

Abstract
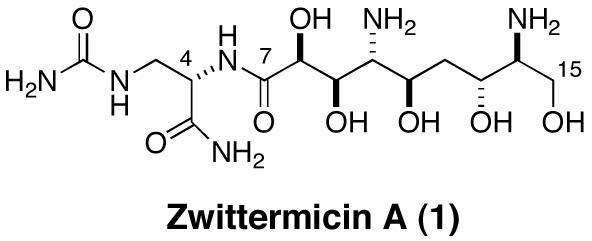
A proposed absolute configuration for the 7 stereocenters in (+)-zwittermicin A is described based on asymmetric synthesis of six diastereomeric 2,6-diamino-1,3,5,7-heptanetetraols corresponding to the C9-C15 segment, pair-wise 13C NMR chemical shift difference analysis of the models with the natural product, interpretation of enantiospecificity of serine loading domain of the zwittermicin A biosynthetic gene cluster, and degradation of the natural product.
(+)-Zwittermicin A (1), a water-soluble natural antibiotic isolated from fermentation of the soil-borne bacterium Bacillus cereus.1 Compound 1 is of significant interest for control of crop diseases both as an antifungal agent and an adjuvant with BT toxin for biocontrol.2
Despite the appearance of its structure, 1 is not a sugar, but a polyketide derived from a serine starter unit followed by consecutive additions of aminomalonate, malonate and two hydroxymalonates, each with a concomitant loss of CO2.3 Although the original isolation and structure elucidation — with partial relative configuration at C8-C10 — were reported twelve years ago, the complete relative and absolute configuration remained unsolved. Neither the natural product or any of the derivatives prepared to date exhibit crystallinity suitable for X-ray analysis. Applications of ‘J-based’ NMR methods for assignment of relative configuration in 1 failed due to lack of stereorelayed scalar couplings across the C12 methylene group; a problem related to the stereotopicity of the corresponding 1H NMR signals (vide infra).4 Thus, this rare diamino-polyol represents a significant challenge for stereochemical elucidation. Herein, we assign the configuration of 1 using a combination of model synthesis, paired 13C NMR chemical shift comparisons, Marfey’s analyis,5 and a bioinformatic interpretation of the gene sequence for zwittermicin A synthase.3 A flexible preparation of the C9-C15 core of 1 is revealed that exploits Miyashita conditions for regioselective 2,3-epoxy-1-alkanol ring opening by azide and is amenable for the total synthesis of the natural product.
The pseudo-symmetry present in the C9-C15 portion of 1 suggested a unified strategy for construction of six model compounds that embody all possible relative configurations for the pair of diads, C10,11 and C13,14.
With the exception of C11, the 13C NMR chemical shifts of these remote centers are expected to be relatively independent of the remainder of the molecule. Consequently, pair-wise comparison of chemical shifts in the models with the corresponding values in 1 should converge upon a unique configurational assignment.
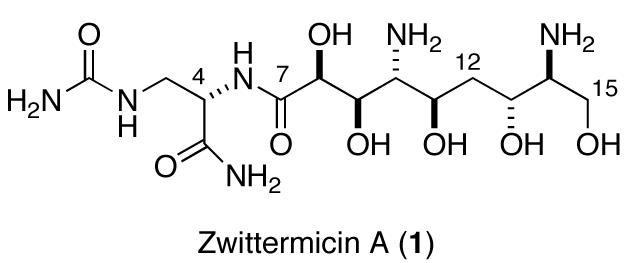
The six models 2-7 were synthesized starting with serine (Scheme 1). O-TBS-N,N-dibenzylserinal (8), prepared from L-serine, was converted to epoxide 9 using the method of Concellón.6,7 Carbon chain extension of 9 by a BF3•Et2O-mediated epoxide opening with the anion derived from O-TBS propargyl ether afforded 10. Protecting group adjustment followed by Red-Al reduction of the triple bond gave E-olefin 11, which was treated with m-CPBA to give diastereomeric epoxides 12 and 13 in a ratio of 1:1.8.
Scheme 1.
Separation of the diastereomers required protection of the primary alcohol and HPLC separation followed by deprotection to give the pure epoxides. Regioselective elaboration of the contiguous 2-amino-1,3-diol motif was projected based on Miyashita’s boron-directed azide opening of 1,2-epoxy-alkanols.8 In the event, separate azide opening of epoxides 12 and 13 using Miyashita’s method provided 1,3-diols 14 (dr 9:1) and 15 (dr 2.3:1), respectively, in good yields.9 Acid catalyzed deprotection of 14 and 15, with concomitant hydrogenolysis of the benzyl and azido groups afforded models 2 and 3, respectively, as their hydrochloride salts. The configurations of the two diastereomers were readily apparent by 1H and 13C NMR spectroscopy which revealed C2v symmetry in 2.
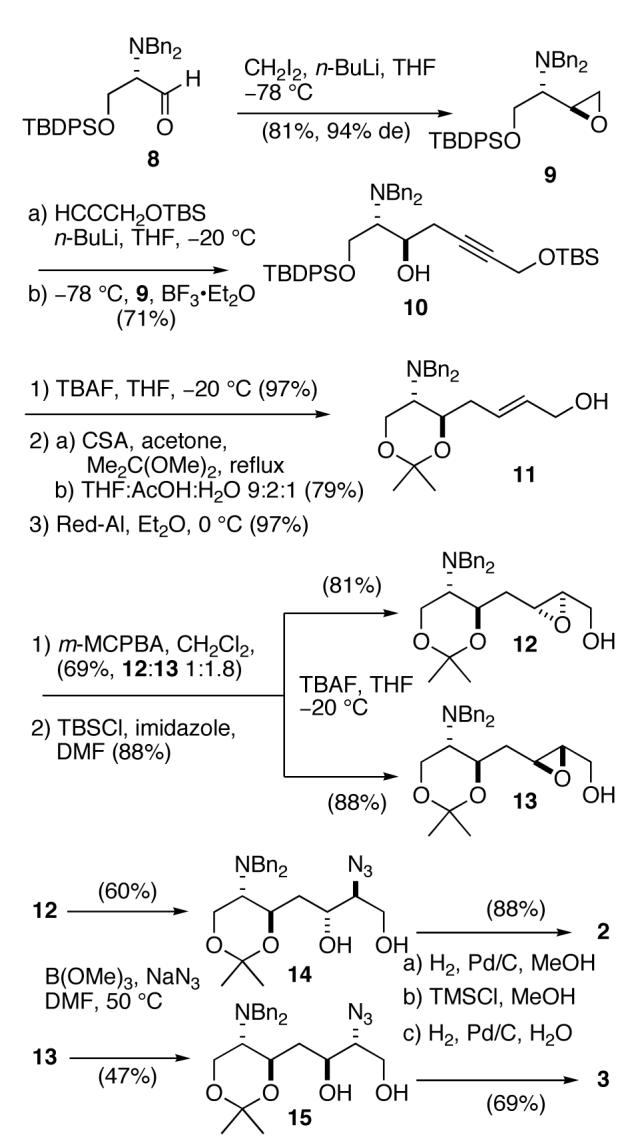
The remaining four models were synthesized from the L-serine methyl ester derivative 16 (Scheme 2) using the complementary syn-selective epoxide formation7 to provide 17, the C2 epimer of 9. Chain extension of 17 was achieved as before to give propargyl alcohol 18 which was separately converted to E- and Z-allylic alcohols 19 and 20 by Red-Al reduction or hydrogenation over Lindlar catalyst, respectively. Epoxidation of olefin 19 using m-CPBA was less successful due to the lability of the diastereomeric products, however, oxidation of 19 with methyltrioxorhenium gave a mixture of distereomeric epoxides which was carried forward using Miyashita’s method followed by acetonide protection to give azides 21 and 22. Since neither of the two diaminotetraols anticipated from conversion of 21 and 22 were expected to show symmetry (C1 space group), the configurational assignments of these molecules from NMR were in doubt. Fortunately, azide 22 crystallized as colorless needles (m.p. 138 °C) and X-ray analysis (Figure 2) provided the configuration of the 4-substitued (4R,5S)-2,2-dimethyl-5-azidodioxane ring [(13R,14S), zwittermicin A numbering]. It follows that 21 is the (4S,5R)-diastereomer.
Scheme 2.
Figure 2.
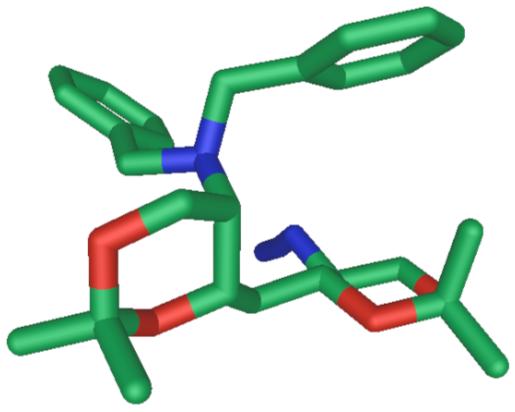
X-ray structure of 22.
To further verify stereochemical assignments of the models, azide 15 was converted to the acetonide 23 (Scheme 3). The 1H NMR spectrum of 23 showed the expected large diaxial vicinal couplings (δ 4.14, ddd, J 10.4, 8.0, 2.4 Hz; δ 3.83, ddd, J= 11.6,6.4, 2.4 Hz) for a syn-4,6-disubstituted 1,3-dioxane and large 13C chemical shift differences for the gem CH3 signals of the isopropylidene group (δ 29.9, q; 19.7, q).10
Scheme 3.
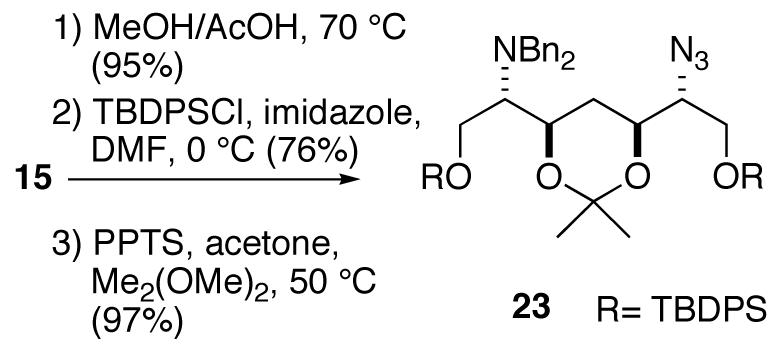
Acid-catalyzed global deprotection and hydrogenolysis of 21 and 22 compounds provided models 4 and 5, respectively, as their HCl salts. Models 6 and 7 were synthesized from olefin 20 using the same approach.
The diastereomeric family of model compounds comprise two meso compounds (3 and 6), two C2 isomers (2 and 7) and two isomers lacking symmetry (C1, 4 and 5). As expected, the 1H NMR signal of the C4 methylene protons in each C2 isomer (e.g. 2, δ 1.66 m, 2H) appeared as complex second-order multiplet owing to the fact that the H4 protons were chemical-shift equivalent but magnetically in-equivalent. Conversely, the 4-CH2 protons in the meso isomer 3 are both chemical shift inequivalent and magnetically inequivalent and appear as diastereotopic protons exhibiting a first-order ABX2 pattern (δ 1.79 dt 1H, J=14.4, 8.4 Hz; δ 1.84, dt, 14.4, 4.7 Hz, 1H). Analogous patterns were observed for C2-symmetrical 7 and meso-6. Interestingly, the 1H NMR signal of corresponding C12 methylene group in 1 also exhibited a complex second order pattern (400, 500 and 600 MHz) similar to those of 2 and 7, but dissimilar to the 4-CH2 signals of 3 and 6 of suggesting that the spin systems in 1, 2 and 7 reflected local C2 or pseudo-C2 symmetry, largely dictated by an anti- relationship of the C11 and C13 OH groups.
An unequivocal assignment of relative configuration for the diaminotetraol segement in 1 was made by pairwise comparisons of the differences in the 13C chemical shifts (Δδ) for C10-C15 of 1 and model compounds (Figure 1).11 There are only six diastereomers of the symmetrically substituted diaminoheptanetetraol models but eight diastereomers of the C10-C15 segment in 1. To complete the comparison, the 13C δ assignments of C1 isomers 4 and 5 were reversed to give the remaining two isomers — virtual compounds “4b” and “5b”. The C2v symmetric 2 is the only model compound with a close match to 1 for every carbon (Figure 1) except C9, which is the point of difference between 1 and the models and expected to show an ‘outlying’ Δδ in every case.
Figure 1.
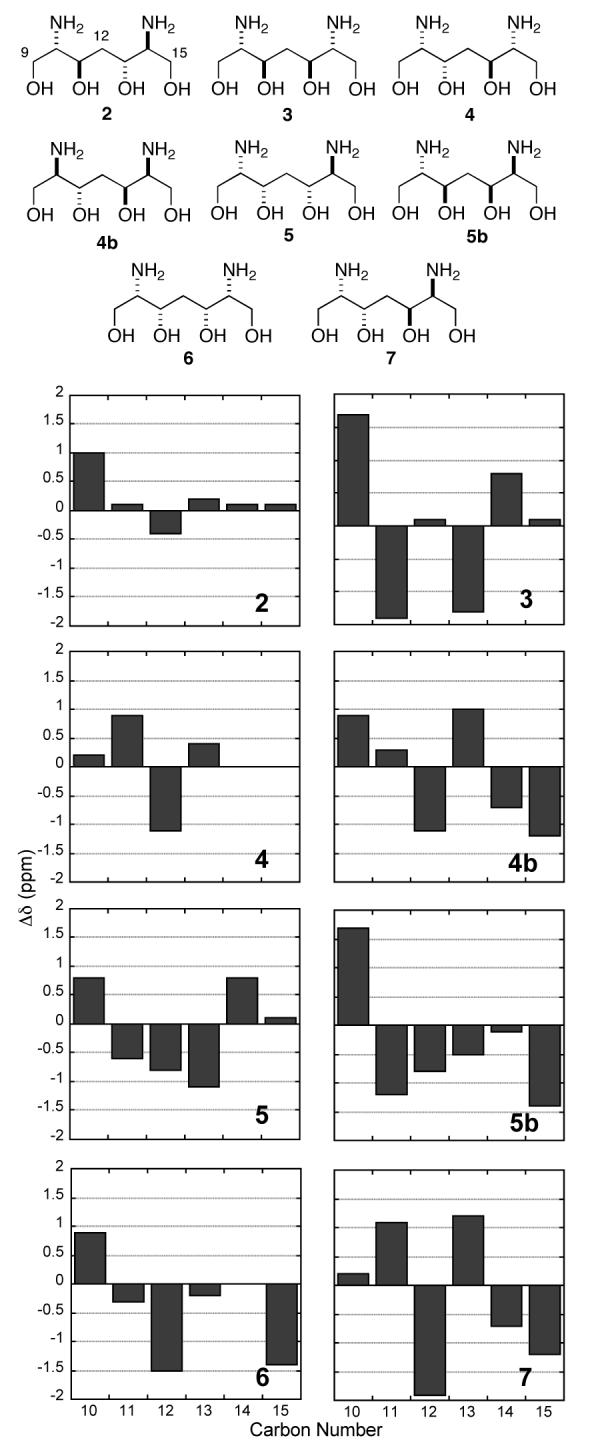
13C NMR (100 MHz, D2O, ref. internal CH3CN, δ1.47 ppm) Δδ values (δC model — δC 1) of model compounds 2-7. “4b” and “5b” are ’virtual isomers’ of 4 and 5, respectively, by reversing the order of 13C δ assignments for the purpose of comparison with 1.
Importantly, the other C2v isomer 7 had the largest mismatch which secures confidence for assignment of erythro relationships in each of the C10,11 and C13,14 diads. Elimination of the mismatched meso isomers 3 and 6, as suggested by 1H NMR and stereotopicity analysis of the 12-CH2 signal (above), is now corroborated by 13C NMR.
Therefore, compounds 1 and 2 share the same relative configuration at the stereogenic centers corresponding to C10, C11, C13 and C14 of 1. The data in Figure 1, in conjunction with the relative configurations at C8-C101 now allow us to extend the assignment of relative configuration of 1 to C8-C15.
Although no direct evidence for the absolute configuration of C8-C15 is yet available, analysis of the published sequence of the gene cluster for biosynthesis of zwittermicin A highly suggests that the C14 shares the same configuration as L-serine.3 Zwittermicin A is synthesized by a hybrid polyketide synthase-nonribosomal peptide synthase (PKS-NRPS) that comprises nine open reading frames including a loading domain for the starter unit that is homologous with serine adenylation domains found in gene clusters for biosynthesis of iturin A and mycosubtilin. Since the proposed gene sequence for production of 1 shows C13-C15 originating from L-serine and epimerase domains are absent, it is highly likely that C14 is L- and the absolute stereochemistry for C8-C14 in 1 is as depicted.
The absolute configuration at the remaining C4 stereocenter in 1 was determined as 4S by Marfey’s analysis.5 Acid hydrolysis of authentic 1 (6 N HCl, 24 h, 110 °C) and derivatization of the products with 2,4-dinitrophenyl-5-fluoro-L-alaninamide (Marfey’s reagent) under standard conditions, followed by analysis (C18 HPLC-MS) gave one peak that matched the peak (coinjection, MS spectrum) obtained by similar treatment of commercially available (-)-(S)-N3-ureido-2,3-diaminopropionic acid (S-albizziin).
In conclusion we have assigned the configuration of 1 as (4S,8S,9R,10R,11R,13R,14S). using an integrated approach based on synthesis and pairwise comparisons with model compounds, Marfey’s analysis, and published data. This sets the stage for completion of 1 by chain extension of a suitably protected derivative of 2 and attachment of the N3-ureido-2,3-diaminopropionamide side chain, which is the subject of current research in our laboratories.
Supplementary Material
Acknowledgment
We thank Mark Zabriskie (Oregon State University) for helpful discussions, and Entotech, Inc. (Davis, CA) for a sample of authentic zwittermicin A. X-ray analysis was carried out by A. Rheingold (UCSD). HRMS measurements were carried out by R. New (UC Riverside MS Facility), Y. Su (UC San Diego MS Facility), and the Scripps Center for Mass Spectrometry (La Jolla, CA). The UC Davis 400 MHz NMR and LCMS were purchased with funds provided by instrument grants NSF CHE-9808183 and RR14701-01 respectively. This work was supported by a grant from the NIH (to TFM, RO1 AI39987) and a fellowship from the Ecotoxicology Lead Campus Program (to EWR, UC Davis).
Footnotes
Supporting Information Available: Experimental procedures, X-ray data for 22 and selected 1H and 13C NMR. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1)(a).He H, Silo-Suh LA, Handelsman J, Clardy J. Tetrahedron Lett. 1994;35:2499. [Google Scholar]; (b) Silo-Suh LA, Lethbridge BJ, Raffel SJ, He H, Clardy J, Handelsman J. Appl. Environ. Microbiol. 1994;60:2023. doi: 10.1128/aem.60.6.2023-2030.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2)(a).Silo-Suh LA, Stabb EV, Raffel SJ, Handelsman J. Curr. Microbiol. 1998;37:6. doi: 10.1007/s002849900328. [DOI] [PubMed] [Google Scholar]; (b) Broderick NA, Goodman RM, Raffa KF, Handelsman J. Environ. Entomol. 2000;29:101. [Google Scholar]; (c) Stohl EA, Brady SF, Clardy J, Handelsman J. J. Bacteriol. 1999;181:5455. doi: 10.1128/jb.181.17.5455-5460.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Broderick NA, Goodman RM, Raffa KF, Handelsman J. Environ. Entomol. 2000;29:101. [Google Scholar]
- (3)(a).Emmert EA, Kilmowicz AK, Thomas MG, Handelsman J. Appl. Environ. Microbiol. 2004;70:104. doi: 10.1128/AEM.70.1.104-113.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Stohl EA, Milner JL, Handelsman J. Gene. 1999;237:403. doi: 10.1016/s0378-1119(99)00315-7. [DOI] [PubMed] [Google Scholar]
- (4).Murata M, Nakamura H, Tachibana K. J. Org. Chem. 1999;64:866. doi: 10.1021/jo981810k. HETLOC or HMBC experiments (600 MHz, D2O) did not extract useful 2,3JCH couplings across the C11-C12-C13 sequence, while 1H-1H couplings to the C12 methylene group showed second-order effects as discusssed later in the text.
- (5).Marfey P. Carlsberg Res. Commun. 1984;49:591. [Google Scholar]
- (6)(a).Hulme AN, Montgomery CH, Henderson DK. Chem. Soc., Perkin. Trans. 1. 2000:1837. [Google Scholar]; (b) Laïb T, Chastanet J, Zhu J. J. Org. Chem. 1998;63:1709. [Google Scholar]
- (7).Concellón JM, Riego E, Rodríguez-Solla H, Plutín AM. J. Org. Chem. 2001;66:8661. doi: 10.1021/jo015920u. [DOI] [PubMed] [Google Scholar]
- (8).Sasaki M, Tanino K, Hirai A, Miyashita M. Org. Lett. 2003;5:1789. doi: 10.1021/ol034455f. [DOI] [PubMed] [Google Scholar]
- (9).Under standard conditions, in the absence of B(OMe)3 yields and regioselectivity were poor (e.g. 12-> 14, (NH4Cl, NaN3, DMF, 44%, dr 1:1.4)
- (10).Rychnovsky SD, Rogers B, Yang G. J. Org. Chem. 1993;58:3511–3515. [Google Scholar]
- (11).13C NMR measurements of the of the hydrochloride salts of 1 and models 2-7 were carried out under essentially identical conditions (D2O, 25 °C) with internal CH3CN as reference.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.