irreversible inhibition (9). Given the ubiquitous role of phosphate-esters and carboxylic acids in biology the number of potential targets for phosphonate and phosphinate inhibitors is very large. Further, numerous regulatory events are controlled by protein phosphorylation (phosphate-esters) and proteolysis (carboxylate chemistry) and these processes are also subject to interference by C-P inhibitors.
An area of particular interest, which has revealed a wealth of novel biochemistry, is the molecular and biochemical characterization of phosphonate and phosphinate metabolism. In the fifty years since the discovery of C-P natural products our knowledge of their biosynthesis has greatly expanded. Although completely characterized biosynthetic pathways are rare, we are beginning to understand the commonalities and differences involved in the synthesis of several prominent phosphonates and phosphinates. As might be expected from their unique structures, the biosynthetic pathways include a number of unusual and interesting reactions. In this review we will discuss the biosynthesis of several well-characterized bioactive C-P compounds, with particular emphasis on these unusual reactions. We will also cover the utility of these studies in natural product discovery and highlight remaining unanswered questions and future directions of the field.
C-P Containing Macromolecules: Phosphono- Lipids, Glycans and Proteins
Discovery, distribution and diversity of phosphonolipids
In 1959 Horiguchi and Kandastu reported the first observation of a naturally produced C-P compound, 2-aminoethylphosphonate (AEP, Figure 1), which was isolated from acid hydrolyzed extracts of protozoa found in the rumen of sheep (3). AEP was subsequently found in a variety of organisms and shown to be present as the headgroup of membrane lipids analogous to phosphatidylethanolamine. These early reports led to a wave of studies demonstrating the presence of phosphonolipids in a wide diversity of organisms (reviewed in (1, 10, 11)). Phosphonolipids are especially abundant in ciliated protozoa, coelenerates, gastropods and bivalves. They have also been observed in plants, bacteria and several vertebrates, including humans; however, in the latter cases these are almost certainly assimilated from dietary sources, rather than synthesized de novo. Nevertheless, this observation indicates that phosphonates play a role in the metabolism of these organisms as well. The phosphonolipids contain a variety of fatty acid side chains and different phosphonate headgroups, including choline analogs with one, two or three N-linked methyl groups on AEP. Phosphonolipids with either 2-hydroxyethylphosphonate (HEP, Figure 1) or 1-hydroxy-2-aminoethylphosphonate have also been identified. 2-Amino-3-phosphonopropionic acid, known as phosphonoalanine (P-Ala or PALA), was detected in acid-hydrolysed cell extracts of Tetrahymena, but it is unclear whether it is derived from a lipid or some other macromolecule (see below).
Other C-P containing macromolecules
Phosphonates have also been observed as a component of exopolysaccharides, which have been generically referred to as phosphonoglycans (reviewed in (1, 10)). Unfortunately, the detailed structure of these complex molecules has rarely been determined, leaving open many questions regarding the nature and role of phosphonate-modified carbohydrates. Most phosphonoglycans contain AEP, although N-methylated AEP derivatives and HEP have also been observed. Phosphonoglycans have been identified in protozoa, invertebrates, snails and bacteria. Phosphonoglycans from sea anemone and Tetrahymena do not react with reagents that target primary amines, suggesting that the linkage to the sugar chain was via the amine of AEP; however, it is also possible that the nitrogen of AEP is acetylated. In contrast, the capsular polysaccharide of B. fragilis is modified with AEP via an ester linkage of the phosphonate moiety to the sugar (12).
Both AEP and HEP containing phosphonoproteins have been characterized from a number of lower eukaryotes (10). In cases where it has been established, these are invariably glycoproteins carrying a phosphonoglycan. Thus, although the conclusion is not firmly established, direct phosphonate attachment to the peptide backbone seems not to occur. Finally, phosphonate lipopolysaccharides, with the phosphonate on either the lipid, or the glycan, or both, have been identified in some organisms.
The Biological Role of C-P containing Macromolecules
The function of phosphonate-containing macromolecules has not been well established in any organism. Based on their abundance and distribution they may play an important role in the organisms that produce them. It has been speculated that the non-hydrolyzable C-P bond enhances the stability of lipids, especially with respect to phospholipases (1, 10). Alternatively, the phosphonate moiety could alter the fluidity of membranes containing phosphonolipids or the structural properties of phosphonoglycans. Others have suggested that the molecules may play a role in cell-cell signaling as specific receptor molecules or as phosphorus storage molecules in P-limited environments. To our knowledge only one experimental study addresses these questions directly, albeit with inconclusive results. Purified AEP-containing polysaccharide (CPS B) from Bacteroides fragilis is known to promote abscess formation; however, deletion mutants lacking the ability to produce this phosphonoglycan are viable and retain virulence in a rodent model system (13). Thus, the function of these macromolecules remains enigmatic.
Biosynthesis of C-P macromolecules
The biosynthesis of phosphonolipids and other C-P containing macromolecules has been extensively reviewed elsewhere (10). With the possible exception of K-26 discussed below, all phosphonate/phosphonite biosynthetic pathways involve the same initial steps, which were first solved by analysis of AEP biosynthesis in Tetrahymena. Therefore, the biosynthesis of this common component of C-P containing macromolecules and small molecules will be discussed first.
2-Aminoethylphosphonate
Biosynthetic pathway
The biosynthesis of AEP from phosphoenolpyruvate (PEP) is the shortest known pathway for the construction of a natural phosphonate, requiring just three enzymes: PEP mutase, phosphonopyruvate decarboxylase, and AEP transaminase (Figure 2A). Although orthologs of all three proteins from various sources have been characterized in some detail, at present no studies have reported the characterization of a complete set of enzymes from one organism. Given the absence of AEP biosynthetic enzymes in humans and the noted importance of AEP for various pathogens, these enzymes could be attractive targets for inhibition.
Figure 2.
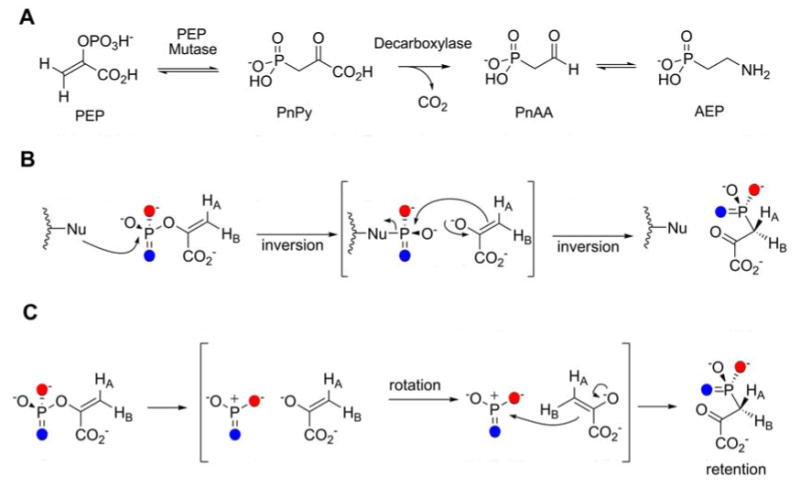
A. Biosynthetic pathway for AEP. B. Originally proposed mechanism for PEP mutase involving nucleophilic catalysis. C. Most recent proposed mechanism, involving a dissociative process with a metaphosphate intermediate.
Phosphoenolpyruvate mutase
In vivo labeling studies of the source of the phosphorus atom in AEP in phosphonolipids in Tetrahymena pyriformis suggested PEP as a potential precursor, but numerous early attempts to experimentally confirm the putative conversion of PEP to phosphonopyruvate (PnPy) failed (14). In 1988, the groups of Knowles, Seto, and Dunaway-Mariano first succeeded in isolating the enzyme responsible for this transformation (15-17) (Figure 2A). The thermodynamics of the reaction were shown to favor PnPy by more than 500-fold (16), providing an explanation for the failure of earlier investigations. The equilibrium was not anticipated to lie towards PnPy given the general notion of PEP as a high energy metabolite, but the inherently stronger P-O bond compared to a P-C bond accounts for much of the lower thermodynamic energy of PEP (16). For the enzyme from T. pyriformis the reaction occurs with retention of stereochemistry at phosphorus (18, 19), which suggested the intermediacy of a phosphoenzyme intermediate. This intermediate would be formed with inversion of configuration in the reaction of PEP with an enzyme residue (Figure 2B). Subsequent attack of the pyruvate enolate onto the phosphoenzyme intermediate, again with inversion of configuration, would account for the observed net retention. Indeed, when the first crystal structure of a PEP mutase in complex with the inhibitor oxalate was solved, Asp58 in the active site appeared to be in an orientation in which it could act as the phosphate carrier (20). However, attempts to trap the phosphoenzyme and/or enolate intermediates have not been successful (21), and in a later structure in complex with the product analog sulfopyruvate, Asp58 was located almost 6 Å from the sulfur atom of the inhibitor (22). Furthermore, although the Asp58Asn mutant displayed greatly reduced turnover, it was active and therefore inconsistent with a role of Asp58 as nucleophile. Other candidate nucleophiles in the active site have also been investigated by site-directed mutagenesis and collectively they have not identified such a residue (22, 23). Dunaway-Mariano and Herzberg and coworkers have therefore proposed that PEP mutase catalyzes its reaction through a dissociative process with metaphosphate as an intermediate (Figure 2C) (22), which would be the first example of such a mechanism. Interactions with active site residues are proposed to hold the metaphosphate in place as rotation occurs around the C1-C2 bond of the pyruvate enolate to allow attack of C3 onto the metaphosphate. This proposal would account for the observed retention of stereochemistry at phosphorus as well as the stereochemistry at C1 of PnPy (24).
Phosphonopyruvate decarboxylase
A corollary to the highly unfavorable equilibrium of the PEP mutase reaction is that the biosynthesis of phosphonates via this enzyme requires a highly exergonic ensuing step, which in the case of AEP consists of decarboxylation of PnPy to phosphonoacetaldehyde (PnAA) by phosphonopyruvate decarboxylase (14, 25, 26). The protein from T. pyriformis is membrane bound and has been difficult to characterize, but the enzyme from Bacteroides fragilis has proven to be more amenable to in vitro analysis (27). The orthologous enzyme involved in phosphinothricin biosynthesis in S. hygroscopicus has also been purified from the native host and characterized biochemically (28). Both proteins are members of the α-ketodecarboxylase family utilizing thiamin pyrophosphate and a divalent metal ion for catalysis.
AEP transaminase
At present, aminotransferases involved in the anabolism of AEP have not been characterized biochemically, but several enzymes catalyzing the same reaction in organisms that use AEP as a source of nutrients have been studied (29-31). These proteins from Salmonella enterica (31) and Pseudomonas aeruginosa (32) utilize pyridoxal 5′-diphosphate (PLP) as cofactor and pyruvate as ammonium acceptor. The mechanism of the transformation and structure of the enzyme from S. typhimurium (30, 31) is similar to that of the α-family of PLP-dependent enzymes (33).
Fosfomycin
(1R,2S)-Epoxypropylphosphonic acid known as fosfomycin (previously called phosphonomycin) was originally isolated by Merck researchers from Streptomyces fradiae, S. wedmorensis, and S. viridochromogenes (34) and was later shown to also be produced by Pseudomonas syringae (35), and Pseudomonas viridiflava (36). In the United States, fosfomycin tromethamine (tris(hydroxymethyl)aminomethane) is used under the name Monurol® and is an FDA-approved drug for the treatment of uncomplicated acute cystitis (urinary tract infections) (37-40). In other countries like Japan, Germany, Spain and France the compound is also used for gastrointestinal infections. The compound has broad-spectrum activity and is effective against both Gram-negative and Gram-positive bacterial infections in mammals (41). It has activity against methicillin- and vancomycin resistant Staphylococcus aureus (42, 43) and enterococci (44) and its successful clinical use beyond urinary tract and gastrointestinal infections was recently reviewed (45). Fosfomycin inactivates UDP-N-acetyl-glucosamine-3-O-enolpyruvyltransferase (MurA), an essential enzyme that catalyzes the first committed step in cell wall biosynthesis (46-48), by covalent alkylation of an active site cysteine (49, 50).
Biosynthetic pathway
Pioneering studies from the Seto laboratory on the biosynthesis of fosfomycin, using genetic techniques (51) complemented with in vivo experiments using isotopically labeled precursors provided evidence for the biosynthetic pathway shown in Figure 3 (8). The first two enzymatic steps are the same as in the biosynthesis of AEP, but the one-carbon homologation of PnAA is unprecedented in biology. On the basis of labeling studies, the methyl group in 2-hydroxypropyl phosphonic acid (HPP) was suggested to be derived from methylcobalamin (MeCbl) (8, 51-53). Supporting the involvement of MeCbl, a mutant S. wedmorensis strain that lacked the ability to synthesize B12 could only produce fosfomycin after addition of methylcobalamin (52). Initially a direct conversion of PnAA to HPP via nucleophilic addition of a methyl anion of methylcobalamin (MeCbl) to the aldehyde was proposed (Figure 3, dashed arrow) (8, 53, 54). This proposed role of MeCbl would be fundamentally different from known MeCbl dependent methyltransferases in which methylation takes place via nucleophilic attack by the substrate at the methyl group of MeCbl (i.e. involve a methyl cation equivalent) (55). More recently, analysis of the gene cluster from Streptomyces fradiae suggested an alternative pathway in which PnAA is first reduced to HEP by FomC (vide infra) (56). fomC was essential for heterologous production of fosfomycin in S. lividans, and a fomC disruption mutant was successfully complemented by the addition of HEP to the media, whereas the compound did not complement fosfomycin production in a fom3 disrupted clone (57). These findings suggested that HEP is a productive intermediate as the product of FomC and the substrate of Fom3. The revised pathway shown in Figure 3 is consistent with all previously reported studies and also explains why neither PnAA nor AEP could be converted to fosfomycin by S. lividans expressing fom3 and fom4, whereas HPP was converted (51).
Figure 3.
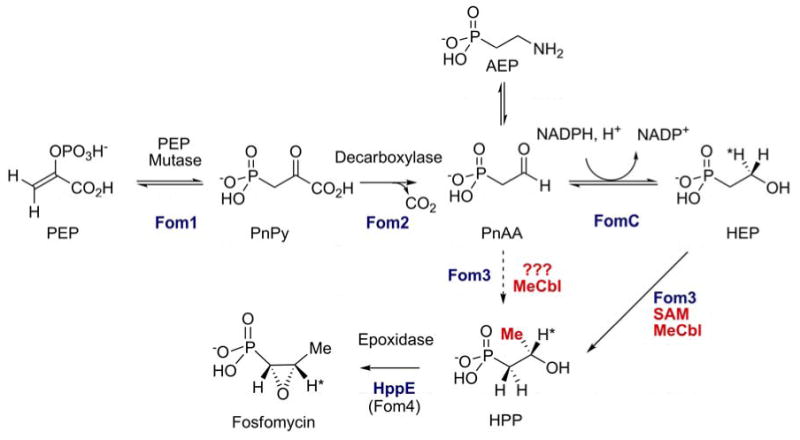
Originally proposed biosynthetic pathway for fosfomycin biosynthesis (dashed arrow) and more recent revised pathway involving FomC and 2-HEP.
Phosphonoacetaldehyde Reductase
The function of FomC from S. wedmorensis was recently confirmed in vitro with heterologously expressed enzyme (58). The protein belongs to the group III iron-dependent alcohol dehydrogenase family and uses NADPH as cofactor. A sequence alignment of FomC with structurally characterized family members revealed several conserved residues, including three ligands to the required Fe2+ (59). Typically, a His serves as a fourth ligand, however, a Gln was found in FomC. The orthologous PnAA reductases from the biosynthetic pathways of phosphinothricin and dehydrophos (vide infra) were also characterized. The former, PhpC, utilizes NADH and Zn2+ for catalysis whereas the latter, DhpG, utilizes NADPH and Fe2+ (58). Sequence comparison and a homology model generated for FomC suggested that phosphonate recognition in these proteins may be achieved by two conserved residues, Ser157 and Tyr261, that form hydrogen bonds to the oxygens of the phosphonate group in HEP.
2-Hydroxyethylphosphonate methyltransferase
The revised biosynthetic pathway shown in Figure 3 incorporated HEP as an additional intermediate, but did not resolve the enigma of how the methyl group present in the final product is introduced. The question has changed from how to add a methyl group to an aldehyde to how to insert a methyl group into a C-H bond α to a primary alcohol (Figure 3), a likewise unprecedented transformation in chemistry and biology. Sequence analysis of Fom3, the protein that putatively carries out this reaction, showed that it has two conserved domains (57). The N-terminal domain was identified as a B12-like binding domain, whereas the C-terminal domain shows homology to the radical-SAM protein family (60), containing three conserved Cys residues that serve as ligands to a [4Fe-4S] cluster.
With this information, a chemically feasible mechanism was proposed as depicted in Figure 4A (61) in which SAM and a reduced [4Fe-4S] cluster form a 5′-deoxyadenosyl radical, as in other radical-SAM proteins (62-64). Based on feeding studies with stereospecifically labeled HEP (65), the adenosyl radical would then abstract the pro-R hydrogen from the C2 position of HEP. Such hydrogen atom abstractions α to a hydroxyl group by a 5′-deoxyadenosyl radical are well precedented in adenosylcobalamin dependent enzymes (55). The resulting organic free radical can then react with the methyl group of MeCbl, yielding HPP and cob(II)alamin. This mechanism finds support in model studies demonstrating that organic radicals react with MeCbl to form methylated products (66, 67).
Figure 4.
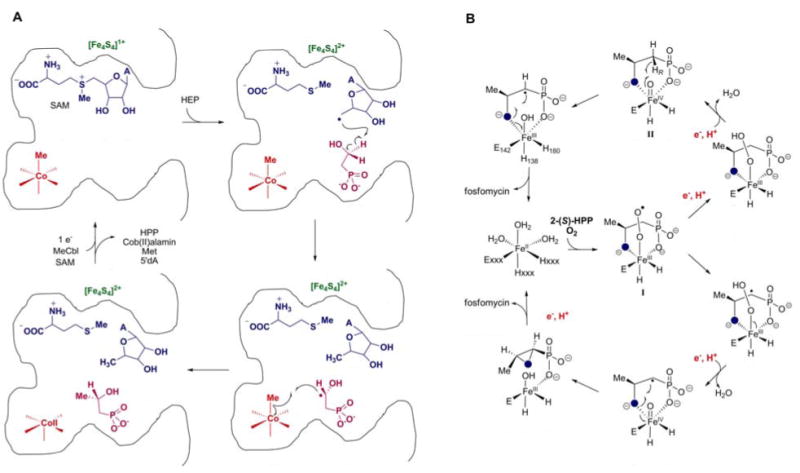
A. Proposed mechanism of methyl transfer to HEP catalyzed by Fom3. B. Proposed mechanisms for conversion of 2-HPP to fosfomycin by HppE (Fom4).
The proposed Fom3 mechanism is consistent with in vivo labeling studies by Hammerschmidt and coworkers. When (2,2-2H2)-HEP was fed to S. fradiae, 34% of the fosfomycin produced contained a deuterium label (54). Feeding (S)-(2-2H)-HEP or (R)-(2-2H)-HEP resulted in 32% labeled fosfomycin from the (S)-isomer whereas no label was retained from (R)-(2H)-HEP (65). Moreover, when HEP 18O-labeled on the hydroxyl oxygen was fed, 50% of the fosfomycin isolated was 18O-labeled (53). These studies revealed for the first time the unusual nature of the epoxidation step discussed in the next section. In the pathway shown in Figure 3, the label would never be in an exchangeable position explaining the high level of retention of the 18O-label with the observed label dilution probably due to endogenously biosynthesized HEP. Determing whether this proposed mechanism to achieve methylation of non-activated carbon centers through stoichiometric use of both SAM and MeCbl is correct will require in vitro reconstitution of enzyme activity.
2-Hydroxypropylphosphonate epoxidase
The last enzyme involved in the biosynthesis of fosfomycin catalyzes another highly unusual reaction, the conversion of (S)-2-hydroxypropylphosphonate (HPP) to fosfomycin. Initially termed Fom4, the protein has been renamed HPP epoxidase (HppE). In vivo labeling studies established that the epoxide oxygen is not derived from molecular oxygen (68). Conversely, HPP with an 18O-labeled hydroxyl group was converted by S. fradiae into 18O-labeled fosfomycin (69). Hence, these studies pointed at a unique mechanism of epoxide formation that involves dehydrogenation of a secondary alcohol. Cloning of the gene for the epoxidase in the Seto group (51) allowed Liu and his coworkers to heterologously express the protein from S. wedmorensis and demonstrate the dehydrogenative epoxidation activity in vitro (70). The transformation required Fe(II), O2, an electron carrier (either a general reductase or catalytic amounts of FMN) and NADH as reductant, but not α-ketoglutarate. At present, the physiological reductase has not been identified. Extensive mechanistic investigations using spectroscopic techniques, substrate analogs, and mutants show that the enzyme is a non-heme mononuclear iron dependent oxidase (71-74) that carries out epoxide formation by one of the two mechanisms shown in Figure 4B (74). The exact timing of entry of the two electrons required for the overall reaction distinguishes the two models in which either a Fe(III)-superoxide (I) or a Fe(IV)-oxo species (II) abstracts a hydrogen atom from C1 of the substrate. In both mechanisms, the substrate radical at C1 is believed to undergo a very interesting intramolecular rebound-like reaction. The overall stereochemistry of the epoxidation occurs with net inversion at C1 (54).
This mechanism is supported by a series of crystal structures of the enzyme from S. wedmorensis (75). The protein is a member of the cupin superfamily with a characteristic jelly roll β-barrel domain with the 2 His/1 Glu facial triad iron ligand set, and a structurally unique smaller domain that consists of 5 short α-helices. Two substrate binding modes were observed in these structures, monodentate coordination of HPP through one of the phosphonate oxygens and a bidentate interaction involving both the phosphonate and hydroxyl groups. The latter binding mode induces a conformational change that effectively closes off the active site and may serve to protect reactive intermediates. Support for bidentate HPP binding during catalysis was provided via EPR studies with 17O-labeled substrate and using NO as surrogate for oxygen (74). It has been noted (75) that this bidentate binding of HPP has similarities with the binding geometry of α-ketoglutarate in the cupin family, which is believed to activate the ferrous iron towards reaction with molecular oxygen. In these enzymes, the α-KG cosubstrate provides two electrons in addition to two electrons typically provided by the substrate allowing the net four-electron reduction of molecular oxygen. In HppE two exogenous electrons must be supplied for the overall conversion.
Phosphinothricin
Discovery and Biological Activity of Phosphinothricin-Containing Peptides
Phosphinothricin (PT), a non-proteinogenic amino acid found in a number of peptide antibiotics, is the only known phosphinic acid natural product, Figure 1. In the early 1970s independent groups in Germany and Japan discovered the compound as a component of a tripeptide antibiotic (PT-Ala-Ala) produced by Streptomyces viridochromogenes (designated phosphinothricin-tripeptide, PTT (76)) or Streptomyces hygroscopicus (designated bialaphos (77)). PT was later found as a component of phosalacine, a PT-Ala-Leu tripeptide produced by Kitasatospora phosalacina (78), and trialaphos (PT-Ala-Ala-Ala), a tetrapeptide produced by Streptomyces hygroscopicus KSB-1285 (79). PT is a structural analog of glutamate and a potent inhibitor of glutamine synthetase. As a free amino acid, PT has relatively poor antibiotic activity, probably due to inefficient transport. Many organisms, however, readily take up the peptide versions that are hydrolyzed by cytoplasmic peptidases releasing the active component, a strategy dubbed a “Trojan Horse” mechanism. Although PTT has excellent antibacterial activity in vitro when minimal media are used, it is a relatively poor therapeutic antibiotic because its activity can easily be counteracted by the presence of glutamine in host tissues. However, because glutamine synthetase plays an essential role in pH homeostasis in plants, PT is an outstanding herbicide and both the tripeptide and synthetic versions of the monomer are widely used in agriculture (80).
Biosynthetic pathway
Interest in the unique C-P-C bond motif and the biotechnological applications of PT has spurred a large number of studies on its biosynthesis (reviewed in (8, 80, 81)). The PTT biosynthetic pathway of S. hygroscopicus was largely solved by the Seto group using an elegant combination of in vivo labeling, in vitro biochemistry, genetics and gene cloning. Subsequent studies in the laboratories of Thompson and Wohlleben using S. viridochromogenes bolstered the understanding of the biosynthetic pathway, especially with regard to synthesis of the tripeptide. In combination, these studies led to a proposed pathway that is substantially similar to that shown in Figure 5, but involving fewer enzymes and intermediates (82).
Figure 5.
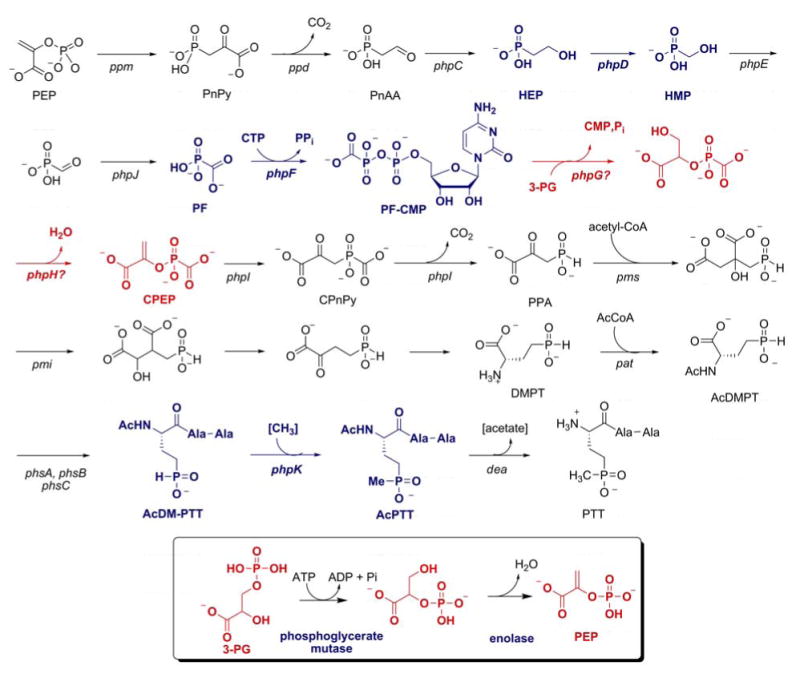
The most recent proposed biosynthetic pathway for the biosynthesis of phosphinothricin. Three highly unusual transformations are highlighted in blue, and the steps with similarities with glycolytic transformations are depicted in red. For comparison, the corresponding reactions in glycolysis are shown in the box.
The complete PTT gene cluster from S. viridochromogenes was recently sequenced and contains 24 genes in a contiguous 33 kbp segment of the chromosome (83, 84). The complete S. hygroscopicus gene cluster has also recently been sequenced and is nearly identical (Metcalf and Blodgett, unpublished data). Although the majority of the genes had previously been identified, three new genes designated phpC, phpD and phpE, whose functions are discussed below, were identified. No other unique genes are required for synthesis of PTT as shown by the ability of the cloned gene cluster to confer antibiotic production on heterologous hosts (84). The availability of these genes allowed genetic and biochemical experiments that led to the revised pathway presented in Figure 5, which includes several unprecendented biochemical transformations (82). The revised pathway involves synthesis of HEP via PEP mutase, PnPy decarboxylase and PnAA reductase using genes and enzymes that are homologous to those discussed in the preceding sections. HEP is then converted to phosphinoalanine (PPA) by a series of unprecedented reactions, some of which are analogous to those of the Embden-Meyerhoff-Parnas pathway for glycolysis, Figure 5, inset.
2-Hydroxyethylphosphonate dioxygenase
The first unique enzyme in the phosphinothricin biosynthetic pathway converts HEP to hydroxymethylphosphonate (HMP), an unprecedented apparent excision of a methylene unit (Figure 5). The product of the phpD gene from S. viridochromogenes was heterologously expressed and purified and shown to carry out this reaction without any requirements for external reductants or cofactors. Thus, all four electrons required for reduction of O2 are provided by HEP. Experiments with isotopically labeled substrate showed that the excised carbon is converted to formate and that the hydrogens on C1 of HEP are retained in HMP (85). Experiments using 18O2 demonstrated that both HMP and formate contained label, but surprisingly, the label in HMP was substoichiometric suggesting an exchange process occurred during catalysis. Indeed, experiments performed in H218O also resulted in incorporation of 18O into HMP. Since both atoms of molecular oxygen end up in the products, the enzyme has been named 2-hydroxyethylphosphonate dioxygenase (HEPD) (85).
The overall structure of HEPD (85) consists of tandem repeats of a bi-domain architecture with each of the repeats consisting of an all α-helical domain linked to a β-barrel fold characteristic of the cupin superfamily (86). In spite of a lack of appreciable similarity in the primary sequence, each of the two repeats is structurally homologous to the HppE monomer discussed above (75). The β-barrel domain contains the metal ligands of the 2 His-1 Glu facial triad with the metal site occupied by a Cd2+ ion in the X-ray structure. The disposition of these ligands within the β-barrel in HEPD is unlike that in HppE as Glu176 is situated on a different β strand than the equivalent Glu142 of HppE (75), and the spacing between the first two metal ligands in HEPD (HX46E) is unique as these residues are closely spaced (HX1-4E/D) in other facial triad enzymes (75). As described for HPP in HppE, bidentate binding was observed for HEP, but ligand binding did not induce the large conformational change seen in HppE, possibly because of the crystallization conditions that required high concentrations of Cd2+.
While oxidative scission of carbon-carbon bonds is well documented, the proteins involved typically act on substrates that contain aromatic (87), alkene (88), or 1,2-dihydroxy functionalities (89). Such activating groups are not present in HEP. A working model has been proposed for the HEPD reaction that is based on the isotope labeling studies (85). Upon substrate binding to Fe(II), reaction with O2 would result in a Fe(III)-[O2-] intermediate (I, Figure 6). This species can abstract a hydrogen atom from C2 of HEP to generate intermediate II, similar to the mechanism proposed for isopenicillin N synthase (IPNS) and myo-inositol oxygenase (89, 90) and one of the two mechanisms presented in Figure 4B for HppE. Two different pathways have been suggested to complete catalysis and account for the labeling studies. The substrate radical may attack the hydroperoxide generating a ferryl intermediate (FeIV=O) and a hemiacetal (Figure 6). The latter can undergo a retro-Claisen type C-C bond scission with the incipient negative charge on C1 attacking the electrophilic ferryl, either in a concerted step as shown or stepwise with a carbanion intermediate stabilized by metal coordination. This type of C-C bond cleavage between sp3-hybridized carbons is unprecedented, but an Fe(IV)-oxo intermediate would account for the observed exchange with solvent. A second mechanism that explains the observed data involves conversion of intermediate II to the hydroperoxide III, which after a Criegee-type rearrangement would provide the formate ester of HMP. Hydrolysis of this ester would normally occur by attack at the carbonyl carbon, but such a mechanism would not account for the incorporation of oxygen derived from solvent into HMP. The solvent exchange can be explained if hydrolysis took place via attack at C1 as shown in Figure 6. Further studies will be required to distinguish between these or alternative mechanisms and to provide insights into the factors that result in epoxide formation by HppE and C-C bond cleavage by HEPD.
Figure 6.
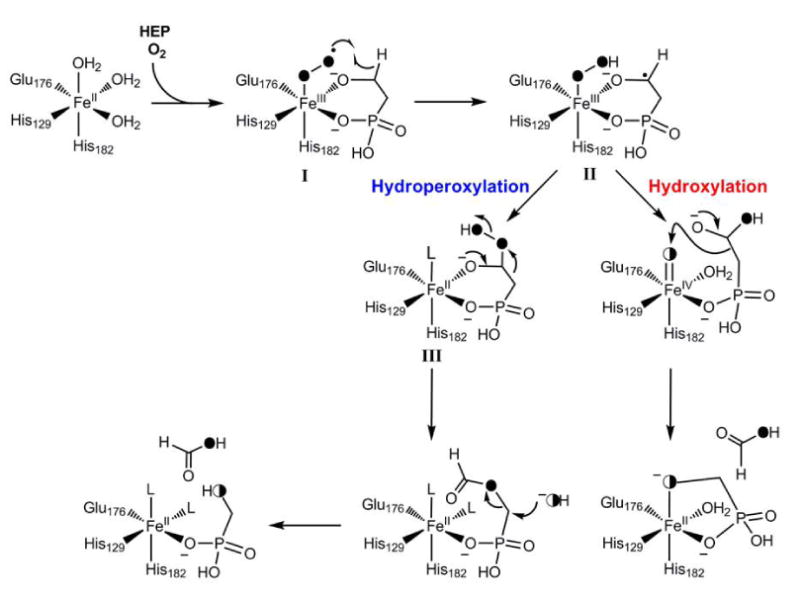
Proposed mechanism for the conversion of HEP to HMP by HEPD. Half-filled circles indicate oxygen atoms derived from O2 that have exchanged with solvent.
Synthesis of Carboxyphosphoenolpyruvate
Conversion of HMP to phosphonoformate (PF) likely occurs via sequential oxidation of HMP to phosphonoformaldehyde and then PF (Figure 5) (82). These reactions are believed to be catalyzed by the products of the phpE and phpJ genes, respectively, which are members of the NAD(P) dependent alcohol and aldehyde dehydrogenase families. Although neither enzyme has been characterized in vitro, genetic experiments provided in vivo support for this conclusion: mutants lacking phpE accumulate HMP, while phpJ mutants accumulate aminomethylphosphonate. The transamination of free aldehydes to the corresponding amine has been commonly observed in blocked mutants (82).
The synthesis of carboxyphosphoenolpyruvate (CPEP) was originally suggested by Hidaka et al to be achieved through a transesterification reaction of PEP using PF (91); however, current evidence suggests that this is unlikely. Mutant studies have implicated the phpH and phpG gene products in the synthesis of CPEP. These proteins are highly homologous to enolase and phosphoglycerate mutase, glycolytic enzymes that are unlikely to catalyze transesterification reactions (82, 92). An alternative model has been proposed based on the sequence similarity in which PhpG catalyzes a Pi/PF exchange reaction similar to that catalyzed by 1,3-diphosphoglycerate independent phosphoglycerate mutase (93) (Figure 5, inset). Subsequently, 2-phosphonoformyl-glycerate would be dehydrated by PhpH to produce CPEP, in a reaction analogous to that catalyzed by the homologous enolase. It is important to note that for this model to hold, PhpG would need to be initially activated by priming with a reactive form of PF. It has been proposed that this reactive form of PF is phosphonoformyl-CMP (PF-CMP, Figure 5), which is a phosphonate analog of CTP. Production of PF-CMP from PF and CTP catalyzed by the PhpF protein has been demonstrated in vitro lending credence to this model (82), which utilizes well-established biochemistry with a little twist.
Carboxyphosphoenolpyruvate mutase
At about the same time that PEP mutase activity was first demonstrated (vide supra), Seto and coworkers discovered the interesting related enzyme carboxyphosphoenolpyruvate mutase (CPEP mutase) (94, 95). The enzyme from Streptomyces hygroscopicus was purified to homogeneity from the producer strain and shown to catalyze the conversion of CPEP isolated from the fermentation broth of a mutant strain to phosphinopyruvate (PPA) (Figure 5). The gene encoding the protein was subsequently cloned and the enzyme expressed heterologously (96). In this study, CPEP was synthesized chemically and shown to be relatively stable. Like the reaction catalyzed by PEP mutase, the conversion of CPEP to PPA first involves a P-O to P-C bond rearrangement reaction that is thought to be energetically unfavorable. For pathways involving PEP mutase, the unfavorable equilibrium is offset by an ensuing energetically favorable or irreversible reaction such as decarboxylation as discussed for the biosynthesis of AEP. With CPEP mutase, the product of the rearrangement step, carboxyphosphono pyruvate (CPnPy, Figure 5), undergoes a decarboxylation without the need of another enzyme and the decarboxylation reaction cleaves a P-C rather than a C-C bond. To test whether the enzyme assists in this process, Knowles and coworkers synthesized CPnPy and showed it to be a stable molecule ruling out spontaneous decarboxylation (97). CPEP mutase did catalyze the decarboxylation of CPnPy to PPA, establishing chemical competence of the compound as an intermediate. However, the catalytic rate constants were much reduced compared to the overall reaction of CPEP to PPA showing the intermediate is not kinetically competent to be a free intermediate in the overall process. It was suggested that CPnPy is an intermediate in the enzyme catalyzed reaction but that it is not released into solution and that its on-rate is slow (97).
Nonribosomal peptide synthetases
The PTT gene cluster contains three genes encoding stand-alone non-ribosomal peptide synthetase modules. PhsA contains an adenylation domain (A-domain) and a peptidyl carrier protein (PCP) domain (also termed thiolation or T-domain) (98). PhsA was demonstrated to adenylate both N-acetylphosphinothricin (AcPT) and N-acetyl-demethylphosphinothricin (AcDMPT) with the latter displaying somewhat higher activity in pyrophosphate exchange assays (99); non-acylated analogs were not substrates. A mutant strain blocked in phsA accumulated AcDMPT suggesting this amino acid is loaded in the initiation step (100). PhsB and PhsC are larger polypeptides that also contain condensation domains, with PhsB possessing a second T-domain with a domain arrangement of T-C-A-T (101). Biochemical studies with all three enzymes isolated from S. viridochromogenes showed that PhsB and PhsC can adenylate alanine as well as other small amino acids and transfer the activated amino acid to the thiol of the phosphopanteteine in the peptide carrier domain (99). Although this finding was expected, it is very interesting that these two proteins that both activate Ala have relatively little sequence identity (38%), that the sequences of their A-domains predict they would activate Ser (PhsB) and Pro (PhsC), and that they cannot substitute for each other in deletion mutants (101). The mechanism of product release is still unclear. Neither PhsB nor PhsC contain a typical C-terminal thioesterase domain. Two other thioesterase genes, theA and theB, have been identified in the PTT gene cluster but disruption of neither gene abolished PTT production (102). Interestingly, a thioesterase motif (GXSXG) was found near the N-terminus of PhsA. Mutation of the Ser in this motif to Ala resulted in a mutant that was unable to restore PTT production in a phsA mutant strain. How this N-terminal thioesterase domain might release the final product is not known.
P-Methyltransferase
P-methyltransferase (Pmet or PhpK), is believed to catalyze the transfer of a methyl group to the phosphorus atom of the tripeptide AcDM-PTT (Figure 5). Similarly to the origin of the methyl group in fosfomycin discussed previously, the methyl donor has been suggested to be methylcobalamin (51, 103). Production of PTT in S. hygroscopicus is enhanced by the addition of cobalt salts to the growth medium (8, 104), and a blocked mutant grown in the absence of Co2+ accumulated DM-PT as well as the tripeptide DM-PTT (105, 106). Subsequent in vivo and whole cell lysate labeling studies using AcDM-PT or AcDM-PTT as substrate revealed a high level of incorporation of radiolabel from 14CH3-MeCbl into the methyl group of AcPT and AcPTT, while the free amino acid DM-PT was not methylated (8, 103, 105). The gene sequence of the P-methyltransferase was subsequently deduced from complementation studies (51). Seto and coworkers concluded that the P-methyltransferase is an unusual MeCbl dependent enzyme.
Alkylphosphinic acids such as AcDM-PTT can exist in the tetracoordinated or the tautomeric tricoordinated form (107) (inset Figure 7A). The equilibrium constant for these compounds usually strongly favors the tetracoordinated tautomer (108, 109). Nevertheless, phosphonous acids and phosphonites are known to carry out both electrophilic and nucleophilic reactions (109, 110). The most straightforward mechanism for the methyl transfer to phosphorus would therefore be attack of a tervalent form of AcDM-PTT onto the methyl group of MeCbl (Figure 7A). This would generate the Co(I) form of the cofactor, which could subsequently accept a methyl group from a cellular methyl donor.
Figure 7.
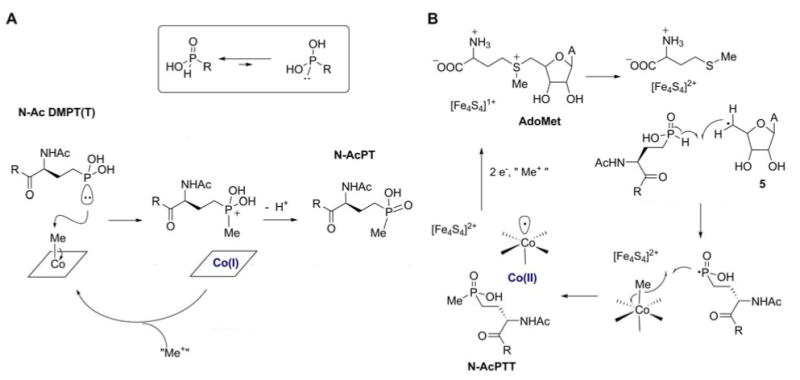
Proposed mechanisms for P-methyltransferase. A. Heterolytic pathway transferring a methyl group from MeCbl as a methyl cation equivalent. B. Homolytic pathway transferring the methyl group as a radical.
However, Pmet has sequence homology with Fom3 (51) with a B12 binding domain and a typical radical-SAM family Fe-S cluster (61). The requirement of these cofactors is not obvious for the mechanism in Figure 7A. Another mechanism can be drawn for Pmet that does require these cofactors and that has similarities with that proposed for Fom3 (Figure 4A). For Pmet, the 5′-deoxyadenosyl radical formed as described in Figure 4A could abstract a hydrogen atom from the phosphinate in AcDM-PTT breaking the relatively weak P-H bond (Figure 7B) (61). The resulting phosphorus based radical could then attack MeCbl to cleave the Co-C bond homolytically and provide the methylated product. Reduction of the oxidized FeS cluster and the cob(II)alamin formed and subsequent methylation of the resulting cob(I)alamin would reset the enzyme for another turnover.
PT acetyl-transferase
Antibiotic producing organisms typically require a mechanism of self-resistance. For PTT, this is conferred by the enzyme phosphinothricin acetyl-transferase, which N-acetylates either PT or demethylphosphinothricin using acetyl-CoA. The enzyme is encoded by the bar gene in S. hygroscopicus (111) and the pat gene in S.viridochromogenes (112), with 85% sequence identity. Interestingly, this resistance mechanism is also required for biosynthesis of PTT. Demethylphosphinothricin is acetylated prior to peptide bond formation and P-methylation.
The potent herbicidal activity of both PTT and PT have led to the development of the pat gene as a selectable marker for genetic engineering of plants and it has been widely used in this capacity. Further, the availability of crops carrying this resistance allele has led to a recent boom in the paired use of PT with recombinant plants in agriculture.
FR900098
Discovery and Biological Activity
FR900098 is one of a group of N-hydroxypropylphosphonic acids that also includes fosmidomycin, FR33289 and FR32863 (Figure 1), which are produced by strains of Streptomyces rubellomurinus and Streptomyces lavendulae (113-115). These compounds were discovered by researchers at Fujisawa Pharmaceutical Co. in the late 1970s and block the nonmevalonate pathway for isoprene biosynthesis. They inhibit the first committed step catalyzed by deoxyxylulose phosphate reductoisomerase (DXR) (116). Both FR900098 and fosmidomycin have very low toxicity in animals and humans and excellent antibacterial activity against most gram-negative bacteria (117-119), but neither compound has achieved widespread clinical use. Renewed interest in these antibiotics came with the genome sequence of Plasmodium falciparum (the causative agent of malaria), which revealed the unexpected presence of the target pathway in this eukaryote. Fosmidomycin and FR900098 are effective anti-malarial agents in animal models and have shown promise in early human trails, including against drug-resistant strains (120-122).
The FR900098 gene cluster and biosynthetic pathway
The FR900098 biosynthetic gene cluster was identified by screening a large insert fosmid library from S. rubellomurinus with degenerate PCR primers designed to amplify PEP mutase-encoding (ppm) genes (123). Further screening of ppm-positive clones led to the identification of a clone that conferred production of FR900098 to the heterologous host Streptomyces lividans and, therefore, contains all of the unique genes needed for synthesis of the antibiotic. Sequence analysis of the clone revealed the presence of genes encoding a putative PEP mutase and a number of genes related to those encoding enzymes of the TCA cycle. The predicted functions of these genes, in combination with in vitro biochemistry and mass spectrometric identification of intermediates provide strong support for the proposed biosynthetic pathway shown in Figure 8.
Figure 8.
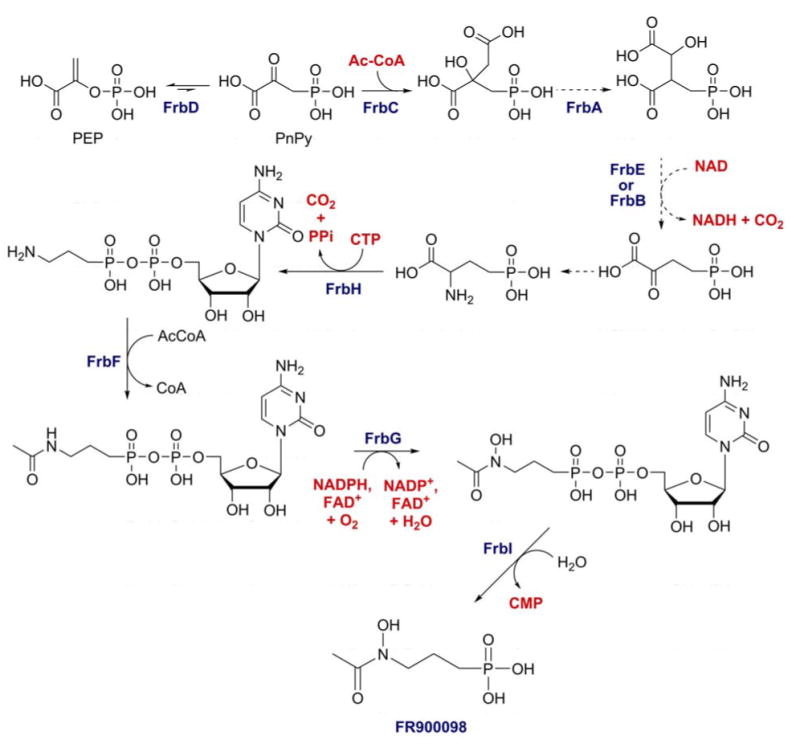
Proposed biosynthetic pathway of FR900098 based on gene homologies and in vitro biochemistry.
Unlike other phosphonate biosynthetic pathways, PnPy decarboxylase does not provide the thermodynamic driving force needed to pull the unfavorable PEP mutase reaction. Instead, a homolog of homocitrate synthase, FrbC, catalyzes the exergonic condensation of acetyl-CoA and phosphonopyruvate to form 2-phosphonomethylmalate. This activity has been verified in vivo using synthetic PnPy as substrate, and also in a coupled reaction with the S. rubellomurinus PEP mutase (FrbD) using PEP as the starting material (123). Subsequent steps similar to those found in the TCA cycle are predicted to convert this intermediate into 2-oxo-4-phosphonobutyrate, an analog of 2-oxo-glutarate, although this has yet to be demonstrated. An unknown host encoded transaminase is thought to convert this intermediate to 2-amino-4-phosphonobutyrate. FrbH is a two-domain protein containing discrete nucleotidyl-transferase and PLP-dependent decarboxylase/aminotranserase domains that was predicted to convert 2-amino-4-phosphonobutyrate into 3-aminopropylphosphonate. In vitro decarboxylation activity of FrbH requires CTP and produces 5′-CMP-3-aminopropylphosphonate (H. Zhao, personal communication). The biological rationale for this nucleotide modification is unclear; however, the CMP-modified product is competent as a substrate for in vitro acetylation with FrbF and N-hydroxylation by FrbG, whereas free 3-aminopropylphosphonate is not. Removal of the CMP is catalyzed by FrbI in vitro, although this gene is not required in vivo, suggesting that other cellular phosphodiesterases can fulfill this function as well.
Immunity
Interestingly, the FR900098 gene cluster also includes a gene (dxrB) that encodes a homolog of DXR, the target of the antibiotic. The dxrB gene was proposed to encode an FR900098-insensitive allele of DXR, and thus to be involved in self-resistance to the antibiotic (123). This idea has yet to be tested experimentally, but if true, the structure of this protein may lend insight into the nature of the interaction of the antibiotic and the enzyme allowing design of more effective derivatives.
Dehydrophos
Structure reassignment
Scientists at Eli Lilly first isolated a phosphonate from Streptomyces luridus and named the compound A53868. It has broad spectrum antibiotic activity, acting on both Gram-positive and Gram-negative bacteria (124). A structure was reported, but recent synthesis of this structure showed that it could not be correct since it displayed different 1H and 31P NMR spectra than the compound isolated from the producing strain (125). Subsequent spectroscopic studies on the compound purified from bacteria grown on various isotopically labeled media resulted in the structure shown in Figure 1; this proposed structure was verified by synthesis (125). Interestingly, the revised structure contains a phosphonate analog of dehydroalanine, which is found in many natural products such as the lantibiotics (126), microcystins (127), and thiostrepton (128). On the basis of the revised structure the compound was therefore renamed dehydrophos. The reassignment of the structure raises interesting questions with respect to its mode of action. As mentioned for PTT, the peptidic phosphonate antibiotics promote uptake by the target organism(s). Upon translocation, these peptides are hydrolyzed to release the active phosphonate-containing amino acids (129-133). Hydrolysis of the C-terminal peptide linkage of dehydrophos would result in an enamine that is expected to hydrolyze to methyl acetylphosphonate, which is a structural analog of pyruvate and could inhibit pyruvate utilizing enzymes. On the other hand, the reactive vinyl phosphonate moiety in dehydrophos may inhibit a cellular process directly, analogous to the activity of the phosphonotripeptide K-26 (vide infra) that inhibits angiotensin converting enzyme without the requirement of hydrolysis (134). Future studies should shed light onto these questions.
Biosynthetic gene cluster sequence
The dehydrophos biosynthetic gene cluster has been cloned and sequenced using a strategy similar to that described above for PTT and FR900098 (Eliot and Metcalf, unpublished data). The minimal gene cluster, as defined by the smallest fragment capable of conferring dehydrophos production upon heterologous hosts, is comprised of twenty genes, including ones whose products are highly homologous to PEP mutase, PnPy decarboxylase and PnAA reductase. Thus, like PTT and fosfomycin, it appears that this antibiotic is synthesized through HEP. Interestingly, the gene cluster does not contain canonical NRPS proteins nor any other proteins with homology to enzymes known to be involved in peptide synthesis. This suggests that a novel mechanism for peptide antibiotic synthesis is used.
K-26
The tripeptide K-26 was isolated from Astrosporangium hypotensionis in a screen for inhibitors of angiotensin converting enzyme (ACE). The compound is a potent inhibitor and shows promise in the treatment of high blood pressure (134). K-26 contains a phosphonate analog of tyrosine as its C-terminal residue that is shared in several analogs produced by Actinomadura strains (135, 136) (Figure 1). At present, the biosynthesis of (R)-1-amino-2-(4-hydroxyphenyl)ethylphosphonic acid (AHEP) is not well understood and its gene cluster has not yet been located. Feeding studies with isotopically labeled tyrosines provided a labeling pattern suggesting that unlike the other phosphonates discussed in this chapter, K-26 may not be biosynthesized using a PEP mutase reaction to install the C-P bond. Instead, AHEP appears to be derived from Tyr (137). Both the nitrogen of Tyr as well as the hydrogens on the aromatic ring and β-carbon were incorporated into the AHEP moiety of K-26 isolated from the producing strain. Furthermore, synthetic racemic 1,2-13C2-AHEP was incorporated with high efficiency into K-26 suggesting it is a discrete precursor. On the other hand, isotopically labeled tyramine, the decarboxylation product of tyrosine, was not incorporated (138). Future studies may reveal the molecular logic of P-C bond formation in this interesting compound.
Conclusions and Outlook
Metabolic diversity of PEPM initiated pathways
Examination of the phosphonate and phosphinate biosynthetic pathways described above reveals a number of common themes, Figure 9. First, due to the unfavorable thermodynamics of C-P bond formation, a favorable reaction is required in subsequent steps. This driving force can be supplied by decarboxylation, as in the CPEP mutase and PnPy decarboxylase reactions, or hydrolysis of acetyl-CoA as in the FrbD-catalyzed step of the FR900098 pathway. This energetic requirement places constraints on the evolution of potential C-P producing pathways and leaves unanswered questions regarding the synthesis of some known C-P natural products. For example, it has been suggested that phosphonoalanine is made by transamination of phosphonopyruvate (139); however, the freely reversible nature of this reaction suggests that net synthesis of C-P bonds via this pathway would be problematic unless transamination is followed by another exergonic reaction. Whether additional driving reactions exist is an open question that is relevant to the potential chemical structures of as yet unknown C-P compounds.
Figure 9.
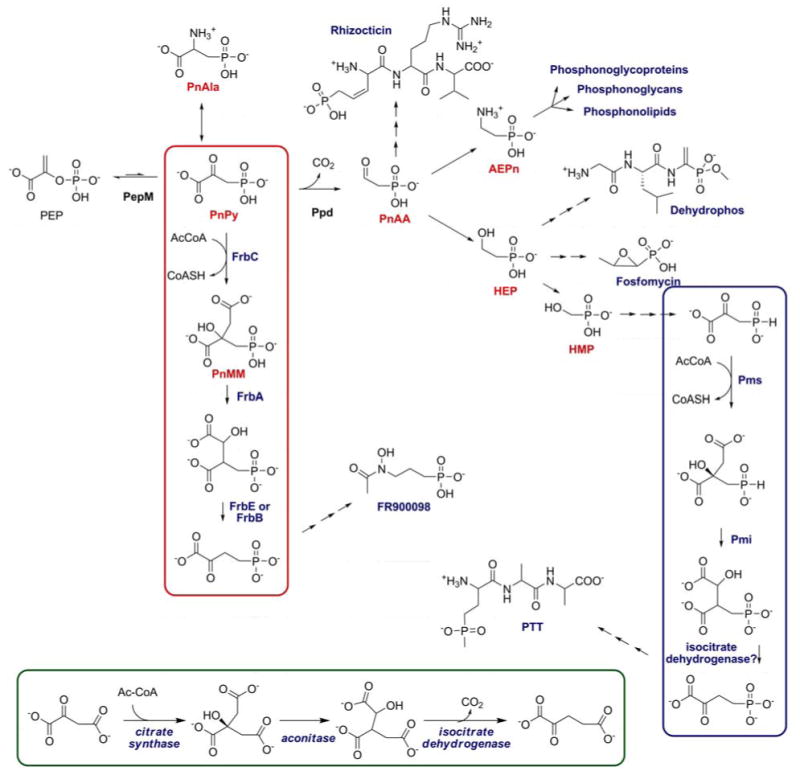
Overview of currently known biosynthetic pathways of phosphonates and phosphonates starting from PEP. The steps resembling TCA cycle reactions for phosphonates (red) and phosphinates (blue) are compared with the reactions in the TCA cycle (inset).
A second common theme is the apparent borrowing of reactions from central metabolic pathways. These include the glycolysis-like reactions of the PTT pathway described above and two distinct, but similar, borrowings from the TCA cycle that occur in the PTT and FR900098 pathways (boxed insets, Figure 9). Accordingly, these pathways include steps for both phosphonates and phosphinates that are analogous to those catalyzed by citrate synthase, aconitase and isocitrate dehydrogenase. Given the known molecular mimicry of C-P compounds, this is perhaps not surprising. The similarity of these molecules to the analogous carboxylates allows them to be substrates for the TCA cycle enzymes. In some cases, this has led to the evolution of dedicated enzymes for catalyzing these reactions (FrbA, FrbB, FrbD, FrbE, Pmi, and Pms), but in other cases it is likely that the C-P molecules are sufficiently similar that normal cellular enzyme catalyze the reaction (140-142). A similar argument involving phosphate ester mimicry can be made for the glycolytic pathway discussed above. The ubiquitous pathways for carboxylate and phosphate ester synthesis in biology therefore provide a rich source for the evolution of pathways for the synthesis of novel C-P natural products. In this regard, it is tempting to speculate that nature produces a large number of phosphonate inhibitors simply by evolution from the target pathways. It seems very likely that additional examples will emerge with continued study of these interesting biosynthetic pathways.
Genome mining of PEPM biosynthetic gene clusters
Finally, the PEPM reaction has potential utility for discovery of C-P biosynthetic gene clusters. As noted above, the homology of known PEP mutases allows design of degenerate PCR primers that can be used to identify ppm-containing clones from genomic libraries. Coupled with the fact that most antibiotic producers cluster the biosynthetic genes, this PCR assay has allowed the cloning of the genes needed to make PTT, fosfomycin and FR900098 (56, 84, 123). Moreover, we have recently cloned and sequenced the genes needed for production of dehydrophos, fosmidomycin, SF2312, rhizocticin and plumbemycin via this technique (unpublished data of W.W. Metcalf, A.C. Eliot, R. Woodyer, T. Johannes and H. Zhao, University of Illinois). Only the K26 genes were not identified by this means; however, given the recent data suggesting that PEPM is not involved in this pathway (137), this is not surprising. Of particular interest for the discovery of new antibiotics is the observation that ca. 5% of randomly isolated actinomycetes possess the ppm gene, as defined by the PCR assay (B. Circello and W.W. Metcalf, unpublished). This suggests that a wealth of new C-P natural products and metabolic pathways await discovery. Thus, the field of phosphonate and phosphinate biology continues to expand and ample research opportunities remain for interested scientists.
Acknowledgments
This work was supported by the National Institutes of Health (GM PO1 GM077596). We thank the coworkers in our laboratories as well as collaborators in the groups of Huimin Zhao, Neil Kelleher, and Satish Nair (University of Illinois) that have contributed to the work described in this review.
Key Terms/definitions List
- C-P compounds
natural products containing one or more carbon-phosphorus bonds
Abbreviations and Acronyms
- AEP
2-aminoethylphosphonate
- AcDM-PT
N-acetyl demethylphosphinothricin
- HEP
2-hydroxyethylphosphonate
- HMP
hydroxymethylphosphonate
- HPP
2-hydroxypropylphosphonate
- MeCbl
methylcobalamin
- PnAA
phosphono acetaldehyde
- PnPy
phosphono pyruvate
- PT
phosphinothricin
- PTT
phosphinothricin tripeptide
- PT
phosphinothricin
Footnotes
- Phosphonates and phosphinates function by mimicking phosphate esters or anhydrides or carboxylate groups in enzyme substrates. As such, a large number of enzymes can be potential targets of this class of compounds.
- The number of unprecedented reactions involved in the biosynthesis of fosfomycin, phosphinothricin, and FR900098 is an indication of the wealth of novel biochemistry used in the biosynthesis of this class of compounds.
- Phosphoenolpyruvate (PEP) mutase catalyzes the C-P bond-forming step in all naturally occurring phosphonates for which the gene clusters are currently known. Hence, degenerate primers for PEPM can be used for the discovery of new phosphonate encoding gene clusters and hence new natural products.
- Given the current commercial use of phosphonates and phosphinates in medicine and agriculture, discovery of new naturally occurring compounds beyond the twenty or so currently known structures may provide an important untapped source of new products for human use.
- Based on the large number of metabolic processes that can be targeted with phosphonates, numerous phosphonate and phosphinate containing natural products likely remain to be discovered. Future studies will confirm or refute this hypothesis.
- The fundamentally new radical-SAM strategy for methylation of unactivated carbon centers must be confirmed or disproven by in vitro studies.
- The molecular logic that dictates epoxidation by HppE and C-C bond cleavage in HEPD is not understood and requires further investigation. Fine-tuning the activities of these enzymes may result in useful catalysts for synthetic purposes.
- What is the cellular target of dehydrophos?
- What alternative strategies are used in Nature to install a C-P bond that do not rely on the PEP mutase-like reaction?
NOTE: this article has been accepted by Ann. Rev. Biochem. in a revised form. This is the submitted version of the manuscript
Literature Cited
- 1.Horiguchi M. Occurence, identification and properties of phosphonic and phosphinic acids. In: Hori T, Horiguchi T, Hayashi A, editors. Biochemistry of natural C-P compounds. Chapter 3. Shiga, Japan: Japanese Association for Research on the Biochemistry of C-P Compounds; 1984. pp. 24–52. [Google Scholar]
- 2.Kafarski P, Lejczak B. Aminophosphonic acids of potential medical importance. Curr Med Chem Anti-Cancer Agents. 2001;1:301–12. doi: 10.2174/1568011013354543. [DOI] [PubMed] [Google Scholar]
- 3.Horiguchi M, Kandatsu M. Isolation of 2-aminoethane phosphonic acid from rumen protozoa. Nature. 1959;184 12:901–2. doi: 10.1038/184901b0. The first observation of a C-P compound in a living system. [DOI] [PubMed] [Google Scholar]
- 4.Miceli MV, Henderson TO, Myers TC. 2-Aminoethylphosphonic Acid Metabolism During Embryonic Development of the Planorbid Snail Helisoma. Science. 1980;209:1245–47. doi: 10.1126/science.209.4462.1245. [DOI] [PubMed] [Google Scholar]
- 5.Quin LD. The presences of compounds with a carbon-phosphorus bond in some marine invertebrates. Biochemistry. 1965;4:324–30. [Google Scholar]
- 6.Kennedy KE, Thompson GA., Jr Phosphonolipids: localization in surface membranes of Tetrahymena. Science. 1970;168:989–91. doi: 10.1126/science.168.3934.989. [DOI] [PubMed] [Google Scholar]
- 7.Clark LL, Ingall ED, Benner R. Marine organic phosphorus cycling: Novel insights from nuclear magnetic resonance. Am J Sci. 1999;299:724–37. [Google Scholar]
- 8.Seto H, Kuzuyama T. Bioactive Natural Products with Carbon-Phosphorus Bonds and Their Biosynthesis. Nat Prod Rep. 1999;16:589–96. doi: 10.1039/a809398i. [DOI] [PubMed] [Google Scholar]
- 9.Wanke C, Amrhein N. Evidence that the reaction of the UDP-N-acetylglucosamine 1-carboxyvinyltransferase proceeds through the O-phosphothioketal of pyruvic acid bound to Cys115 of the enzyme. Eur J Biochem. 1993;218:861–70. doi: 10.1111/j.1432-1033.1993.tb18442.x. [DOI] [PubMed] [Google Scholar]
- 10.Hilderbrand RL, editor. The Role of Phosphonates in Living Systems. Boca Raton: CRC Press; 1983. p. 207. [Google Scholar]
- 11.Moschidis MC. Phosphonolipids. Prog Lipid Res. 1985;23:223–46. doi: 10.1016/0163-7827(84)90012-2. [DOI] [PubMed] [Google Scholar]
- 12.Baumann H, Tzianabos AO, Brisson JR, Kasper DL, Jennings HJ. Structural elucidation of two capsular polysaccharides from one strain of Bacteroides fragilis using high-resolution NMR spectroscopy. Biochemistry. 1992;31:4081–9. doi: 10.1021/bi00131a026. [DOI] [PubMed] [Google Scholar]
- 13.Coyne MJ, Kalka-Moll W, Tzianabos AO, Kasper DL, Comstock LE. Bacteroides fragilis NCTC9343 produces at least three distinct capsular polysaccharides: cloning, characterization, and reassignment of polysaccharide B and C biosynthesis loci. Infect Immun. 2000;68:6176–81. doi: 10.1128/iai.68.11.6176-6181.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Warren WA. Biosynthesis of phosphonic acids in Tetrahymena. Biochim Biophys Acta. 1968;156:340–6. doi: 10.1016/0304-4165(68)90263-8. [DOI] [PubMed] [Google Scholar]
- 15.Seidel HM, Freeman S, Seto H, Knowles JR. Phosphonate Biosynthesis: Isolation of the Enzyme Responsible for the Formation of a Carbon-Phosphorus Bond. Nature. 1988;335:457–58. doi: 10.1038/335457a0. First in vitro demonstration that the C-P bond in phosphonates is derived from PEP. [DOI] [PubMed] [Google Scholar]
- 16.Bowman E, McQueney M, Barry RJ, Dunaway-Mariano D. Catalysis and Thermodynamics of the Phosphoenolpyruvate/Phosphopyruvate Rearrangement. Entry into the Phosphonate Class of Naturally Occurring Organophosphorus Compounds. J Am Chem Soc. 1988;110:5575–76. [Google Scholar]
- 17.Hidaka T, Mori M, Imai S, Hara O, Nagaoka K, Seto H. Studies on the biosynthesis of bialaphos (SF-1293). 9. Biochemical mechanism of C-P bond formation in bialaphos: discovery of phosphoenolpyruvate phosphomutase which catalyzes the formation of phosphonopyruvate from phosphoenolpyruvate. J Antibiot. 1989;42:491–4. doi: 10.7164/antibiotics.42.491. [DOI] [PubMed] [Google Scholar]
- 18.Freeman S, Seidel HM, Schwalbe CH, Knowles JR. Phosphonate biosynthesis: the sterochemical course of phosphoenolpyruvate phosphomutase. J Am Chem Soc. 1989;111:9233–34. [Google Scholar]
- 19.McQueney MS, Lee SL, Swartz WH, Ammon HL, Mariano PS, Dunaway-Mariano D. Evidence for an intramolecular, stepwise reaction pathway for PEP phosphomutase catalyzed phosphorus-carbon bond formation. J Org Chem. 1991;56:7121–30. [Google Scholar]
- 20.Huang K, Li Z, Jia Y, Dunaway-Mariano D, Herzberg O. Helix swapping between two alpha/beta barrels: crystal structure of phosphoenolpyruvate mutase with bound Mg(2+)-oxalate. Structure Fold Des. 1999;7:539–48. doi: 10.1016/s0969-2126(99)80070-7. [DOI] [PubMed] [Google Scholar]
- 21.Kim J, Dunaway-Mariano D. Phosphoenolpyruvate mutase catalysis of phosphoryl transfer in phosphoenolpyruvate: kinetics and mechanism of phosphorus-carbon bond formation. Biochemistry. 1996;35:4628–35. doi: 10.1021/bi952944k. [DOI] [PubMed] [Google Scholar]
- 22.Liu S, Lu Z, Jia Y, Dunaway-Mariano D, Herzberg O. Dissociative phosphoryl transfer in PEP mutase catalysis: structure of the enzyme/sulfopyruvate complex and kinetic properties of mutants. Biochemistry. 2002;41:10270–6. doi: 10.1021/bi026024v. [DOI] [PubMed] [Google Scholar]
- 23.Jia Y, Lu Z, Huang K, Herzberg O, Dunaway-Mariano D. Insight into the mechanism of phosphoenolpyruvate mutase catalysis derived from site-directed mutagenesis studies of active site residues. Biochemistry. 1999;38:14165–73. doi: 10.1021/bi990771j. [DOI] [PubMed] [Google Scholar]
- 24.Hammerschmidt F, Kählig H. Incorporation of D-[1-2H1]glucose into 2-aminoethylphsophonic acid in Tetrehymena thermophila and into fosfomycin in Streptomyces fradiae. - The stereochemical course of a phosphoenolpyruvate mutase-catalyzed reaction. Liebigs Ann Chem. 1992:1201–03. [Google Scholar]
- 25.Horiguchi M. Biosynthesis of 2-aminoethylphosphonic acid in cell-free preparations from Tetrahymena. Biochim Biophys Acta. 1972;261:102–13. doi: 10.1016/0304-4165(72)90319-4. [DOI] [PubMed] [Google Scholar]
- 26.Barry RJ, Bowman E, McQueney M, Dunaway-Mariano D. Elucidation of the 2-aminoethylphosphonate biosynthetic pathway in Tetrahymena pyriformis. Biochem Biophys Res Commun. 1988;153:177–82. doi: 10.1016/s0006-291x(88)81205-1. [DOI] [PubMed] [Google Scholar]
- 27.Zhang G, Dai J, Lu Z, Dunaway-Mariano D. The phosphonopyruvate decarboxylase from Bacteroides fragilis. J Biol Chem. 2003;278:41302–8. doi: 10.1074/jbc.M305976200. [DOI] [PubMed] [Google Scholar]
- 28.Nakashita H, Watanabe K, Hara O, Hidaka T, Seto H. Studies on the biosynthesis of bialaphos. Biochemical mechanism of C-P bond formation: discovery of phosphonopyruvate decarboxylase which catalyzes the formation of phosphonoacetaldehyde from phosphonopyruvate. J Antibiot. 1997;50:212–9. [PubMed] [Google Scholar]
- 29.Lacoste AM, Dumora C, Balas L, Hammerschmidt F, Vercauteren J. Stereochemistry of the reaction catalysed by 2-aminoethylphosphonate aminotransferase. A 1H-NMR study. Eur J Biochem. 1993;215:841–4. doi: 10.1111/j.1432-1033.1993.tb18100.x. [DOI] [PubMed] [Google Scholar]
- 30.Chen CC, Zhang H, Kim AD, Howard A, Sheldrick GM, et al. Degradation pathway of the phosphonate ciliatine: crystal structure of 2-aminoethylphosphonate transaminase. Biochemistry. 2002;41:13162–9. doi: 10.1021/bi026231v. [DOI] [PubMed] [Google Scholar]
- 31.Kim AD, Baker AS, Dunaway-Mariano D, Metcalf WW, Wanner BL, Martin BM. The 2-aminoethylphosphonate-specific transaminase of the 2-aminoethylphosphonate degradation pathway. J Bacteriol. 2002;184:4134–40. doi: 10.1128/JB.184.15.4134-4140.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dumora C, Lacoste AM, Cassaigne A. Purification and properties of 2-aminoethylphosphonate:pyruvate aminotransferase from Pseudomonas aeruginosa. Eur J Biochem. 1983;133:119–25. doi: 10.1111/j.1432-1033.1983.tb07436.x. [DOI] [PubMed] [Google Scholar]
- 33.Alexander FW, Sandmeier E, Mehta PK, Christen P. Evolutionary relationships among pyridoxal-5′-phosphate-dependent enzymes. Regio-specific alpha, beta and gamma families. Eur J Biochem. 1994;219:953–60. doi: 10.1111/j.1432-1033.1994.tb18577.x. [DOI] [PubMed] [Google Scholar]
- 34.Hendlin D, Stapley EO, Jackson M, Wallick H, Miller AK, et al. Phosphonomycin, a new antibiotic produced by strains of streptomyces. Science. 1969;166:122–3. doi: 10.1126/science.166.3901.122. [DOI] [PubMed] [Google Scholar]
- 35.Shoji J, Kato T, Hinoo H, Hattori T, Hirooka K, et al. Production of fosfomycin (phosphonomycin) by Pseudomonas syringae. J Antibiot. 1986;39:1011–12. doi: 10.7164/antibiotics.39.1011. [DOI] [PubMed] [Google Scholar]
- 36.Katayama N, Tsubotani S, Nozaki Y, Harada S, Ono H. Fosfadecin and fosfocytocin, new nucleotide antibiotics produced by bacteria. J Antibiot. 1990;43:238–46. doi: 10.7164/antibiotics.43.238. [DOI] [PubMed] [Google Scholar]
- 37.Stein GE. Single-dose treatment of acute cystitis with fosfomycin tromethamine. Ann Pharmacother. 1998;32:215–9. doi: 10.1345/aph.17227. [DOI] [PubMed] [Google Scholar]
- 38.Bailey RR. Management of lower urinary tract infections. Drugs. 1993;45 3:139–44. doi: 10.2165/00003495-199300453-00023. [DOI] [PubMed] [Google Scholar]
- 39.Reeves DS. Treatment of bacteriuria in pregnancy with single dose fosfomycin trometamol: a review. Infection. 1992;20 4:S313–6. doi: 10.1007/BF01710022. [DOI] [PubMed] [Google Scholar]
- 40.Bailey RR. Brief overview of single-dose therapy for uncomplicated urinary tract infections. Chemotherapy. 1990;36 1:27–30. doi: 10.1159/000238812. [DOI] [PubMed] [Google Scholar]
- 41.Reeves DS. Fosfomycin trometamol. J Antimicrob Chemother. 1994;34:853–8. doi: 10.1093/jac/34.6.853. [DOI] [PubMed] [Google Scholar]
- 42.Nakazawa H, Kikuchi Y, Honda T, Isago T, Nozaki M. Enhancement of antimicrobial effects of various antibiotics against methicillin-resistant Staphylococcus aureus (MRSA) by combination with fosfomycin. J Infect Chemother. 2003;9:304–9. doi: 10.1007/s10156-003-0266-2. [DOI] [PubMed] [Google Scholar]
- 43.Cassone M, Campanile F, Pantosti A, Venditti M, Stefani S. Identification of a variant “Rome clone” of methicillin-resistant Staphylococcus aureus with decreased susceptibility to vancomycin, responsible for an outbreak in an intensive care unit. Microb Drug Resist. 2004;10:43–9. doi: 10.1089/107662904323047790. [DOI] [PubMed] [Google Scholar]
- 44.Allerberger F, Klare I. In-vitro activity of fosfomycin against vancomycin-resistant enterococci. J Antimicrob Chemother. 1999;43:211–7. doi: 10.1093/jac/43.2.211. [DOI] [PubMed] [Google Scholar]
- 45.Falagas ME, Giannopoulou KP, Kokolakis GN, Rafailidis PI. Fosfomycin: use beyond urinary tract and gastrointestinal infections. Clin Infect Dis. 2008;46:1069–77. doi: 10.1086/527442. [DOI] [PubMed] [Google Scholar]
- 46.Skarzynski T, Mistry A, Wonacott A, Hutchinson SE, Kelly VA, Duncan K. Structure of UDP-N-acetylglucosamine enolpyruvyl transferase, an enzyme essential for the synthesis of bacterial peptidoglycan, complexed with substrate UDP-N-acetylglucosamine and the drug fosfomycin. Structure. 1996;4:1465–74. doi: 10.1016/s0969-2126(96)00153-0. [DOI] [PubMed] [Google Scholar]
- 47.Schonbrunn E, Svergun DI, Amrhein N, Koch MH. Studies on the conformational changes in the bacterial cell wall biosynthetic enzyme UDP-N-acetylglucosamine enolpyruvyltransferase (MurA) Eur J Biochem. 1998;253:406–12. doi: 10.1046/j.1432-1327.1998.2530406.x. [DOI] [PubMed] [Google Scholar]
- 48.De Smet KA, Kempsell KE, Gallagher A, Duncan K, Young DB. Alteration of a single amino acid residue reverses fosfomycin resistance of recombinant MurA from Mycobacterium tuberculosis. Microbiology. 1999;145:3177–84. doi: 10.1099/00221287-145-11-3177. [DOI] [PubMed] [Google Scholar]
- 49.Marquardt JL, Brown ED, Lane WS, Haley TM, Ichikawa Y, et al. Kinetics, stoichiometry, and identification of the reactive thiolate in the inactivation of UDP-GlcNAc enolpyruvoyl transferase by the antibiotic fosfomycin. Biochemistry. 1994;33:10646–51. doi: 10.1021/bi00201a011. [DOI] [PubMed] [Google Scholar]
- 50.Kim DH, Lees WJ, Kempsell KE, Lane WS, Duncan K, Walsh CT. Characterization of a Cys115 to Asp substitution in the Escherichia coli cell wall biosynthetic enzyme UDP-GlcNAc enolpyruvyl transferase (MurA) that confers resistance to inactivation by the antibiotic fosfomycin. Biochemistry. 1996;35:4923–8. doi: 10.1021/bi952937w. [DOI] [PubMed] [Google Scholar]
- 51.Hidaka T, Goda M, Kuzuyama T, Takei N, Kidaka M, Seto H. Cloning and Nucleotide Sequence of Fosfomycin Biosynthetic Genes of Streptomyces wedmorensis. Mol Gen Genet. 1995;249:274–80. doi: 10.1007/BF00290527. [DOI] [PubMed] [Google Scholar]
- 52.Kuzuyama T, Hidaka T, Kamigiri K, Imai S, Seto H. Studies on the biosynthesis of fosfomycin. 4. The biosynthetic origin of the methyl group of fosfomycin. J Antibiot. 1992;45:1812–4. doi: 10.7164/antibiotics.45.1812. [DOI] [PubMed] [Google Scholar]
- 53.Hammerschmidt F. Incorporation of L-[Methyl-2H3]methionine and 2-[Hydroxy-18O]hydroxyethylphosphonic Acid into Fosfomycin in Streptomyces fradiae-An Unusual Methyl Transfer. Angew Chem Int Ed Engl. 1994;33:341–42. [Google Scholar]
- 54.Hammerschmidt F, Kählig H. Biosynthesis of Natural Products with a P-C Bond. 7. Synthesis of [1,1-2H2]-, [2,2-2H2]-, (R)- and (S)-[1-2H1](2-Hydroxyethyl)phosphonic Acid and (R,S)-[1-2H1](1,2-Dihydroxyethyl)phosphonic Acid and Incorporation Studies into Fosfomycin in Streptomyces fradiae. J Org Chem. 1991;56:2364–70. [Google Scholar]
- 55.Banerjee R, editor. The Chemistry and Biochemistry of B12. New York: Wiley; 1999. [Google Scholar]
- 56.Woodyer RD, Shao Z, Metcalf WM, Thomas PM, Kelleher NL, et al. Heterologous Production of Fosfomycin and Identification of the Minimal Biosynthetic Cluster. Chem Biol. 2006;13:1171–82. doi: 10.1016/j.chembiol.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 57.Woodyer RD, Li G, Zhao H, van der Donk WA. New Insight into the Biosynthesis of Fosfomycin: Discovery of the Missing Link Illuminates the Mechanism of Methyl Transfer. Chem Commun. 2007:359–61. doi: 10.1039/b614678c. [DOI] [PubMed] [Google Scholar]
- 58.Shao Z, Blodgett JA, Circello BT, Eliot AC, Woodyer R, et al. Biosynthesis of 2-hydroxyethylphosphonate, an unexpected intermediate common to multiple phosphonate biosynthetic pathways. J Biol Chem. 2008;283:23161–8. doi: 10.1074/jbc.M801788200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Montella C, Bellsolell L, Perez-Luque R, Badia J, Baldoma L, et al. Crystal structure of an iron-dependent group III dehydrogenase that interconverts L-lactaldehyde and L-1,2-propanediol in Escherichia coli. J Bacteriol. 2005;187:4957–66. doi: 10.1128/JB.187.14.4957-4966.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sofia HJ, Chen G, Hetzler BG, Reyes-Spindola JF, Miller NE. Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods. Nucleic Acids Res. 2001;29:1097–106. doi: 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van der Donk WA. Rings, Radicals, and Regeneration: the Early Years of a Bioorganic Laboratory. J Org Chem. 2006;71:9561–71. doi: 10.1021/jo0614240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Layer G, Heinz DW, Jahn D, Schubert WD. Structure and function of radical SAM enzymes. Curr Opin Chem Biol. 2004;8:468–76. doi: 10.1016/j.cbpa.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 63.Frey PA. Radical mechanisms of enzymatic catalysis. Annu Rev Biochem. 2001;70:121–48. doi: 10.1146/annurev.biochem.70.1.121. [DOI] [PubMed] [Google Scholar]
- 64.Frey PA, Magnusson OT. S-Adenosylmethionine: a wolf in sheep's clothing, or a rich man's adenosylcobalamin? Chem Rev. 2003;103:2129–48. doi: 10.1021/cr020422m. [DOI] [PubMed] [Google Scholar]
- 65.Hammerschmidt F. Biosynthesis of natural products with a P-C bond 9. Synthesis and incorporation of (S)-2-hydroxy-[2-2H-1]ethylphosphonic and (R)-2-hydroxy-[2-H-2]ethylphosphonic acid into fosfomycin by Streptomyces fradiae. Liebigs Ann Chem. 1992:553–57. [Google Scholar]
- 66.Mosimann H, Kräutler B. Methylcorrinoids methylate radicals - Their second biological mode of action? Angew Chem Int Ed. 2000;39:393–95. doi: 10.1002/(sici)1521-3773(20000117)39:2<393::aid-anie393>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 67.Glasenapp-Breiling M, Montforts FP. Unusual Alkylation Reactions in the Biosynthesis of Natural Products and Elucidation of Their Reaction Mechanisms. Angew Chem Int Ed Engl. 2000;39:721–23. [PubMed] [Google Scholar]
- 68.Hammerschmidt F, Bovermann G, Bayer K. Das Oxiran-Sauerstoff-Atom des Fosfomycins entstammt nicht dem Luft-Sauerstoff. Liebigs Ann Chem. 1990:1055–61. [Google Scholar]
- 69.Hammerschmidt F. Biosynthesis of Natural Products with a P-C Bond. Part 8: On the Origin of the Oxirane Oxygen Atom of Fosfomycin in Streptomyces fradiae. J Chem Soc Perkin Trans. 1991;1:1993–96. [Google Scholar]
- 70.Liu P, Murakami K, Seki T, He X, Yeung SM, et al. Protein purification and function assignment of the epoxidase catalyzing the formation of fosfomycin. J Am Chem Soc. 2001;123:4619–20. doi: 10.1021/ja004153y. First dehydrogenative epoxide forming reaction by a non-heme iron enzyme. [DOI] [PubMed] [Google Scholar]
- 71.Zhao Z, Liu P, Murakami K, Kuzuyama T, Seto H, Liu HW. Mechanistic Studies of HPP Epoxidase: Configuration of the Substrate Governs Its Enzymatic Fate. Angew Chem Int Ed Engl. 2002;41:4529–32. doi: 10.1002/1521-3773(20021202)41:23<4529::AID-ANIE4529>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 72.Liu P, Liu A, Yan F, Wolfe MD, Lipscomb JD, Liu HW. Biochemical and spectroscopic studies on (S)-2-hydroxypropylphosphonic acid epoxidase: a novel mononuclear non-heme iron enzyme. Biochemistry. 2003;42:11577–86. doi: 10.1021/bi030140w. [DOI] [PubMed] [Google Scholar]
- 73.Yan F, Li T, Lipscomb JD, Liu A, Liu HW. Site-directed mutagenesis and spectroscopic studies of the iron-binding site of (S)-2-hydroxypropylphosphonic acid epoxidase. Arch Biochem Biophys. 2005;442:82–91. doi: 10.1016/j.abb.2005.07.024. [DOI] [PubMed] [Google Scholar]
- 74.Yan F, Moon SJ, Liu P, Zhao Z, Lipscomb JD, et al. Determination of the substrate binding mode to the active site iron of (S)-2-hydroxypropylphosphonic acid epoxidase using 17O-enriched substrates and substrate analogues. Biochemistry. 2007;46:12628–38. doi: 10.1021/bi701370e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Higgins LJ, Yan F, Liu P, Liu HW, Drennan CL. Structural insight into antibiotic fosfomycin biosynthesis by a mononuclear iron enzyme. Nature. 2005;437:838–44. doi: 10.1038/nature03924. Structures illuminating the mechanism of dehydrogenative epoxide formation. [DOI] [PubMed] [Google Scholar]
- 76.Bayer H, Gugel KH, Hagele K, Hagenmaier H, Jessipow S, et al. Phosphinothricin und phosphinothricyl-alanyl-alanin. Helv Chim Acta. 1972;55 doi: 10.1002/hlca.19720550126. [DOI] [PubMed] [Google Scholar]
- 77.Ogawa H, Tsuruoka T, Inouye S, Niida T. Studies on a new antibiotic SF-1293. Sci Rep Meiji Seika Kaisha. 1973;13:42–48. [Google Scholar]
- 78.Omura S, Murata M, Hanaki H, Hinotozawa K, Oiwa R, Tanaka H. Phosalacine, a new herbicidal antibiotic containing phosphinothricin. Fermentation, isolation, biological activity and mechanism of action. J Antibiot. 1984;37:829–35. doi: 10.7164/antibiotics.37.829. [DOI] [PubMed] [Google Scholar]
- 79.Kato H, Nagayama K, Abe H, Kobayashi R, Ishihara E. Isolation, structure and biological activity of trialaphos. Agric Biol Chem. 1991;55:1133–34. [Google Scholar]
- 80.Thompson CJ, Seto H. Bialaphos. Biotechnology. 1995;28:197–222. doi: 10.1016/b978-0-7506-9095-9.50014-9. [DOI] [PubMed] [Google Scholar]
- 81.Thompson CJ, Seto H. Bialaphos. In: Vining LC, Stuttard C, editors. Genetics and Biochemistry of Antibiotic Production. Newton, MA: Butterworth-Heinemann; 1995. pp. 197–222. [Google Scholar]
- 82.Blodgett JA, Thomas PM, Li G, Velasquez JE, van der Donk WA, et al. Unusual transformations in the biosynthesis of the antibiotic phosphinothricin tripeptide. Nat Chem Biol. 2007;3:480–5. doi: 10.1038/nchembio.2007.9. Revision of the PTT pathway and evidence for PhpD and PhpF reactions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schwartz D, Berger S, Heinzelmann E, Muschko K, Welzel K, Wohlleben W. Biosynthetic gene cluster of the herbicide phosphinothricin tripeptide from Streptomyces viridochromogenes Tu494. Appl Environ Microbiol. 2004;70:7093–102. doi: 10.1128/AEM.70.12.7093-7102.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Blodgett JA, Zhang JK, Metcalf WW. Molecular cloning, sequence analysis, and heterologous expression of the phosphinothricin tripeptide biosynthetic gene cluster from Streptomyces viridochromogenes DSM 40736. Antimicrob Agents Chemother. 2005;49:230–40. doi: 10.1128/AAC.49.1.230-240.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cicchillo RM, Zhang H, Blodgett JAV, Whitteck JT, Li G, et al. An Unusual Carbon-Carbon Bond Cleavage Reaction in the Biosynthesis of the Antibiotic Phosphinothricin. submitted for publication. [Google Scholar]
- 86.Dunwell JM, Purvis A, Khuri S. Cupins: the most functionally diverse protein superfamily? Phytochemistry. 2004;65:7–17. doi: 10.1016/j.phytochem.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 87.Costas M, Mehn MP, Jensen MP, Que L., Jr Dioxygen activation at mononuclear nonheme iron active sites: enzymes, models, and intermediates. Chem Rev. 2004;104:939–86. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 88.Grogan G. Emergent mechanistic diversity of enzyme-catalysed beta-diketone cleavage. Biochem J. 2005;388:721–30. doi: 10.1042/BJ20042038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Xing G, Diao Y, Hoffart LM, Barr EW, Prabhu KS, et al. Evidence for C-H cleavage by an iron-superoxide complex in the glycol cleavage reaction catalyzed by myo-inositol oxygenase. Proc Natl Acad Sci USA. 2006;103:6130–5. doi: 10.1073/pnas.0508473103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Burzlaff NI, Rutledge PJ, Clifton IJ, Hensgens CM, Pickford M, et al. The reaction cycle of isopenicillin N synthase observed by X-ray diffraction. Nature. 1999;401:721–4. doi: 10.1038/44400. [DOI] [PubMed] [Google Scholar]
- 91.Hidaka T, Hidaka M, Uozumi T, Seto H. Nucleotide sequence of a carboxyphosphonoenolpyruvate phosphonomutase gene isolated from a bialaphos-producing organism, Streptomyces hygroscopicus, and its expression in Streptomyces lividans. Mol Gen Genet. 1992;233:476–8. doi: 10.1007/BF00265446. [DOI] [PubMed] [Google Scholar]
- 92.Babbitt PC, Hasson MS, Wedekind JE, Palmer DR, Barrett WC, et al. The enolase superfamily: a general strategy for enzyme-catalyzed abstraction of the alpha-protons of carboxylic acids. Biochemistry. 1996;35:16489–501. doi: 10.1021/bi9616413. [DOI] [PubMed] [Google Scholar]
- 93.Potters MB, Solow BT, Bischoff KM, Graham DE, Lower BH, et al. Phosphoprotein with phosphoglycerate mutase activity from the archaeon Sulfolobus solfataricus. J Bacteriol. 2003;185:2112–21. doi: 10.1128/JB.185.7.2112-2121.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hidaka T, Seto H, Imai S. Biosynthetic Mechanism of C-P Bond Fromation. Isolation of Carboxyphosphoenolpyruvate and Its Conversion to Phosphinopyruvate. J Am Chem Soc. 1989;111:8012–13. [Google Scholar]
- 95.Hidaka T, Imai S, Hara O, Anzai H, Murakami T, et al. Carboxyphosphonoenolpyruvate phosphonomutase, a novel enzyme catalyzing C-P bond formation. J Bacteriol. 1990;172:3066–72. doi: 10.1128/jb.172.6.3066-3072.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Freeman S, Pollack SJ, Knowles JR. Synthesis of the unusual metabolite carboxyphosphonoenolpyruvate. Cloning and expression of carboxyphosphonoenolpyruvate mutase. J Am Chem Soc. 1992;114:377–8. [Google Scholar]
- 97.Pollack SJ, Freeman S, Pompliano DL, Knowles JR. Cloning, overexpression and mechanistic studies of carboxyphosphonoenolpyruvate mutase from Streptomyces hygroscopicus. Eur J Biochem. 1992;209:735–43. doi: 10.1111/j.1432-1033.1992.tb17342.x. [DOI] [PubMed] [Google Scholar]
- 98.Schwartz D, Alijah R, Nussbaumer B, Pelzer S, Wohlleben W. The peptide synthetase gene phsA from Streptomyces viridochromogenes is not juxtaposed with other genes involved in nonribosomal biosynthesis of peptides. Appl Environ Microbiol. 1996;62:570–7. doi: 10.1128/aem.62.2.570-577.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Grammel N, Schwartz D, Wohlleben W, Keller U. Phosphinothricin-tripeptide synthetases from Streptomyces viridochromogenes. Biochemistry. 1998;37:1596–603. doi: 10.1021/bi9719410. [DOI] [PubMed] [Google Scholar]
- 100.Alijah R, Dorendorf J, Talay S, Puhler A, Wohlleben W. Genetic analysis of the phosphinothricin-tripeptide biosynthetic pathway of Streptomyces viridochromogenes Tu494. Appl Microbiol Biotechnol. 1991;34:749–55. doi: 10.1007/BF00169345. [DOI] [PubMed] [Google Scholar]
- 101.Schwartz D, Grammel N, Heinzelmann E, Keller U, Wohlleben W. Phosphinothricin tripeptide synthetases in Streptomyces viridochromogenes Tu494. Antimicrob Agents Chemother. 2005;49:4598–607. doi: 10.1128/AAC.49.11.4598-4607.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Eys S, Schwartz D, Wohlleben W, Schinko E. Three thioesterases are involved in the biosynthesis of phosphinothricin tripeptide in Streptomyces viridochromogenes Tu494. Antimicrob Agents Chemother. 2008;52:1686–96. doi: 10.1128/AAC.01053-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kamigiri K, Hidaka T, Imai S, Murakami T, Seto H. Studies on the biosynthesis of bialaphos (SF-1293) 12. C-P bond formation mechanism of bialaphos: discovery of a P-methylation enzyme. J Antibiot. 1992;45:781–7. doi: 10.7164/antibiotics.45.781. [DOI] [PubMed] [Google Scholar]
- 104.Takebe H, Imai S, Ogawa H, Satoh A, Tanaka H. Studies on the production of bialaphos. (I). Breeding of bialaphos producing strains from a biochemical engineering viewpoint. J Ferment Bioeng. 1989;67 [Google Scholar]
- 105.Imai S, Seto H, Sasaki T, Tsuruoka T, Ogawa H, et al. Studies on the biosynthesis of bialaphos (SF-1293). 6. Production of N-acetyl-demethylphosphinothricin and N-acetylbialaphos by blocked mutants of Streptomyces hygroscopicus SF-1293 and their roles in the biosynthesis of bialaphos. J Antibiot. 1985;38:687–90. doi: 10.7164/antibiotics.38.687. [DOI] [PubMed] [Google Scholar]
- 106.Seto H, Sasaki T, Imai S, Tsuruoka T, Ogawa H, et al. Studies on the biosynthesis of bialaphos (SF-1293). 2. Isolation of the first natural products with a C-P-H bond and their involvement in the C-P-C bond formation. J Antibiot. 1983;36:96–8. doi: 10.7164/antibiotics.36.96. [DOI] [PubMed] [Google Scholar]
- 107.Hartley FR. Introduction. In: Hartley FR, editor. The Chemistry of Organophosphorus Compounds. John Wiley & Sons, Ltd; 1990. [Google Scholar]
- 108.Guthrie P. Tautomerization Equilibria for Phosphorous acid and its Ethyl Esters, Free Energies of Formation of Phosphorous and Phosphonic Acids and their Ethyl Esters, and pKa Values for Ionization of the P-H Bond in Phosphonic Bond in Phosphonic Acid and Phosphonic Esters. Can J Chem. 1979;57:236–39. [Google Scholar]
- 109.Dahl O. The Preparation and Properties of Tervalent Phosphorus Acid Derivatives. In: Hartley FR, editor. The Chemistry of Organophosphorus Compounds. New York: John Wiley; 1996. pp. 1–45. [Google Scholar]
- 110.Edmunson RS. The Synthesis of Phosphonic and Phosphinic Acids and Their Derivatives. In: Hartley FR, editor. The Chemistry of Organophosphorus Compounds. New York: John Wiley; 1996. pp. 48–144. [Google Scholar]
- 111.Thompson CJ, Movva NR, Tizard R, Crameri R, Davies JE, et al. Characterization of the herbicide-resistance gene bar from Streptomyces hygroscopicus. EMBO J. 1987;6:2519–24. doi: 10.1002/j.1460-2075.1987.tb02538.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wohlleben W, Arnold W, Broer I, Hillemann D, Strauch E, Puhler A. Nucleotide sequence of the phosphinothricin N-acetyltransferase gene from Streptomyces viridochromogenes Tu494 and its expression in Nicotiana tabacum. Gene. 1988;70:25–37. doi: 10.1016/0378-1119(88)90101-1. [DOI] [PubMed] [Google Scholar]
- 113.Iguchi E, Okuhara M, Kohsaka M, Aoki H, Imanaka H. Studies on new phosphonic acid antibiotics. II. Taxonomic studies on producing organisms of the phosphonic acid and related compounds. J Antibiot. 1980;33:19–23. [PubMed] [Google Scholar]
- 114.Okuhara M, Kuroda Y, Goto T, Okamoto M, Terano H, et al. Studies on new phosphonic acid antibiotics. I. FR-900098, isolation and characterization. J Antibiot. 1980;33:13–7. doi: 10.7164/antibiotics.33.13. [DOI] [PubMed] [Google Scholar]
- 115.Okuhara M, Kuroda Y, Goto T, Okamoto M, Terano H, et al. Studies on new phosphonic acid antibiotics. III. Isolation and characterization of FR-31564, FR-32863 and FR-33289. J Antibiot. 1980;33:24–8. doi: 10.7164/antibiotics.33.24. [DOI] [PubMed] [Google Scholar]
- 116.Shigi Y. Inhibition of bacterial isoprenoid synthesis by fosmidomycin, a phosphonic acid-containing antibiotic. J Antimicrob Chemother. 1989;24:131–45. doi: 10.1093/jac/24.2.131. [DOI] [PubMed] [Google Scholar]
- 117.Kuemmerle HP, Murakawa T, De Santis F. Pharmacokinetic evaluation of fosmidomycin, a new phosphonic acid antibiotic. Chemioterapia. 1987;6:113–9. [PubMed] [Google Scholar]
- 118.Tsuchiya T, Ishibashi K, Terakawa M, Nishiyama M, Itoh N, Noguchi H. Pharmacokinetics and metabolism of fosmidomycin, a new phosphonic acid, in rats and dogs. Eur J Drug Metab Pharmacokin. 1982;7:59–64. doi: 10.1007/BF03189544. [DOI] [PubMed] [Google Scholar]
- 119.Murakawa T, Sakamoto H, Fukada S, Konishi T, Nishida M. Pharmacokinetics of fosmidomycin, a new phosphonic acid antibiotic. Antimicrob Agents Chemother. 1982;21:224–30. doi: 10.1128/aac.21.2.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Missinou MA, Borrmann S, Schindler A, Issifou S, Adegnika AA, et al. Fosmidomycin for malaria. Lancet. 2002;360:1941–2. doi: 10.1016/S0140-6736(02)11860-5. [DOI] [PubMed] [Google Scholar]
- 121.Wiesner J, Borrmann S, Jomaa H. Fosmidomycin for the treatment of malaria. Parasitol Res. 2003;90 2:S71–6. doi: 10.1007/s00436-002-0770-9. [DOI] [PubMed] [Google Scholar]
- 122.Jomaa H, Wiesner J, Sanderbrand S, Altincicek B, Weidemeyer C, et al. Inhibitors of the nonmevalonate pathway of isoprenoid biosynthesis as antimalarial drugs. Science. 1999;285:1573–6. doi: 10.1126/science.285.5433.1573. comment. [DOI] [PubMed] [Google Scholar]
- 123.Eliot AC, Griffin BM, Thomas PM, Johannes TW, Kelleher NL, et al. Cloning, expression, and biochemical characterization of Streptomyces rubellomurinus genes required for biosynthesis of antimalarial compound FR900098. Chem Biol. 2008;15:765–70. doi: 10.1016/j.chembiol.2008.07.010. The biosynthetic gene cluster of FR900098 and the first driving reaction different than PnPy decarboxylase. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hunt AH, Elzey TK. Revised structure of A53868A. J Antibiot. 1988;41:802. doi: 10.7164/antibiotics.41.802. [DOI] [PubMed] [Google Scholar]
- 125.Whitteck JT, Ni W, Griffin BM, Eliot AC, Thomas PM, et al. Reassignment of the structure of the antibiotic A53868 reveals an unusual amino dehydrophosphonic acid. Angew Chem Int Ed Engl. 2007;46:9089–92. doi: 10.1002/anie.200703810. [DOI] [PubMed] [Google Scholar]
- 126.Chatterjee C, Paul M, Xie L, van der Donk WA. Biosynthesis and Mode of Action of Lantibiotics. Chem Rev. 2005;105:633–84. doi: 10.1021/cr030105v. [DOI] [PubMed] [Google Scholar]
- 127.Rinehart KL, Harada K, Namikoshi M, Chen C, Harvis C, et al. Nodularin, microcystin, and the configuration of Adda. J Am Chem Soc. 1988;110:8557–58. [Google Scholar]
- 128.Anderson B, Hodgkin DC, Viswamitra MA. The structure of thiostrepton. Nature. 1970;225:233–35. doi: 10.1038/225233a0. [DOI] [PubMed] [Google Scholar]
- 129.Leason M, Cunliffe D, Parkin D, Lea PJ, M BJ. Inhibition of pea leaf glutamine synthetase by methionine sulfoximine, phosphinothricin, and other glutamate analogs. Phytochemistry. 1982;21:855. [Google Scholar]
- 130.Tachibana K, Watanabe T, Sekizawa Y, Takematsu T. Action mechanism of bialaphos. I. Inhibition of glutamine synthetase and quantitative changes of free amino acids in shoots of bialaphos-treated Japanese barnyard millet Nippon Noyaku Gakkaishi. J Pestic Sci. 1986;11:27–31. [Google Scholar]
- 131.Atherton FR, Hall MJ, Hassall CH, Lambert RW, Lloyd WJ, Ringrose PS. Phosphonopeptides as antibacterial agents: mechanism of action of alaphosphin. Antimicrob Agents Chemother. 1979;15:696–705. doi: 10.1128/aac.15.5.696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Allen JG, Atherton FR, Hall MJ, Hassall CH, Holmes SW, et al. Phosphonopeptides, a new class of synthetic antibacterial agents. Nature. 1978;272:56–8. doi: 10.1038/272056a0. [DOI] [PubMed] [Google Scholar]
- 133.Park BK, Hirota A, Sakai H. Studies on new antimetabolite produced by microorganism. Part III. Structure of plumbemycin A and B, antagonists of L-threonine from Streptomyces plumbeus. Agric Biol Chem. 1977;41:573–9. [Google Scholar]
- 134.Yamato M, Koguchi T, Okachi R, Yamada K, Nakayama K, et al. K-26, a novel inhibitor of angiotensin I converting enzyme produced by an actinomycete K-26. J Antibiot. 1986;39:44–52. doi: 10.7164/antibiotics.39.44. [DOI] [PubMed] [Google Scholar]
- 135.Koguchi T, Yamada K, Yamato M, Okachi R, Nakayama K, Kase H. K-4, a novel inhibitor of angiotensin I converting enzyme produced by Actinomadura spiculosospora. J Antibiot. 1986;39:364–71. doi: 10.7164/antibiotics.39.364. [DOI] [PubMed] [Google Scholar]
- 136.Kido Y, Hamakado T, Anno M, Miyagawa E, Motoki Y, et al. Isolation and characterization of I5B2, a new phosphorus containing inhibitor of angiotensin I converting enzyme produced by Actinomadura sp. J Antibiot. 1984;37:965–9. doi: 10.7164/antibiotics.37.965. [DOI] [PubMed] [Google Scholar]
- 137.Ntai I, Manier ML, Hachey DL, Bachmann BO. Biosynthetic origins of C-P bond containing tripeptide K-26. Org Lett. 2005;7:2763–5. doi: 10.1021/ol051091d. [DOI] [PubMed] [Google Scholar]
- 138.Ntai I, Phelan VV, Bachmann BO. Phosphonopeptide K-26 biosynthetic intermediates in Astrosporangium hypotensionis. Chem Commun. 2006:4518–20. doi: 10.1039/b611768f. [DOI] [PubMed] [Google Scholar]
- 139.Roberts E, Simonsen DG, Horiguchi M, Kittredge JS. Transamination of Aminoalkylphosphonic Acids with Alpha Ketoglutarate. Science. 1968;159:886–88. doi: 10.1126/science.159.3817.886. [DOI] [PubMed] [Google Scholar]
- 140.Shimotohno K, Seto H, Otake N, Imai S, Satoh A. Studies on the biosynthesis of bialaphos (SE-1293). 7. The absolute configuration of 2-phosphinomethylmalic acid, a biosynthetic intermediate of bialaphos. J Antibiot. 1986;39:1356–9. doi: 10.7164/antibiotics.39.1356. [DOI] [PubMed] [Google Scholar]
- 141.Seto H, Imai S, Sasaki T, Shimotohno K, Tsuruoka T, et al. Studies on the biosynthesis of bialaphos (SF-1293). 5. Production of 2-phosphinomethylmalic acid, an analogue of citric acid by Streptomyces hygroscopicus SF-1293 and its involvement in the biosynthesis of bialaphos. J Antibiot. 1984;37:1509–11. doi: 10.7164/antibiotics.37.1509. [DOI] [PubMed] [Google Scholar]
- 142.Hidaka T, Shimotohno KW, Morishita T, Seto H. Studies on the biosynthesis of bialaphos (SF-1293). 18. 2-phosphinomethylmalic acid synthase: a descendant of (R)-citrate synthase? J Antibiot. 1999;52:925–31. doi: 10.7164/antibiotics.52.925. [DOI] [PubMed] [Google Scholar]