

Abstract
There are several roadblocks that hinder systemic delivery of oncolytic viruses to the sites of metastatic disease. These include the tumor vasculature, which provides a physical barrier to tumor-specific virus extravasation. Although interleukin-2 (IL-2) has been used in antitumor therapy, it is associated with endothelial cell injury, leading to vascular leak syndrome (VLS). Here, we demonstrate that IL-2-mediated VLS, accentuated by depletion of regulatory T cells (Treg), facilitates localization of intravenously (IV) delivered oncolytic virus into established tumors in immune-competent mice. IL-2, in association with Treg depletion, generates “hyperactivated” natural killer (NK) cells, possessing antitumor activity and secreting factors that facilitate virus spread/replication throughout the tumor by disrupting the tumor architecture. As a result, the combination of Treg depletion/IL-2 and systemic oncolytic virotherapy was found to be significantly more therapeutic against established disease than either treatment alone. These data demonstrate that it is possible to combine biological therapy with oncolytic virotherapy to generate systemic therapy against established tumors.
INTRODUCTION
The issue of how to deliver viral and/or nonviral vectors to the sites of metastatic disease in immune-competent patients, in the absence of direct access by a needle, remains one of fundamental importance.1,2 In a virus-naive host, antiviral activities in the circulation1,2 include complement, preimmune immunoglobulin M (IgM), and other components of the innate immune system which inhibit replication within tumors.3-5 The circulating vector must also avoid nonspecific adhesion to multiple cells as well as specific sequestration in organs such as the liver. In a virus-immune host, neutralizing antibody (NAb) is a major additional inhibitor,1 although NAb can also provide a safety barrier to the widespread dissemination/toxicity of such viruses. Transient immune suppression can remove, or reduce, levels of NAb, and other immune effectors,4,6 and we7,8 as well as others9-13 have used cells to chaperone viral vectors into tumors as a means of protecting them from the hazards of exposure to the circulation.2
One major impediment is the tumor vasculature, which provides a physical barrier to systemic delivery.14 Therefore, we hypothesized that disruption of the vasculature might enhance the efficiency of viral delivery to tumors. Several agents can induce increased permeability of the vasculature to virus, including vascular endothelial growth factor15 and a combination of histamine and papaverine.16 In addition, although interleukin-2 (IL-2) has been developed as a biological therapy, it is associated with endothelial cell injury leading to vascular leak syndrome (VLS).17,18 VLS is mediated in part by effector lymphocytes activated in vivo by IL-2 (refs. 19,20), which bind and lyse endothelial cells.21,22 Therefore, although VLS is classically viewed as a toxic side effect of IL-2, we hypothesized that the associated vascular permeability/disruption might allow access of viruses from the circulation into tumors. If this is indeed the case, the use of biological therapy (IL-2) along with systemic delivery of oncolytic virotherapy could have offer a considerable advantage in the treatment of metastatic disease.
Even when it has been possible to inject vectors directly into human tumors, the vectors could not migrate far from the end of the needle. These findings inspired the development of replication-competent vectors which would, in theory, require only low levels of seeding to initiate the spread of infection to cover the tumor comprehensively.23-25 One such example is vesicular stomatitis virus (VSV), a negative-strand RNA Rhabdovirus, which replicates in the cytoplasm and is highly lytic.26,27 VSV infection of normal cells induces a potent type I interferon (IFN) response (IFN-α/β) that blocks viral replication and extinguishes the infection. However, many tumor cells have defects in their IFN response and are nonresponsive to exogenous IFN;26,28 consequently, VSV infection induces little or no IFN response, allowing free-ranging spread, infection, and lysis of tumors.29-31 However, significant problems continue to persist, because stromal and immune barriers still hinder effective intratumoral spread.4,32,33
In this study, we selected a nontoxic dose of IL-2 which nevertheless induces VLS, and showed that the intensity of the VLS can be increased, without increasing systemic toxicity, by depleting regulatory T cells (Treg).21 Induction of enhanced VLS facilitated the localization of intravenously (IV) delivered oncolytic virus into established subcutaneous and lung metastatic tumors in fully immune-competent mice. These effects were dependent upon activated natural killer (NK) cells, which both mediate enhanced virus spread through the tumor and allow for continued delivery of virus to tumors even in mice previously vaccinated against the virus. Finally, the combination of enhanced VLS and systemic oncolytic virotherapy was significantly more therapeutic against established disease than either treatment used alone. Therefore, a combination of systemic oncolytic virotherapy and a biological therapy leads to tumor cure under conditions where neither therapy alone is effective.
RESULTS
Systemic IL-2 induces vascular leak
We first determined a protocol in which systemic IL-2 was nontoxic in C57Bl/6 mice. Twelve or more injections of recombinant IL-2 (75,000 U/injection) induced elevated body temperatures and some morbidity; however, 10 injections (three/day) did not induce significant toxicities apart from occasional mild fevers which resolved within 24 hours (Figure 1a). In addition, this regimen was rarely therapeutically superior to phosphate-buffered saline (PBS) in controlling the growth of established B16 tumors (data not shown).
Figure 1. Interleukin-2 (IL-2) induces vascular leak and immune infiltration into tumors.

(a) C57Bl/6 mice were seeded with B16 tumors subcutaneously (SC). After 10 days, (when tumors were typically 0.5–0.6cm in the longest diameter), the mice were injected intraperitoneally with rhIL-2 at 75,000U/injection three times a day for 3 days. On the fourth day, a single further injection of IL-2 was given. (b) Forty-eight hours after the final injection of phosphate-buffered saline (PBS) or IL-2 (in a), vascular leakage into the lungs was measured34 (n = 4; representative of two separate experiments). (c) C57Bl/6 mice treated with intraperitoneal injections of either IL-2 or PBS (as described in a; three/group) were killed 48 hours after the last injection. Immune cells positive for the markers shown are plotted as a percentage of CD45+ gated cells. NK cells, natural killer cells; NKT cells, NK T cells; rhIL-2, recombinant human IL-2.
Mice treated with 10 injections of IL-2 showed significant increases in the ratio of wet/dry lung weights (P < 0.03),34 demonstrating induction of VLS as characterized by the efflux of fluid from the circulation into the body organs34 (Figure 1b). These mice also showed significant increases in the number of NK and NKT cells (Figure 1c), effectors previously shown to mediate VLS (Figure 1c).19-21,35,36 Although we observed consistent trends toward increases in other cell types, such as CD8+ T cells, these did not reach significance (data not shown), except for significant elevations in the number of Treg (Figure 1c).
Depletion of Treg enhances VLS
We investigated whether elevated levels of Treg regulate the extent of VLS. In the protocol of Figure 1a, we included an injection of the α-CD25, Treg-depleting PC61 antibody37 24 hours before the first injection of IL-2. IL-2 with Treg depletion had a small, but significant, effect on tumor growth (Figure 1a), but did not lead to any cures (see later text). Treg depletion also increased the severity of VLS into the lungs of IL-2-treated mice (P < 0.01 as compared with untreated mice and P < 0.03 as compared with IL-2 treatment alone) (Figure 2a). Treg depletion did not further increase the numbers of NK or NKT cells induced by IL-2 treatment alone (data not shown). However, depletion of NK cells from mice treated either with IL-2 alone (data not shown), or with Treg depletion and IL-2, prevented the induction of VLS (Figure 2a). Additional concomitant depletion of CD8 or CD4 cells had no effect on the magnitude of PC61/IL-2-induced VLS (data not shown). These findings indicate that IL-2-activated NK cells are major effectors of vascular damage/leakage.21,36
Figure 2. Depletion of regulatory T cells (Treg) enhances activity of interleukin-2 (IL-2) activated natural killer (NK)/lymphokine-activated killer cells.

(a) Nontumor-bearing C57Bl/6 mice were treated as described for Figure 1a, with the addition of an intraperitoneal injection of PC61 24 hours before the first intraperitoneal injection of IL-2 or phosphate-buffered saline (PBS). An additional group of mice received an injection of the NK cell-depleting asialo GM-1 antibody 24 hours before treatment with PC61 and then IL-2. Forty-eight hours after the final injection of IL-2 or PBS, vascular leakage into the lungs was measured. The results shown are from two separate experiments per treatment. (b) Splenocytes were recovered from mice treated as in a, and were plated with YAC cells (1 splenocyte effector:1 YAC target cell) in triplicate wells. After 48 hours, the surviving cells were counted and the result is expressed as a percentage of the controls (YAC + splenocytes from PBS-treated mice). (c) Splenocytes were recovered from mice treated as in a, and were plated with YAC cells (1:1). After 48 hours, the supernatants were assayed for interferon-γ (IFN-γ) using enzyme-linked immunosorbent assay (ELISA). (d) Splenocytes from the same samples as in c were also plated with B16 targets (1:1), and 48 hours later the supernatants were assayed for IFN-γ using ELISA. The results are representative of two separate experiments.
Splenocytes from IL-2-treated mice lysed (Figure 2b) and also secreted IFN-γ in response to culture in vitro with NK-sensitive YAC target cells (Figure 2c). However, these splenocytes neither lysed (data not shown) nor secreted IFN-γ when co-cultured with B16 tumor targets (Figure 2d). However, in mice treated with both PC61 and IL-2, both of these effector functions were significantly enhanced in splenocytes against YAC targets (Figure 2c), and now these splenocytes also secreted IFN-γ in response to B16 tumor targets (Figure 2d). However, no IFN-γ secretion was observed in mice that had been depleted of NK cells before PC61/IL-2 treatment (Figure 2b-d).
IL-2-mediated VLS increases virus localization to tumors
We tested the hypothesis that a combination of Treg depletion and IL-2 could increase efflux of circulating oncolytic virus into a well-established subcutaneous B16 tumor (Figure 3a). A single IV injection of VSV (108 plaque forming units) was not found to be therapeutic (data not shown), consistent with minimal levels of virus being recovered from the tumor (Figure 3b,c, and f). Pretreatment with IL-2 alone (Figure 3d and f), but not with PC61 alone (data not shown), significantly increased the virus infection of tumors. However, treatment with a combination of PC61, IL-2, and VSV (Figure 3a) facilitated very high levels of infection of subcutaneous tumors (Figure 3e and f). Histological analysis of tumors from mice treated with PC61/IL-2/VSV reproducibly revealed areas of extensive destruction of tumor architecture. These areas usually contained green fluorescent protein-positive (GFP+) staining, and were sometimes located near blood vessels, where they could still be discerned. However, such green-staining areas were by no means exclusively perivascular. Therefore, we cannot be certain, from these studies, that the viral infection was associated solely with foci of increased vascular leak.
Figure 3. Regulatory T cells depletion combined with interleukin-2 (IL-2) permits tumor localization of vesicular stomatitis virus (VSV).

(a) C57Bl/6 mice (three/group) were seeded with subcutaneous (SC) B16 tumors. After 9 days, the mice received an intraperitoneal (IP) injection of PC61 or control immunoglobulin G (IgG). After a further 24hours, the mice were injected IP with phosphate-buffered saline (PBS) or with recombinant human (75,000U/injection three times a day for 3 days). On the fourth day, a single further injection of IL-2 was given. Two hours after this last injection of IL-2/PBS, the mice received an intravenous (IV) injection of 108 plaque forming units (pfu) of VSV-GFP. (b–e) Thirty-six hours after in vivo virus delivery, the tumors were explanted and dissociated in vitro. The cells were left to adhere to the culture dish for 6–12 hours and then washed three times gently in PBS to remove nonadherent cells. These cultures were examined visually using phase microscopy (upper panels) or fluorescence for GFP expression (lower panels). The treatments received by the mice from which the tumors were explanted are shown between the panels. This protocol reproducibly generated cultures which were >98% positive for gp100, a B16-specific melanoma/melanocyte-associated antigen. This shows that GFP positivity is derived almost exclusively from recovered tumor cells rather than from infiltrating (nonadherent) immune cells [such as natural killer (NK) cells]. (f) Samples of the explanted B16 tumors obtained from a–e above were analyzed using flow cytometry for GFP. The results from a–f are representative of three separate experiments. GFP, green fluorescent protein. FSC, forward scatter.
Consistent with the GFP and fluorescence-activated cell sorting data (Figure 3b-f), only very low levels of viral titers could be detected in B16 tumors from mice treated with VSV (Table 1). The levels increased in mice that had been pretreated with IL-2 (P < 0.03), and increased dramatically in mice pretreated with both PC61 and IL-2 (Table 1) (P < 0.001 with respect to controls).
Table 1.
Treg depletion combined with IL-2 facilitates replication of intravenously delivered VSV within the tumor
| Treatment | VSV titer (pfu/mg tumor) | ||
|---|---|---|---|
| PC61 | 0 | 0 | 0 |
| VSV | 500 | 100 | 500 |
| IL-2/VSV | 5 × 103 | 103 | 8 × 103 |
| PC61/IL-2/VSV | 2 × 106 | 3 × 105 | 9 × 105 |
Abbreviations: IL-2, interleukin-2; pfu, plaque forming units; Treg, regulatory T cells; VSV, vesicular stomatitis virus.
Tumor-bearing C57Bl/6 mice (three/group) were treated as shown in Figure 3a. Thirty-six hours after the injection of VSV, the tumors were explanted and analyzed; the viral titers recovered from the freeze–thaw lysates for each tumor are shown.
These findings indicate that treatment with PC61 + IL-2 facilitates access of circulating oncolytic virus into-well established subcutaneous B16 tumors.
IL-2-mediated VLS enhances virus extravasation, but not replication, in normal tissues
VSV was detected only at low levels in normal (nontumor bearing) lungs after treatment with IL-2 (Figure 4a), and even this disappeared after 48 hours, suggesting that it represented input virus rather than viral replication in the lungs. Treatment with PC61 + IL-2 did not result in recovery of higher levels of virus from normal lungs (Figure 4a). In contrast, lungs of tumor-bearing mice contained infectious titers of VSV between three and four logs higher than those from tumor-free lungs when the mice were pretreated with PC61 and IL-2 (Figure 4a). In mice depleted of NK cells before the PC61/IL-2/VSV treatment, no virus was recovered from tumor-bearing lungs (Figure 4a), consistent with a similar finding of dependence upon NK cells for maximal VLS (Figure 2a).
Figure 4. Regulatory T cells depletion with interleukin-2 (IL-2) enhances viral delivery to systemic metastatic disease.

(a and b) C57Bl/6 mice were left uninjected (nontumor bearing) or were injected intravenously (IV) with B16 cells to seed metastases in the lung. After 9 days, the mice received an intraperitoneal injection of PC61 or a control immunoglobulin G. One group received the natural killer (NK) cell-depleting asialo GM-1 antibody 24 hours before PC61 (no NK cell). After a further 24 hours, the mice were injected intraperitoneally with phosphate-buffered saline (PBS) or recombinant human IL-2 (75,000 U/injection for 10 injections). Two hours after this last injection of IL-2/PBS, the mice received 108 plaque forming units (pfu) of VSV-GFP IV. After a further 36 hours the mice were killed, the lungs were removed, and viral titers were measured from freeze–thaw lysates of (a) the lungs or (b) hearts (two mice/group). One group of mice, that had been injected IV with 108 pfu of VSV-GFP 3 weeks before the seeding of the B16 lung metastases, was treated with PC61/IL-2 and VSV (VSV, PC61/IL-2, preimmune). (c) Dissociated heart tissues and (d) spleen tissues from the mice described in were analyzed using flow cytometry for GFP expression. GFP, green fluorescent protein; VSV, vesicular stomatitis virus.
Interestingly, relatively high levels of VSV could still be recovered from tumor-bearing lungs of mice immunized against VSV and treated with PC61/IL-2 and VSV (Figure 4a); these effects too were not observed under conditions of prior NK depletion (data not shown). However, no virus was recovered from tumor-bearing lungs (or from subcutaneous B16 tumors) from mice preimmunized with VSV and treated with VSV alone, or with IL-2 plus VSV (data not shown). It is evident, therefore, that NK cells activated by PC61/IL-2 treatment offer some degree of protection to systemically VSV delivered against high levels of circulating NAb.
VSV at very low levels was recovered from the hearts of mice treated with IL-2 + VSV, or with PC61/IL-2 + VSV, and these levels did not increase if the mice carried lung metastases (Figure 4b). These low levels of virus probably represent input virus that had extravasated after IL-2-mediated VLS, rather than replicating VSV (Figure 4b), a hypothesis supported by the lack of GFP cells in dissociated heart tissue (Figure 4c). The depletion of NK cells from mice treated with PC61/IL-2/VSV consistently increased the amount of VSV recovered from the heart by approximately one log (Figure 4b), which was unlike the situation with tumor-bearing lungs of mice that had received similar treatment. No virus could be recovered from the hearts of mice that were preimmune to VSV and were treated with VSV ± PC61/IL-2 (Figure 4b), and NK cell depletion did not alter this. Also, we were unable to recover detectable VSV from the spleens of mice treated with VSV, IL-2 + VSV, or PC61/IL-2/VSV either by GFP fluorescence-activated cell sorting analysis (Figure 4d) or by viral titration assays (data not shown). The mice treated with PC61/IL-2 and VSV did not lose weight or manifest any other overt signs of systemic toxicity.
PC61/IL-2-activated NK/lymphokine-activated killer cells facilitate intratumoral viral replication
We investigated further how the activity of NK/lymphokine-activated killer (LAK) cells induced by PC61/IL-2 contributes to the three to four log increase in virus titers recovered from B16 tumors in vivo (Table 1). B16 tumors freshly excised from C57Bl/6 mice, but not dissociated, generally formed a very poor substrate for de novo replication of VSV (Figure 5a and b) in vitro. Co-culture of tumors with splenocytes from mice treated with IL-2, but not PC61, facilitated viral replication from the tumor mass to a small, but significant (P < 0.01), degree (Figure 5a and b). However, co-culture of intact tumors with splenocytes from mice treated with PC61/IL-2 led to increased viral replication in the tumors by several logs as compared with co-culture with normal splenocytes (Figure 5a and b). These increases were not observed in co-cultured splenocytes from mice depleted of NK cells prior to PC61/IL-2 treatment (Figure 5a and b). Co-culture of B16-cultured cells (as distinct from intact B16 tumors) with splenocytes from mice treated with IL-2 and PC61 actually decreased the titer of virus produced from the B16 cultures as compared with co-culture with splenocytes from IL-2- or PC61-treated or untreated mice (Figure 5c). We attribute these effects to the increased cytotoxicity of these splenocytes against B16 targets, thereby leading to a reduction in the number of cells that are able to support viral replication.
Figure 5. PC61/interleukin-2 (IL-2)-activated natural killer/lymphokine-activated killer cells facilitate intratumoral viral replication and spread.

(a) Virus replication and spread throughout intact, dissociate B16 tumors was measured as described in Materials and Methods. Splenocytes recovered from mice treated with PC61/control immunoglobulin G (IgG)/± asialo GM-1 and then with phosphate-buffered saline (PBS) or with recombinant human IL-2 were added to the wells containing explanted B16 tumors along with vesicular stomatitis virus (VSV) [4 × 108 plaque forming units (pfu)/well]. One set of tumors was treated with splenocytes from mice that had received PC61 + IL-2 with added EDTA (1 mmol/l). Virus titers from freeze–thaw lysates of the tumors 48 hours later are shown (pfu/mg tumor; three/group). (b) The experiment described in a was repeated, except that, after co-culture with splenocytes (from mice treated as shown over each panel) and VSV, tumors were dissociated in vitro and analyzed for the extent of viral spread/infection, using flow cytometry for green fluorescent protein expression, 24 hours after plating. Dissociated cultures were almost exclusively tumor cells as assessed by fluorescence-activated cell sorting for the melanoma marker gp100. (c) 5 × 105 B16 cells were plated in vitro and 24 hours later, 5 × 105 splenocytes from mice treated with PC61/IL-2/asialo-GM-1 as in a were added along with VSV at a density of 5 × 104 pfu/well. After a further 48 hours, the supernatants and surviving cells were recovered. Virus titers from freeze–thaw lysates are shown as pfu/ml (three/group). (d) C57Bl/6 mice received an intraperitoneal injection of PC61 or control immunoglobulin G. One group of these mice received asialo GM-1 antibody 24 hours before treatment with PC61. After 24 hours, the mice were injected intraperitoneally with PBS or with rhIL-2 (75,000 U/injection for 10 injections). After a further 24 hours, splenocytes from these mice were recovered, cDNA was prepared, and the expression of the metalloproteinase-2 (MMP-2) gene was analyzed using PCR as shown. Equal loading of RNA was demonstrated using amplification of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as a control. (e) The experiment described in a was repeated with 107 splenocytes from mice treated with PBS, IL-2, PC61, or PC61/IL-2 added to wells containing explanted B16 tumors along with VSV at a density of 4 × 108 pfu/well. One set of tumors (n = 2) was not treated with splenocytes; instead they were incubated with VSV (4 × 108 pfu/well) along with recombinant MMP-2 (12.5 ng/ml). After a further 48 hours, the tumors were recovered from the wells and dissociated, and virus titers were determined from freeze–thaw lysates (shown as pfu/mg tumor; two/group).
These data suggest that NK/LAK cells induced by PC61/IL-2 treatment secrete a factor(s) which promote viral replication and/or viral spread through a tumor having an established three-dimensional architecture. We hypothesized that such a factor(s) may facilitate the breakdown of the stromal barriers within an established tumor.38,39 In order to test this hypothesis, we used EDTA as an inhibitor of the matrix metalloproteinases (MMPs).40 Increased viral titers from intact B16 tumors induced by splenocytes from PC61/IL-2-treated mice were largely (but not completely) inhibited by 1 mmol/l EDTA (Figure 5a and b). EDTA per se did not inhibit VSV replication, as shown by the finding that no similar reduction was seen in viral output from cultured B16 tumor cells in the presence of EDTA (Figure 5c). Consistent with these data, normal splenocytes express very low levels of MMP-2 (Figure 5d). Although treatment with IL-2 increases MMP-2 expression to a small degree, splenocytes from mice treated with PC61 + IL-2 express very high levels of MMP-2, as assessed at the levels both of mRNA (using reverse transcriptase PCR) (Figure 5d) and of protein activity. Significantly, MMP-2 expression in splenocytes from mice depleted of NK cells and subsequently treated with PC61 + IL-2 was no longer detectable (Figure 5d).
These data indicate that NK/LAK cells activated in vivo by IL-2, in the absence of Treg, secrete MMPs. However, recombinant MMP alone was never able to increase the titers of VSV released from explanted B16 tumors to levels comparable with those induced by splenocytes from PC61/IL-2-treated mice (Figure 5e). It is therefore clear that other factors, in addition to MMP-2, must also be contributing to the disruption of tumor architecture and enhanced virus spread and replication.38,39,41
Treg depletion–enhanced VLS promotes therapeutic systemic oncolytic virotherapy
The administration of IV VSV or PC61 alone to mice bearing well-established B16 tumors had no significant therapeutic effect as compared with the administration of PBS (Figure 6a and b). IL-2 alone (no VSV), PC61/IL-2 (no VSV) and IL-2 + VSV all extended the survival times significantly as compared with the controls (P < 0.04), though only by a few days (Figure 6b). Of all the treatments, only PC61/IL-2 + VSV led to long-term survival in mice (with the exception of one survivor mouse in the IL-2 + VSV group in Figure 6b), with typically between 25 and 90% of mice surviving tumor-free for 75 days after challenge (Figure 6b). None of the treated animals showed VSV-associated toxicity, and an examination of the lungs of the mice treated with PC61/IL-2 indicated no gross pathological changes or infiltration when compared with controls (Figure 6c). In addition, there was no evidence that VSV, under the influence of PC61/IL-2-mediated VLS, leads to increased infection in the brain.42 When these therapeutic experiments were repeated in mice depleted of NK cells, there were no long-term survivors among the mice, even after treatment with PC61/IL-2 and VSV (Figure 6d).
Figure 6. Systemic oncolytic virotherapy in combination with regulatory T cells depletion–enhanced interleukin-2 (IL-2) therapy.
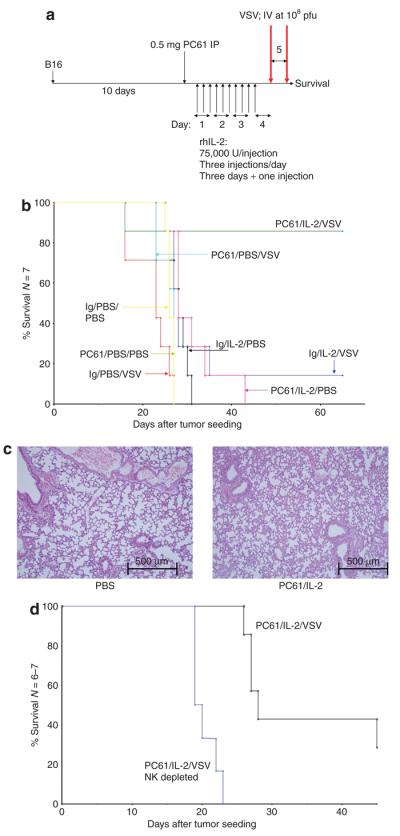
(a) C57Bl/6 mice (six to seven per group) were seeded subcutaneously with B16 tumors. After 9 days, the mice received an intraperitoneal (IP) injection of PC61 or control immunoglobulin G (IgG). After a further 24 hours, the mice were injected IP with phosphate-buffered saline (PBS) or with recombinant human IL-2 (75,000 U/injection three times a day for 3 days). On the fourth day, a single further injection of IL-2 was given. Two hours after this last injection of IL-2/PBS, the mice received the first of two intravenous (IV) injections of 108 plaque forming units (pfu) of VSV-GFP, followed 24 hours later by the second IV virus injection. (b) The survival of the mice treated as shown is plotted against the days elapsed after tumor seeding. (c) The lungs taken from the mice that had received the treatments described in a or b were examined 7 days after the final injection. Hematoxylin and eosin–stained sections, examined by two independent pathologists, showed no abnormalities in any group. Representative sections of lungs from mice treated with PBS or PC61/IL-2 are shown. (d) The experiment described in b was repeated in two groups of mice, both of which received PC61/IL-2 and VSV. One group also received an injection of the natural killer (NK) cell-depleting asialo GM-1 antibody 24 hours before treatment with PC61 (NK depleted). The survival of the mice treated as shown is plotted against the days elapsed after tumor seeding. GFP, green fluorescent protein; VSV, vesicular stomatitis virus.
DISCUSSION
Our data suggest a model in which IL-2 expands a population of NK/LAK cells in vivo (Figure 1c). However, these cells are restrained from maximal effector activity by Treg cells, which are themselves expanded in vivo by IL-2 (Figure 1c).21 We hypothesize that depletion of Treg generates “hyperactivated” NK/LAK cells, as compared with IL-2-activated NK/LAK cells; this is manifested by the induction of more VLS (Figure 2a),21 by increased cytokine production against YAC target cells (Figure 2b) and, significantly, by de novo effector functions against B16 tumor cells (Figure 2c). The mechanisms by which IL-2-expanded Treg suppress the effector functions of concomitantly expanded NK/LAK cells are still unclear.21
Consistent with enhanced VLS, IV administration of oncolytic virus after PC61/IL-2 was shown to promote infection of established subcutaneous or lung-resident tumors to much higher levels than with the IV administration of virus into normal mice, or into mice treated with IL-2 alone (Figures 3 and 4 and Table 1). These levels of transduction were completely dependent on NK cells. Moreover, even when VSV was administered to mice previously immunized against the virus (under a protocol that we have shown induces high levels of circulating NAb, data not shown), virus could still be detected in tumor-bearing lungs at higher levels than in nontumor-bearing lungs (Figure 4a). Prior depletion of NK cells in preimmunized mice resulted in the total absence of transduction of tumors. Preliminary studies suggest that VSV particles may physically bind to the surfaces of NK/LAK cells activated in vivo by IL-2, perhaps even through interaction of virus–antibody complexes with Fc receptors on the NK cells. This may help to chaperone antibody-bound virus into tumors directly on the NK cells, although such a mechanism remains to be confirmed.
Although viral efflux into normal lungs and hearts also occurred, these levels were low, and probably represent input virus rather than active viral replication. In contrast, high levels of virus could be recovered from tumor-bearing lungs, confirming the oncolytic preference of VSV for tumor cells over normal cells43 (Figure 4a). The depletion of NK cells from mice treated with PC61/IL-2/VSV consistently increased the amount of VSV recovered from the heart (Figure 4b), which was unlike the situation with tumor-bearing lungs of mice that had received similar treatment. The depletion of Treg in IL-2-treated mice did not further enhance the levels of virus recovered from the heart. Taking these observations together, it may be that the physical association of virus with NK/LAK cells in vivo increases viral access to tumors (perhaps associated with NK/LAK intratumoral infiltration), but simultaneously prevents viral access to other tissues such as the heart (where no NK/LAK cell trafficking is observed).
We also observed that, in recently excised tumors that still retain their full three-dimensional architecture, NK/LAK cells generated in vivo by IL-2 (in the absence of Treg) are able to condition the tumors to enhance replication and spread of VSV. That is, “hyperactivated” NK/LAK cells not only facilitate increased viral localization to tumors by the induction of VLS, but also elaborate the effector molecules which allow virus to replicate in, and/or spread through, the tumor more effectively. Tumor “conditioning” was reduced, but not eliminated, in vitro by EDTA, which inhibits MMPs40 and PC61/IL-2-activated NK/LAK cells express high levels of MMP (Figure 5d). This probably allows their directed migration to kill target cells in vivo.44,45 These high levels of proteases, such as MMP and others, induced in “hyperactivated” NK cells, may mediate the destruction of the tumor architecture, especially of the stromal barriers,38,41 leading to the greatly enhanced intratumoral viral spread that we observed both in vivo (Table 1) and in vitro (Figure 5). We do not believe that increased expression of MMP-2 in these hyperactivated NK/LAK cells is, by itself, sufficient to explain the increased viral spread/replication through tumors that we have observed both in vivo (Figure 3) and in vitro (Figure 5). Ongoing experiments using gene chip analysis of NK/LAK cells from PC61/IL-2-treated mice will reveal a more complete spectrum of induced proteases and other proteins that contribute to the effects we describe here. An understanding of the mechanism by which Treg suppress effector functions of NK/LAK cells will help to develop more potent antitumor cellular therapies and to understand the etiology/treatment of conditions wherein de-regulated MMP activity leads to disease.46 Moreover, the identification of the MMP and other factors secreted by “hyperactivated” NK/LAK cells that prime the tumors to permit high-level VSV replication/spread41 will help in the design of more effective oncolytic viruses that can propagate through established tumors.38,39 Earlier, we have shown that the therapeutic efficacy of intratumoral VSV on subcutaneous B16 tumors depends on both CD8+ T cells and NK cells.47 Moreover, the depletion of Treg had an antitherapeutic effect because it relieved suppression of the antiviral immune response, leading to early viral clearance.47 It is evident, therefore, that Treg depletion can potentially have opposing therapeutic effects. On the one hand, Treg depletion, in combination with IL-2, permits the expansion of hyperactivated NK/LAK cells (Figure 1c). These NK/LAK cells are important for antitumor therapy per se47 and also mediate enhanced access of IV delivered VSV to the tumor, along with increased spread of virus through the established tumor architecture (Figures 3-6). On the other hand, Treg depletion by itself relieves the in vivo suppression of the antiviral immune response, contributes to faster viral clearance, and can reduce the oncolytic therapeutic action of VSV.47 Therefore, while seeking to enhance the efficacy of oncolytic virotherapy through immune conditioning interventions, one needs seek to take into consideration the multiple interactions between antitumor and antiviral immune effectors that make the outcome of such interventions difficult to predict.
Finally, we show that the combination of PC61/IL-2 and IV administered oncolytic virus leads to a very significant increase in therapeutic effect as compared to treatment with either of these approaches alone. Consistent with the role of NK/LAK cells in generating VLS, directing antitumor effector recognition, and creating an environment conducive to viral replication/spread, NK cell depletion negated this therapeutic effect. Encouragingly, although PC61/IL-2 treatment clearly changes the homeostasis of normal tissues (evidenced by increased VLS in normal lung, Figure 2a), this was not reflected in any detectable toxicity on account of virotherapy. There was no increase in the levels of virus replication in the heart (Figure 4b), no observable toxicities in mice that had been cured of tumors (Figure 6), and no pathological abnormalities in their lungs (Figure 6c). Importantly, these data suggest that the cumulative activity of PC61/IL-2-activated NK/LAK cells is predominantly tumor specific, as seen from the finding that normal tissues remained free of significant toxicity whereas tumor growth was significantly inhibited.
In summary, although other roadblocks to successful systemic delivery of viruses to metastatic tumors remain,1,25,48 our data suggest that an IV administered oncolytic virus can survive in the circulation long enough to reach tumor sites. We show in this study that treatment with IL-2, along with Treg depletion, is a viable method of localizing IV administered oncolytic virus to tumors, so as to effect tumor regression in a fully immune-competent host through multiple contributory components. That is, the effector functions of a population of “hyperactivated” NK/LAK cells lead to enhanced virus localization at the tumor site through induction of VLS; they also have direct antitumor activity; and these same cells condition the tumor to facilitate increased viral replication, viral spread, and oncolysis. Taken together, these results are significant because they demonstrate that it is possible to combine biological therapy with oncolytic virotherapy in order to facilitate a genuinely systemic therapeutic approach against established tumors in a fully immune-competent host.
MATERIALS AND METHODS
Cells and viruses
B16 are murine melanoma cells (H2-Kb). They were grown in Dulbecco's modified Eagle's minimal essential medium (Life Technologies, Rockville, MD) supplemented with 10% (vol/vol) fetal calf serum (Life Technologies, Rockville, MD) and l-glutamine (Life Technologies, Rockville, MD). YAC-1, an NK-sensitive cell line, was grown in Roswell Park Memorial Institute medium containing 10% fetal calf serum. Cell lines were monitored routinely and confirmed to be free of Mycoplasma infection.
VSV-GFP was generated by cloning the cDNA for GFP into the plasmid pVSV-XN2, as described earlier.29 VSV-GFP is referred to as “VSV”. Monoclonal VSV was obtained by plaque purification on BHK-21 cells. Concentration and purification were performed using sucrose gradient centrifugation. Virus stock titers were measured using standard plaque assays of serially diluted samples on BHK-21 cells.29
Flow cytometry
For analysis of phenotype, organs/tumors were removed from mice and dissociated in vitro to achieve single-cell suspensions. 1 × 106 cells were washed in PBS containing 0.1% bovine serum albumin (wash buffer), re-suspended in 50 μl of wash buffer, and exposed to directly conjugated primary Abs for 30 minutes at 4 °C. The cells were then washed and re-suspended in 500 μl of PBS containing 4% formaldehyde. The cells were analyzed using flow cytometry, and the data were analyzed using CellQuest software (BD Biosciences, San Jose, CA). Anti-CD8β fluorescein isothiocyanate, anti-CD4 fluorescein isothiocyanate, anti-NK1.1 PE, anti-Mac-3 PE, anti-CD11c PE, anti-Gr-1 PE, anti-CD45 PerCP, and their respective isotype controls were purchased from BD Pharmingen (San Diego, CA). Treg cells were analyzed using anti-CD4, anti-CD25, and anti-FOXP3 antibodies (E-Bioscience, San Diego, CA).
Antigen priming assays—splenocyte preparation and antigen presentation
Splenocytes enriched in lymphocytes were prepared from spleens from treated/vaccinated animals using standard techniques.49 Freshly purified splenocyte populations were washed in PBS, and 250,000 cells/well were incubated with target tumor cells (YAC or B16), typically at ratios of 100:1; 10:1, or 1:1. After 48–72 hours, cell-free supernatants were tested for IFN-γ using enzyme-linked immunosorbent assay (BD Pharmingen, San Diego, CA).
In vivo studies
All procedures were approved by the Mayo Foundation Institutional Animal Care and Use Committee. C57Bl/6 mice of 6–8 weeks of age were purchased from Jackson Laboratories (Bar Harbor, ME). In order to establish subcutaneous tumors, 2 × 105 B16-OVA cells in 100 μl of PBS were injected into the flanks of mice. For survival studies, the tumor diameter was measured three times weekly in two dimensions using calipers, and the mice were killed when the tumor size was ∼1.0 × 1.0 cm2 in two perpendicular directions.
For the purpose of establishing systemic metastatic disease, C57Bl/6 mice were injected IV with 2 × 105 B16-OVA cells to form lung metastases. At the first sign of any distress shown by the mice, they were killed.
Immune cell depletions were performed by intraperitoneal injections (0.1 mg/mouse) of anti-CD8 (Lyt 2.43) and anti-CD4 (GK1.5), both from the Monoclonal Antibody Core Facility, Mayo Clinic; and IgG control (ChromPure Rat IgG; Jackson ImmunoResearch Laboratories, West Grove, PA). For Treg depletion, 0.5 mg of PC61 antibody (Monoclonal Antibody Core Facility, Mayo Clinic) per mouse was administered intraperitoneally as described. We used flow cytometry to confirm that, by day 3 after a single intraperitoneal injection of 0.5 mg of PC61, >95% of CD4+ CD25Hi cells were depleted from the spleens and LN of C57Bl/6 mice.
Measurement of vascular leak
The extent of pulmonary edema induced by IL-2 ± PC61 was determined by measuring wet/dry lung weight ratios as described by Queluz et al.34 The mice were treated with recombinant human IL-2 (Mayo Clinic Pharmacy) at a dose of 75,000 U/injection for a total of 10 intraperitoneal injections as described in the text. The treated animals were killed 24–72 hours after the final injection of IL-2, the lungs were excised, and wet weights were recorded. The lungs were then dried for 72 hours and weighed again to determine the wet/dry weight ratio ± SD.
NK cell depletion
Anti-asialo-GM1 was purchased (CEDARLANE, Burlington, NC) and resuspended in 1 ml. An amount of 25 μl (∼0.75 mg/mouse) was injected intraperitoneally once at the time-points indicated in the text. The mice treated with isotype received the same protein concentration of Rabbit IgG (Jackson ImmunoResearch Laboratories). NK depletion was verified using spleen NK1.1 flowcytometry analysis (R&D Systems, Minneapolis, MN).
Virus titration from tumors and organs
The tumors and organs removed from the mice were weighed and lysed with three cycles of freeze/thawing. Virus was recovered from the lysates, and the titers were determined on BHK-21 cells as described earlier, and expressed as plaque forming units of VSV/mg tissue.
Reverse transcriptase PCR
Splenocytes were removed as described earlier and RNA was prepared using the Qiagen RNA extraction kit. An amount of 1 μg total cellular RNA was reverse transcribed in a 20 μl volume using oligo-(dT) as a primer. A cDNA equivalent of 1 ng RNA was amplified with PCR for the MMP-2 gene using the MMP-2 primer pair purchased from R&D Systems, Minneapolis, MN (RDP-84).
Virus spread throughout explanted, intact tumors
C57Bl/6 mice were seeded with subcutaneous B16 tumors. After 15 days, established tumors were excised and, without being dissociated, placed intact into wells of a 24-well tissue culture plate in Dulbecco's modified Eagle's medium. Separately, C57Bl/6 mice received an intraperitoneal injection of PC61 or control IgG. One group received asialo GM-1 antibody 24 hours before the treatment with PC61. After 24 hours, the mice were injected intraperitoneally with PBS or with recombinant human IL-2 (75,000 U/injection for 10 injections). Splenocytes (107) were recovered from these mice and added to the wells containing explanted B16 tumors, along with VSV at a density of 4 × 108 plaque forming units/well. One set of tumors was treated with splenocytes from mice treated with PC61 + IL-2 with added EDTA (1mmol/l). After 48 hours, the tumors were removed from the wells and dissociated, and virus titers were determined from freeze–thaw lysates (shown as plaque forming units/mg tumor, three/group). In separate experiments, the tumors were treated with recombinant mouse MMP-2 (Sigma, St. Louis, MO) at various concentrations instead of splenocytes, and 12.5ng/ml was found to be the optimal amount necessary to support intratumoral viral spread.
Histopathology of tumor sections
Lungs were removed from the mice and fixed in 10% formalin in PBS, and then paraffin-embedded and sectioned. Hematoxylin and eosin–stained sections were prepared for analysis of tissue destruction and gross infiltrate. Two independent pathologists, blinded as to the experimental design, examined the hematoxylin and eosin sections.
Statistics
Survival data from the animal studies was analyzed using the log rank test,50 and the two-sample unequal variance student's t-test analysis was applied for in vitro assays. Statistical significance was determined at the level of P < 0.05.
ACKNOWLEDGMENTS
We thank Toni L. Higgins for expert secretarial assistance. This work was supported by the Mayo Foundation, by the National Institutes of Health Grant CA RO1107082-02 and by the Richard M. Schulze Family Foundation.
REFERENCES
- 1.Fisher K. Striking out at disseminated metastases: the systemic delivery of oncolytic viruses. Curr Opin Mol Ther. 2006;8:301–313. [PubMed] [Google Scholar]
- 2.Harrington K, Alvarez-Vallina L, Crittenden M, Gough M, Chong H, Diaz RM, et al. Cells as vehicles for cancer gene therapy: the missing link between targeted vectors and systemic delivery? Hum Gene Ther. 2002;13:1263–1280. doi: 10.1089/104303402760128504. [DOI] [PubMed] [Google Scholar]
- 3.Ikeda K, Wakimoto H, Ichikawa T, Jhung S, Hochberg FH, Louis DN, et al. Complement depletion facilitates the infection of multiple brain tumors by an intravascular, replication-conditional herpes simplex virus mutant. J Virol. 2000;74:4765–4775. doi: 10.1128/jvi.74.10.4765-4775.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fulci G, Breymann L, Gianni D, Kurozomi K, Rhee SS, Yu J, et al. Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proc Natl Acad Sci USA. 2006;103:12873–12878. doi: 10.1073/pnas.0605496103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Friedman A, Tian JP, Fulci G, Chiocca EA, Wang J. Glioma virotherapy: effects of innate immune suppression and increased viral replication capacity. Cancer Res. 2006;66:2314–2319. doi: 10.1158/0008-5472.CAN-05-2661. [DOI] [PubMed] [Google Scholar]
- 6.Hirasawa K, Nishikawa S, Norman KL, Coffey MC, Thompson BG, Yoon CS, et al. Systemic reovirus therapy of metastatic cancer in immune-competent mice. Cancer Res. 2003;63:348–353. [PubMed] [Google Scholar]
- 7.Cole C, Qiao J, Kottke T, Diaz RM, Ahmed A, Sanchez-Perez L, et al. Tumor-targeted, systemic delivery of therapeutic viral vectors using hitchhiking on antigen-specific T cells. Nat Med. 2005;11:1073–1081. doi: 10.1038/nm1297. [DOI] [PubMed] [Google Scholar]
- 8.Thanarajasingam U, Sanz L, Diaz R, Qiao J, Sanchez-Perez L, Kottke T, et al. Delivery of CCL-21 to metastatic disease improves the efficacy of adoptive T-cell therapy. Cancer Res. 2007;67:300–308. doi: 10.1158/0008-5472.CAN-06-1017. [DOI] [PubMed] [Google Scholar]
- 9.Yotnda P, Savoldo B, Charlet-Berguerand N, Rooney C, Brenner M. Targeted delivery of adenoviral vectors by cytotoxic T cells. Blood. 2004;104:2272–2280. doi: 10.1182/blood-2003-11-3803. [DOI] [PubMed] [Google Scholar]
- 10.Thorne SH, Negrin RS, Contag CH. Synergistic antitumor effects of immune cell-viral biotherapy. Science. 2006;311:1780–1784. doi: 10.1126/science.1121411. [DOI] [PubMed] [Google Scholar]
- 11.Ong HT, Hasegawa K, Dietz AB, Russell SJ, Peng KW. Evaluation of T cells as carriers for systemic measles virotherapy in the presence of antiviral antibodies. Gene Ther. 2007;14:324–333. doi: 10.1038/sj.gt.3302880. [DOI] [PubMed] [Google Scholar]
- 12.Gómez-Navarro J, Contreras JL, Arafat W, Jiang XL, Krisky D, Oligino T, et al. Genetically modified CD34+ cells as cellular vehicles for gene delivery into areas of angiogenesis in a rhesus model. Gene Ther. 2000;7:43–52. doi: 10.1038/sj.gt.3301054. [DOI] [PubMed] [Google Scholar]
- 13.Power AT, Wang J, Falls TJ, Paterson JM, Parato KA, Lichty BD, et al. Carrier cell-based delivery of an oncolytic virus circumvents antiviral immunity. Mol Ther. 2007;15:123–130. doi: 10.1038/sj.mt.6300039. [DOI] [PubMed] [Google Scholar]
- 14.Liu Y, Deisseroth A. Tumor vascular targeting therapy with viral vectors. Blood. 2006;107:3027–3033. doi: 10.1182/blood-2005-10-4114. [DOI] [PubMed] [Google Scholar]
- 15.Gregorevic P, Blankinship MJ, Allen JM, Crawford RW, Meuse L, Miller DG, et al. Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat Med. 2004;10:828–834. doi: 10.1038/nm1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greelish JP, Su LT, Lankford EB, Burkman JM, Chen H, Konig SK, et al. Stable restoration of the sarcoglycan complex in dystrophic muscle perfused with histamine and a recombinant adeno-associated viral vector. Nat Med. 1999;5:439–433. doi: 10.1038/7439. [DOI] [PubMed] [Google Scholar]
- 17.Baluna R, Vitetta ES. Vascular leak syndrome: a side effect of immunotherapy. Immunopharmacology. 1997;37:117–132. doi: 10.1016/s0162-3109(97)00041-6. [DOI] [PubMed] [Google Scholar]
- 18.Rosenberg SA, Mulé JJ, Spiess PJ, Reichert CM, Schwarz SL. Regression of established pulmonary metastases and subcutaneous tumor mediated by the systemic administration of high-dose recombinant interleukin 2. J Exp Med. 1985;161:1169–1188. doi: 10.1084/jem.161.5.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakajima T, Schulte S, Warrington KJ, Kopecky SL, Frye RL, Goronzy JJ, et al. T-cell-mediated lysis of endothelial cells in acute coronary syndromes. Circulation. 2002;105:570–575. doi: 10.1161/hc0502.103348. [DOI] [PubMed] [Google Scholar]
- 20.Yoneda O, Imai T, Goda S, Inoue H, Yamauchi A, Okazaki T, et al. Fractalkine-mediated endothelial cell injury by NK cells. J Immunol. 2000;164:4055–4062. doi: 10.4049/jimmunol.164.8.4055. [DOI] [PubMed] [Google Scholar]
- 21.Melencio L, McKallip RJ, Guan H, Ramakrishnan R, Jain R, Nagarkatti PS, et al. Role of CD4+CD25+ T regulatory cells in IL-2-induced vascular leak. Int Immunol. 2006;18:1461–1471. doi: 10.1093/intimm/dxl079. [DOI] [PubMed] [Google Scholar]
- 22.Renkonen R, Ristimäki A, Hävry P. Interferon-γ protects human endothelial cells from lymphokine-activated killer cell-mediated lysis. Eur J Immunol. 1998;18:1839–1842. doi: 10.1002/eji.1830181129. [DOI] [PubMed] [Google Scholar]
- 23.Kelly E, Russell SJ. History of oncolytic viruses: genesis to genetic engineering. Mol Ther. 2007;15:651–659. doi: 10.1038/sj.mt.6300108. [DOI] [PubMed] [Google Scholar]
- 24.Kirn D, Martuza RL, Zwiebel J. Replication-selective virotherapy for cancer: biological principles, risk management and future directions. Nat Med. 2001;7:781–787. doi: 10.1038/89901. [DOI] [PubMed] [Google Scholar]
- 25.Bell JC. Oncolytic viruses: what's next? Curr Cancer Drug Targets. 2007;7:127–131. doi: 10.2174/156800907780058844. [DOI] [PubMed] [Google Scholar]
- 26.Lichty BD, Power AT, Stojdl DF, Bell JC. Vesicular stomatitis virus: re-inventing the bullet. Trends Mol Med. 2004;10:210–216. doi: 10.1016/j.molmed.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 27.Barber GN. Vesicular stomatitis virus as an oncolytic vector. Viral Immunol. 2004;17:516–527. doi: 10.1089/vim.2004.17.516. [DOI] [PubMed] [Google Scholar]
- 28.Barber GN. VSV tumor selective replication and protein translation. Oncogene. 2005;24:7710–7719. doi: 10.1038/sj.onc.1209042. [DOI] [PubMed] [Google Scholar]
- 29.Fernandez M, Porosnicu M, Markovic D, Barber GN. Genetically engineered vesicular stomatitis virus in gene therapy: application for treatment of malignant disease. J Virol. 2002;76:895–904. doi: 10.1128/JVI.76.2.895-904.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ebert O, Harbaran S, Shinozaki K, Woo SL. Systemic therapy of experimental breast cancer metastases by mutant vesicular stomatitis virus in immune-competent mice. Cancer Gene Ther. 2005;12:350–358. doi: 10.1038/sj.cgt.7700794. [DOI] [PubMed] [Google Scholar]
- 31.Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N, et al. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med. 2000;6:821–825. doi: 10.1038/77558. [DOI] [PubMed] [Google Scholar]
- 32.Kirn D. Oncolytic virotherapy for cancer with the adenovirus dl1520 (Onyx-015): results of phase I and II trials. Expert Opin Biol Ther. 2001;1:525–538. doi: 10.1517/14712598.1.3.525. [DOI] [PubMed] [Google Scholar]
- 33.Nemunaitis J, Khuri F, Ganly I, Arseneau J, Posner M, Vokes E, et al. Phase II trial of intratumoral administration of ONYX-015, a replication-selective adenovirus, in patients with refractory head and neck cancer. J Clin Oncol. 2001;19:289–298. doi: 10.1200/JCO.2001.19.2.289. [DOI] [PubMed] [Google Scholar]
- 34.Queluz TT, Brunda M, Vladutiu AO, Brentjens JR, Andres G. Morphological basis of pulmonary edema in mice with cytokine-induced vascular leak syndrome. Exp Lung Res. 1991;17:1095–1108. doi: 10.3109/01902149109064337. [DOI] [PubMed] [Google Scholar]
- 35.Rafi AQ, Zeytun A, Bradley MJ, Sponenberg DP, Grayson RL, Nagarkatti M, et al. Evidence for the involvement of Fas ligand and perforin in the induction of vascular leak syndrome. J Immunol. 1998;161:3077–3086. [PubMed] [Google Scholar]
- 36.McKallip RJ, Fisher M, Do Y, Szakal AK, Gunthert U, Nagarkatti PS, et al. Targeted deletion of CD44v7 exon leads to decreased endothelial cell injury but not tumor cell killing mediated by interleukin-2-activated cytolytic lymphocytes. J Biol Chem. 2003;278:43818–43830. doi: 10.1074/jbc.M304467200. [DOI] [PubMed] [Google Scholar]
- 37.Daniels GA, Sanchez-Perez L, Diaz RM, Kottke T, Thompson J, Lai M, et al. A simple method to cure established tumors by inflammatory killing of normal cells. Nat Biotechnol. 2004;22:1125–1132. doi: 10.1038/nbt1007. [DOI] [PubMed] [Google Scholar]
- 38.Brown E, McKee T, diTomaso E, Pluen A, Seed B, Boucher Y, et al. Dynamic imaging of collagen and its modulation in tumors in vivo using second-harmonic generation. Nat Med. 2003;9:796–800. doi: 10.1038/nm879. [DOI] [PubMed] [Google Scholar]
- 39.Cheng J, Sauthoff H, Huang Y, Kutler DI, Bajwa S, Rom WN, et al. Human matrix metalloproteinase-8 gene delivery increases the oncolytic activity of a replicating adenovirus. Mol Ther. 2007;15:1982–1990. doi: 10.1038/sj.mt.6300264. [DOI] [PubMed] [Google Scholar]
- 40.Fingleton B. Matrix metalloproteinase inhibitors for cancer therapy: the current situation and future prospects. Expert Opin Ther Targets. 2003;7:385–397. doi: 10.1517/14728222.7.3.385. [DOI] [PubMed] [Google Scholar]
- 41.Overall CM, Kleifeld O. Tumour microenvironment—opinion: validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat Rev Cancer. 2006;6:227–239. doi: 10.1038/nrc1821. [DOI] [PubMed] [Google Scholar]
- 42.Johnson JE, Nasar F, Coleman JW, Price RE, Javadian A, Draper K, et al. Neurovirulence properties of recombinant vesicular stomatitis virus vectors in non-human primates. Virology. 2006;360:36–49. doi: 10.1016/j.virol.2006.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lichty BD, Stojdl DF, Taylor RA, Miller L, Frenkel I, Atkins H, et al. Vesicular stomatitis virus: a potential therapeutic virus for the treatment of hematologic malignancy. Hum Gene Ther. 2004;15:821–831. doi: 10.1089/hum.2004.15.821. [DOI] [PubMed] [Google Scholar]
- 44.Kim MH, Kitson RP, Albertsson P, Nannmark U, Basse PH, Kuppen PJ, et al. Secreted and membrane-associated matrix metalloproteinases of IL-2-activated NK cells and their inhibitors. J Immunol. 2000;164:5883–5889. doi: 10.4049/jimmunol.164.11.5883. [DOI] [PubMed] [Google Scholar]
- 45.Ishida Y, Migita K, Izumi Y, Nakao K, Ida H, Kawakami A, et al. The role of IL-18 in the modulation of matrix metalloproteinases and migration of human natural killer (NK) cells. FEBS Lett. 2004;569:156–160. doi: 10.1016/j.febslet.2004.05.039. [DOI] [PubMed] [Google Scholar]
- 46.Hu J, Van den Steen PE, Sang QX, Opdenakker G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat Rev Drug Discov. 2007;6:480–498. doi: 10.1038/nrd2308. [DOI] [PubMed] [Google Scholar]
- 47.Diaz RM, Galivo F, Kottke T, Wongthida P, Qiao J, Thompson J, et al. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res. 2007;67:2840–2848. doi: 10.1158/0008-5472.CAN-06-3974. [DOI] [PubMed] [Google Scholar]
- 48.Harrington K, Alvarez-Vallina L, Crittenden M, Gough M, Chong H, Diaz RM, et al. Cells as vehicles for cancer gene therapy: the missing link between targeted vectors and systemic delivery? Hum Gene Ther. 2002;13:1263–1280. doi: 10.1089/104303402760128504. [DOI] [PubMed] [Google Scholar]
- 49.Coligan JE, Kruisbeek AM, Margulies DH, Shevach EM, Strober W. Current Protocols in Immunology. Wiley and Sons; Hoboken, NJ: 1998. [Google Scholar]
- 50.Altman DG. Practical Statistics for Medical Research. Chapman Hall; London, UK: 1991. Analysis of survival times; pp. 365–395. [Google Scholar]