

Abstract
Amphetamine (AMPH) and its derivatives are regularly used in the treatment of a wide array of disorders such as attention deficit hyperactivity disorder (ADHD), obesity, traumatic brain injury, and narcolepsy1–6. Despite the important medicinal role for AMPH, it is more widely known for its psychostimulant and addictive properties as a drug of abuse. The primary molecular targets of AMPH are both the vesicular monoamine transporters (VMATs) and plasma membrane monoamine—dopamine (DA), norepinephrine (NE), and serotonin (5-HT)—transporters. The rewarding and addicting properties of AMPH rely on its ability to act as a substrate for these transporters and ultimately increase extracellular levels of monoamines. AMPH achieves this elevation in extracellular levels of neurotransmitter by inducing synaptic vesicle depletion, which increases intracellular monoamine levels, and also by promoting reverse transport (efflux) through plasma membrane monoamine transporters7–13. This review will focus on two important aspects of AMPH-induced regulation of the plasma membrane monoamine transporters—transporter mediated monoamine efflux and transporter trafficking.
Monoamine Transporter Structure and Function
The monoamine transporters—dopamine, norepinephrine, and serotonin transporters (DAT, NET, and SERT respectively)—belong to the SLC6 gene family of Na+/Cl− dependent transporters that are critical for regulating extracellular levels of neurotransmitters. These transporters rely mainly on the co-transport of Na+ down its electrochemical gradient to facilitate the uptake of biogenic amines from the inter- and extrasynaptic space. This transporter mediated re-uptake controls both the duration and the intensity of monoamine signaling at the synapse and is hypothesized to occur via an alternating access mechanism14–16. This model of transporter function suggests that substrate and Na+ binding trigger conformational changes that shift the transporter from an “outward-facing” conformation, in which the substrate is exposed extracellularly, to an “inward-facing” conformation where the substrate is exposed to the intracellular milieu15–18. This mechanism enables monoamine transporters to accumulate neurotransmitters back into the intracellular compartment after vesicular release in order to ensure both appropriate regulation and maintenance of synaptic signaling.
Topological predictions and experimental data to date indicate that the monoamine transporters have 12 transmembrane domains (TMD) with intracellular amino and carboxy termini, and these predictions have been confirmed by the crystal structure of the bacterial leucine transporter (LeuT), a close bacterial homolog of the neurotransmitter tansporters19–23. Numerous putative phosphorylation sites and binding domains have been identified within the intracellular domains of the various monoamine transporters. These domains are considered vital for transporter regulation, especially AMPH-induced reverse transport24–26. Another important region is the large extracellular domain, located between TMD3 and TMD4, that is post-translationally modified in order to ensure appropriate targeting of the transporter to the surface27.
Original Model for Transporter Mediated Monoamine Efflux: Facilitated Exchange Diffusion
The molecular mechanism underlying AMPH action remained a mystery until the late 1950’s when the work of Burn and Rand revealed that AMPH acts by “releasing a noradrenaline-like substance”10. Thus the foundation of the field was established, and since then numerous studies have focused intently on discovering the detailed mechanism behind AMPH’s ability to induce monoamine release into the extracellular milieu. Investigations following the work of Burn and Rand implicated both vesicular and plasma membrane monoamine transporters as important conduits for monoamine release. This review, however, will focus on efforts surrounding the plasma membrane monoamine transporters. The reader is directed to our substantial review for a thorough discussion of vesicular monoamine transporter contribution to AMPH-mediated monoamine release1.
Early evidence demonstrated that AMPH-like drugs act as substrates for monoamine transporters and that AMPH-induced monoamine release could be blocked by uptake inhibitors such as cocaine and nomifensine11,13,28–32. In tandem with these studies, Fischer and Cho proposed the facilitated exchange diffusion model as a model for AMPH-induced monoamine release via DAT33,34. Fischer and Cho hypothesized that AMPH is transported as a substrate into the cell via DAT which subsequently results in the counter transport of DA extracellularly. Given that AMPH serves as a substrate for DAT, its transport into the cell increases the number of transporters in the inward facing conformation, and thus increases the probability that intracellular DA will bind to DAT and induce reverse transport. Evidence in support of this model of AMPH-induced efflux demonstrates that AMPH accumulation in rat synaptosomes is saturable, temperature-dependent, and ouabain-sensitive, implicating an active transport mechanism for AMPH35. Additional evidence for an active transport mechanism has been supported by several electrophysiology studies illustrating AMPH’s ability to generate DA-like transporter associated currents36,37.
Since the introduction of facilitated exchange diffusion in 1979, new experimental results have emerged that challenge this model. For example, direct intracellular injections of AMPH into the giant DA neuron of Planorbis corneus, the pond snail, induce reverse transport despite the fact that AMPH has not been taken up via the transporter13. Furthermore, AMPH increases intracellular Na+ concentration and an increase in intracellular Na+ is sufficient to drive DA efflux even in the absence of extracellular AMPH38. In fact, numerous studies to date on AMPH-induced changes in uptake, charge transfer, and efflux all support the notion that the influx of extracellular sodium ions via the transporter triggers reverse transport28,37,39–42. Other experiments, utilizing concatemers of SERT (AMPH sensitive) and GAT (AMPH insensitive) reveal that AMPH-induced efflux also depends upon the oligomeric nature of the monoamine transporters43. Finally, additional studies have revealed that AMPH can cause DAT-mediated DA efflux by a process that results in rapid bursts of DA efflux through a channel-like mode of DAT, a process that is independent from the slow exchange-like mechanism44. This was demonstrated in outside-out patches from both heterologous cells stably expressing DAT and from dopaminergic neurons. Interestingly, this channel-like mode of DA release is approximately equivalent to a quantum of DA release from synaptic vesicle fusion. Therefore, this channel-like burst mode may actually influence the synaptic action and psychostimulant properties of AMPH. These studies suggest that while facilitated exchange diffusion may contribute to AMPH-mediated monoamine release, it cannot account for all experimental observations to date (Figure 1).
Figure 1. Schematic Representation of Transporter Mediated Monoamine Efflux.
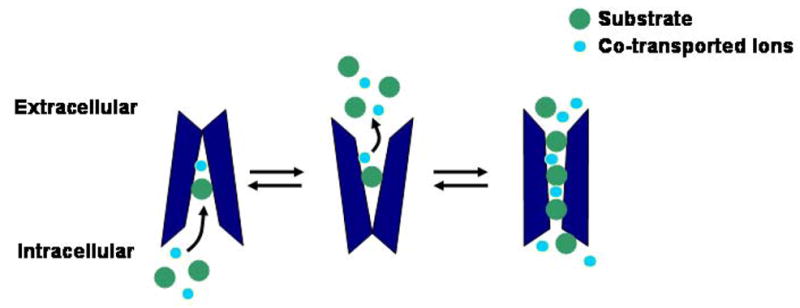
Transporters in the inward facing conformation are capable of mediating monoamine efflux by binding substrate and co-transported ions. Transporter reversal can be enhanced under certain conditions such as in the presence of AMPH or increased intracellular Na+. In addition to the slow exchange-like mechanism of efflux, AMPH can also induce efflux through a channel-like mode of the transporter.
Regulation of Efflux by Second Messenger Systems
While the model of AMPH-induced efflux evolved to include not only facilitated exchange diffusion but also channel-like modes of the transporter, its development is incomplete without considering regulation by second messenger systems. As mentioned previously, numerous putative phosphorylation sites for various protein kinases have been identified within the intracellular regions of the monoamine transporters. In fact, several studies have demonstrated that DAT function is heavily regulated by a plethora of protein kinases24,45–47. As evidence emerged that AMPH is capable of increasing protein kinase C (PKC) activity in vivo, the possibility that kinase regulation may impact AMPH’s ability to induce transporter-mediated monoamine efflux became enticing48. Soon thereafter, strong evidence for the involvement of PKC in AMPH-induced DAT-mediated DA efflux appeared in experiments that utilized specific PKC inhibitors to prevent AMPH-stimulated DA release altogether49,50. A similar role for PKC was also established for AMPH regulation of NET efflux in undifferentiated PC12 cells51. Not surprisingly, these experiments also revealed a requirement for intracellular Ca2+ in AMPH-induced NET efflux, along with the necessity for PKC activity. In addition to a role for intracellular Ca2+, the effects of repeated AMPH exposure on NET were shown to depend on N-type and L-type Ca2+ channel activity52. These studies were further extended by research that demonstrated the ability of intracellular Ca2+ to regulate both AMPH-induced DAT currents and efflux53. Finally, recent studies investigating PKC’s involvement in AMPH mediated DA release have provided evidence for the importance of an actual physical association of PKCβII with DAT in the rat striatum54.
Considering the wealth of data supporting a role for PKC in AMPH stimulated neurotransmitter release, the finding that PKC activation leads to N-terminal phosphorylation of DAT in the rat striatum is not surprising55. Nevertheless, a DAT mutant lacking the first 22 amino acids of the N-terminus, which eliminates 32P incorporation in response to PKC, shows no significant deficits in uptake, inhibitor binding, internalization, or oligomerization24. Given the substantial evidence for PKC involvement in AMPH-induced transporter efflux, Galli and coworkers chose to investigate the role of the N-terminus in regulating DAT-mediated DA efflux by using the 22 amino acid deletion mutant. The study revealed an 80% reduction in AMPH-induced DA efflux in the mutant DAT25. Additionally, they demonstrated that mutation of 5 N-terminal serine residues to alanine residues (S/A) produced an identical phenotype to that of the 22 amino acid deletion DAT mutant. Furthermore, mutation of the same five residues to aspartate (S/D) restored AMPH-induced DA efflux to normal levels. From this study, the authors proposed a novel model for AMPH-induced regulation of DAT efflux that implies a role for N-terminal phosphorylation in shifting DAT from a “reluctant” to a more “willing” state for efflux.
While an abundance of evidence insinuates a role for phosphorylation via PKC in AMPH-induced DAT efflux, additional data implicates the involvement of other kinases in this complex process56,57. In fact, recent data indicates that Ca2+/calmodulin-dependent protein kinase α (CaMKIIα) is a key component of AMPH-induced regulation of DAT efflux26. In this study, the researchers clearly demonstrated that inactivation or inhibition of CaMKIIα dramatically reduced AMPH-induced DA efflux. Furthermore, a physical association between CaMKIIα and the DAT C-terminus was observed, and subsequent disruption of this association was sufficient to diminish AMPH-stimulated DA release. Thus, research clearly indicates a role for both PKC and CaMKIIα signaling in AMPH-induced DAT mediated DA efflux. Whether or not these two signaling pathways contribute to efflux in parallel or sequentially, however, has yet to be determined. Interestingly, recent research focused on the role of CaMKII in AMPH-mediated DA efflux has identified syntaxin1A (SYN1A) as an important link between CaMKII signaling and transporter reversal58. SYN1A is a SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) that is critical for synaptic vesicle release. In addition to its role in vesicular fusion, SYN1A also interacts with and regulates numerous transmembrane proteins including ion channels and importantly, neurotransmitter transporters. In fact, DAT/SYN1A and NET/SYN1A associations have been shown to increase in response to AMPH, and evidence now indicates that this enhancement in SYN1A association is vital for AMPH-mediated DA efflux58,59. Interestingly, SYN1A interaction with NET and DAT has also been shown to influence transporter channel-like activity, a mode of the transporter that is important for AMPH-induced efflux60,61. By utilizing pharmacological and peptide inhibitors of CaMKII, experiments have clearly established that the increase in SYN1A/DAT association in response to AMPH requires CaMKII activity. From these findings, a model for AMPH-induced DA efflux can be formulated, whereby the binding of CaMKIIα to the C-terminus of DAT facilitates the phosphorylation of the N-terminus and the binding of SYN1A, promoting the shift of DAT towards a ‘willing’ state for AMPH-induced DA efflux (Figure 2).
Figure 2. AMPH-Induced DAT Efflux is Regulated by Second Messengers.
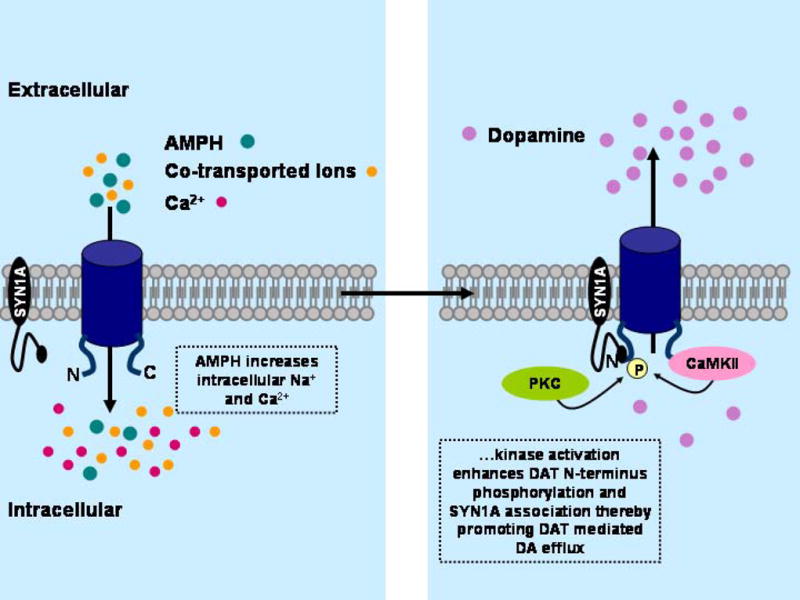
A model of AMPH-induced regulation of DAT mediated DA efflux via intracellular signaling pathways. AMPH transport via DAT into the intracellular milieu results in an increase in both intracellular Na+ and Ca2+ levels. As a consequence, PKC and CaMKII activation initiates both phosphorylation of the N-terminus of DAT and enhancement of DAT/SYN1A association, shifting the transporter from a ‘reluctant’ to a ‘willing’ state for efflux.
AMPH-Induced Trafficking of the Dopamine Transporter
Unlike AMPH-induced neurotransmitter efflux, a phenomenon recognized in the late 1950’s, another aspect of AMPH regulation of monoamine transporters did not emerge from the literature until the late 1990’s. Fleckenstein and coworkers hypothesized that AMPH not only induces transporter efflux, but also may regulate transporter surface expression levels based on their observation that in rats a single high dose injection of AMPH results in a decrease in DAT function one hour later 62. Since then, AMPH-induced regulation of DAT trafficking has been confirmed by numerous studies, and the investigation into the mechanisms underlying this phenomenon has become an area of intense research63–66.
The first comprehensive demonstration of AMPH-induced trafficking of DAT in heterologous systems surfaced a few years after Fleckenstein’s original proposal. In this study, acute treatment with the DAT substrates AMPH and DA not only reduced [3H]DA uptake and AMPH-induced currents, but also clearly decreased DAT cell surface expression63. By utilizing a dominant negative mutant of dynamin I (K44A) to prevent substrate-induced trafficking, the authors also provided evidence to suggest that AMPH-stimulated DAT endocytosis occurs via a dynamin dependent pathway. Following these experiments, a series of investigations verified these results in other heterologous systems (Xenopus oocytes), rat synaptosomal preparations, and finally, indirectly, in vivo via high speed chronoamperometry65,67–70. Interestingly, application of DAT inhibitors such as cocaine, mazindol, and nomifensine is sufficient to prevent the AMPH-induced DAT trafficking, implying that transport of AMPH into the cell may be an important component of this regulation. To address this hypothesis, a mutant DAT (Y335A) capable of substrate binding but impaired in substrate transport, was exposed to AMPH and analyzed for redistribution from the cell surface to the cytosol. Interestingly, extracellular AMPH application did not induce internalization of the uptake-impaired DAT, but when applied directly into the intracellular milieu, AMPH was sufficient for inducing trafficking of the mutant66. From this, the researchers concluded that while the DAT transport cycle is unnecessary for AMPH-induced DAT trafficking, an increase in intracellular AMPH is an essential component of this regulation. An important caveat to consider in these studies is the timing of transporter cell surface redistribution in response to AMPH application. Indeed, recent studies point out that AMPH-induced trafficking of DAT is dependent upon the time of AMPH exposure. For example, a rapid enhancement of DAT surface expression occurs within seconds of AMPH application and diminishes by 2.5 minutes implying that AMPH’s regulation of transporter trafficking differs with respect to acute versus long lasting effects71.
As AMPH-induced DAT trafficking became well established, researchers shifted their focus towards identifying underlying key components of the phenomenon. Considering the wealth of evidence supporting a role for AMPH-induced PKC activation in transporter efflux, the idea that PKC activity may also be involved with transporter trafficking seemed plausible. In fact, numerous studies had already demonstrated that PKC activation leads to the rapid redistribution of DAT from the cell surface in both heterologous and neuronal systems24,47,72–74 However, recent results show that while AMPH application induces N-terminal phosphorylation of DAT via PKC, prevention of this phosphorylation by mutation does not deter AMPH-induced DAT trafficking24,75. Furthermore, more recent studies have demonstrated that residues required for PKC-induced internalization are not critical for AMPH-triggered DAT sequestration, and PKC inhibition also failed to inhibit AMPH-induced DAT redistrubtion76,77. Therefore, while experiments to date clearly implicate a role for PKC in AMPH-induced regulation of DAT efflux, it does not appear to be involved in AMPH-induced transporter trafficking. Interestingly, new work from Vaughan and co-workers indicates that PKC induced DAT regulation may differ depending on the membrane localization of the transporter78. In this study the authors, demonstrate that PKC-stimulated phosphorylation of DAT occurs to a significantly higher level in lipid raft populations of DAT compared to non-lipid raft populations. In fact, the PKC-triggered DAT internalization happens primarily from non-raft population. Thus, PKC regulation of DAT depends heavily upon the discrete membrane localization of the transporter. Given AMPH’s dual effect on transporters, one can imagine a scenario where non-raft populations are primarily responsible for AMPH-induced DAT internalization whereas raft populations may primarily constitute transporters-mediating monoamine efflux. Whether membrane localization is important or not for AMPH-induced efflux and trafficking has yet to be determined, but it will be an interesting question for future studies. In addition to PKC, other kinases have also been implicated in AMPH-induced DAT trafficking. For example, AMPH application results in a time-dependent increase in CaMKII activity which is required for DAT trafficking79. Importantly, this CaMKII activation inhibits an important kinase in the insulin signaling pathway, Akt. This study suggests that insulin signaling is involved in DAT trafficking and as a consequence may impact DA homeostasis. Thus, the current model of AMPH-induced DAT internalization proposes an intersection of AMPH-stimulated signaling and the insulin signaling pathway.
The notion that insulin signaling may be involved in the regulation of DA clearance began with studies that revealed striking alterations in the dopaminergic system of rodents rendered diabetic through streptozotocin (STZ) treatment, which results in necrosis of insulin producing pancreatic β cells80–82. Additionally, tyrosine kinase inhibitors, which block the receptors activated by insulin and insulin-like growth factor were shown to reduce DA clearance due to a decrease in the surface expression levels of DAT83. Concurrently, other investigators expanded these results by illustrating that inhibition of downstream components of the insulin signaling pathway, such as phosphatidylinositol 3-kinase (PI3K) and Akt, also dramatically reduced DA clearance and surface expression of DAT46,84. Thus, the idea that basal insulin signaling is critical for the appropriate maintenance of DA clearance via DAT became firmly established. Given the importance of insulin signaling for maintaining DAT at the plasma membrane, researchers quickly realized the ability of insulin signaling to impact AMPH action in the brain. For example, the reinforcing properties of AMPH are tremendously diminished in the STZ model of diabetes, as demonstrated by the AMPH self-administration paradigm85. Additionally, selective inhibition of PI3K via LY294002 results in a dramatic reduction in AMPH’s ability to elicit DAT-mediated DA efflux in heterologous cells, dopaminergic neurons, and in vivo within the striatum of rats as measured by both in vivo voltammetry and functional magnetic resonance imaging70,86. These data suggest that kinases linked to both glucose homeostasis and food intake regulation are also capable of regulating the reward pathways in the brain that are targeted by psychostimulants such as AMPH.
AMPH-Induced Trafficking of the Norepinephrine Transporter
While evidence for AMPH-induced internalization of DAT has accumulated since the 1990’s, experiments supporting a similar phenomenon with respect to NET did not appear until the 2000’s59,87. The first studies, from Ordway and colleagues, which investigated the effects of chronic AMPH exposure on NET revealed that long term AMPH exposure reduces NET expression in both a time and concentration dependent manner87. Furthermore, a study from 2007 demonstrated that, like DAT, acute AMPH application stimulated a slow, significant reduction in surface levels of NET in a catecholaminergic cell line59. While these results come as no surprise, the study also revealed novel aspects of AMPH-induced regulation of the transporter. First, the authors clearly demonstrated that this process is Ca2+ dependent by utilizing BAPTA-AM and Cd2+ to diminish intracellular Ca2+ levels and prevent AMPH-stimulated NET internalization. Furthermore, inhibition of CaMKII via KN93 also prevented AMPH-induced NET trafficking, implying that this process for NET is both Ca2+ and CaMKII dependent just as it is for DAT. Finally, the study of an N-terminal deletion mutant (hNETΔ28–47) also indicated that AMPH-stimulated regulation of NET internalization may be mediated through the N-terminus59. Interestingly, whether a similar region is critical for AMPH-induced DAT internalization has yet to be determined. In comparison to the wealth of research dedicated to determining how AMPH regulates both trafficking and efflux of DAT, knowledge related to AMPH-induced regulation of NET is rather limited. Despite this gap in the field, much research has been done on β-PMA induced downregulation of NET and evidence supports a role for lipid rafts in NET internalization88,89. Perhaps, similar mechanisms will be elucidated for AMPH-mediated NET trafficking which would suggest a divergence in the regulation of NET compared to DAT given the clathrin dependence of AMPH-induced DAT trafficking. Thus, much research is still needed in the field of AMPH-induced NET internalization and efflux. Many important questions remain unanswered such as which compartment does the transporter travel through during AMPH-stimulated trafficking and what intracellular signals and modifications of NET are required for this trafficking phenomenon? Furthermore, does the insulin signaling pathway play any role in the ability of AMPH to induce NET mediated NE efflux and NET trafficking?
Concluding Remarks
Since the first clues behind AMPH’s mechanism of action began to emerge as early as the late 1950’s, this field of research has seen an exponential amount of growth. After almost 50 years of investigation, the original model for AMPH-induced monoamine efflux has evolved from its simplest form of facilitated exchange diffusion to a multifaceted mechanism that requires not only exchange diffusion and channel-like modes of release but also regulation by second messenger systems. Perhaps, even more remarkable than the transformation of the efflux field, is the discovery of a second mechanism of action for AMPH altogether—transporter trafficking. Despite the advancements over the last few years, both the broad implications and the intricate details of AMPH’s actions continue to elude us. For instance, how does AMPH’s ability to dynamically regulate monoamine transporter membrane expression contribute to its psychostimulant and addictive properties? Which intracellular signaling pathways are critical for AMPH-induced regulation of transporter efflux and trafficking, and how do they differ? Research aimed at addressing these types of questions promises to bring us one step closer to comprehending the basis of not only AMPH abuse and addiction but also its role as a treatment for various pathological conditions. As our understanding of AMPH action continues to progress, so too will our comprehension of monoaminergic regulation in general. For example, recent work involving a DAT coding variant associated with ADHD that effluxes DA as if it were exposed continuously to AMPH, has been shown to have aberrant regulation under normal conditions that closely parallels regulation of wild-type DAT by AMPH90. Thus, uncovering the secrets of AMPH-mediated monoamine transporter regulation promises to enhance our capacity to generate novel therapeutic strategies for treating drug abuse as well as disorders associated with monoaminergic dysfunction such as depression and ADHD.
References
- 1.Sulzer D, Sonders MS, Poulsen NW, Galli A. Mechanisms of neurotransmitter release by amphetamines: a review. Prog Neurobiol. 2005;75:406–33. doi: 10.1016/j.pneurobio.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 2.Prinzmetal M, Bloomberg W. Use of benzedrine for the treatment of narcolepsy. J Am Med Assoc. 1935;105:2051–2054. [Google Scholar]
- 3.Olfson M, Marcus SC, Weissman MM, Jensen PS. National trends in the use of psychotropic medications by children. J Am Acad Child Adolesc Psychiatry. 2002;41:514–21. doi: 10.1097/00004583-200205000-00008. [DOI] [PubMed] [Google Scholar]
- 4.Knutson B, et al. Amphetamine modulates human incentive processing. Neuron. 2004;43:261–9. doi: 10.1016/j.neuron.2004.06.030. [DOI] [PubMed] [Google Scholar]
- 5.Fleckenstein AE, Volz TJ, Riddle EL, Gibb JW, Hanson GR. New insights into the mechanism of action of amphetamines. Annu Rev Pharmacol Toxicol. 2007;47:681–98. doi: 10.1146/annurev.pharmtox.47.120505.105140. [DOI] [PubMed] [Google Scholar]
- 6.Martinsson L, Eksborg S. Drugs for stroke recovery: the example of amphetamines. Drugs Aging. 2004;21:67–79. doi: 10.2165/00002512-200421020-00001. [DOI] [PubMed] [Google Scholar]
- 7.Kirshner N. Uptake of catecholamines by a particulate fraction of the adrenal medulla. J Biol Chem. 1962;237:2311–7. [PubMed] [Google Scholar]
- 8.Carlsson A, Hillarp NA, Waldeck B. A Mg-ATP dependent storage mechanism in the amine granules of the adrenal medulla. Med Exp Int J Exp Med. 1962;6:47–53. [PubMed] [Google Scholar]
- 9.Fon EA, et al. Vesicular transport regulates monoamine storage and release but is not essential for amphetamine action. Neuron. 1997;19:1271–83. doi: 10.1016/s0896-6273(00)80418-3. [DOI] [PubMed] [Google Scholar]
- 10.Burn JH, Rand MJ. The action of sympathomimetic amines in animals treated with reserpine. J Physiol. 1958;144:314–36. doi: 10.1113/jphysiol.1958.sp006104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jones SR, Gainetdinov RR, Wightman RM, Caron MG. Mechanisms of amphetamine action revealed in mice lacking the dopamine transporter. Journal of Neuroscience. 1998;18:1979–86. doi: 10.1523/JNEUROSCI.18-06-01979.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ritz MC, Lamb RJ, Goldberg SR, Kuhar MJ. Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science. 1987;237:1219–23. doi: 10.1126/science.2820058. [DOI] [PubMed] [Google Scholar]
- 13.Sulzer D, et al. Amphetamine redistributes dopamine from synaptic vesicles to the cytosol and promotes reverse transport. Journal of Neuroscience. 1995;15:4102–8. doi: 10.1523/JNEUROSCI.15-05-04102.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Axelrod J. The Metabolism, Storage, and Release of Catecholamines. Recent Prog Horm Res. 1965;21:597–622. [PubMed] [Google Scholar]
- 15.Jardetzky O. Simple allosteric model for membrane pumps. Nature. 1966;211:969–70. doi: 10.1038/211969a0. [DOI] [PubMed] [Google Scholar]
- 16.Forrest LR, et al. Mechanism for alternating access in neurotransmitter transporters. Proc Natl Acad Sci U S A. 2008;105:10338–43. doi: 10.1073/pnas.0804659105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Erreger K, Grewer C, Javitch JA, Galli A. Currents in response to rapid concentration jumps of amphetamine uncover novel aspects of human dopamine transporter function. J Neurosci. 2008;28:976–89. doi: 10.1523/JNEUROSCI.2796-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi L, Quick M, Zhao Y, Weinstein H, Javitch JA. The mechanism of a neurotransmitter:sodium symporter--inward release of Na+ and substrate is triggered by substrate in a second binding site. Mol Cell. 2008;30:667–77. doi: 10.1016/j.molcel.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl--dependent neurotransmitter transporters. Nature. 2005;437:215–23. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- 20.Bruss M, Hammermann R, Brimijoin S, Bonisch H. Antipeptide antibodies confirm the topology of the human norepinephrine transporter. J Biol Chem. 1995;270:9197–201. doi: 10.1074/jbc.270.16.9197. [DOI] [PubMed] [Google Scholar]
- 21.Hersch SM, Yi H, Heilman CJ, Edwards RH, Levey AI. Subcellular localization and molecular topology of the dopamine transporter in the striatum and substantia nigra. J Comp Neurol. 1997;388:211–27. [PubMed] [Google Scholar]
- 22.Chen JG, Liu-Chen S, Rudnick G. Determination of external loop topology in the serotonin transporter by site-directed chemical labeling. J Biol Chem. 1998;273:12675–81. doi: 10.1074/jbc.273.20.12675. [DOI] [PubMed] [Google Scholar]
- 23.Androutsellis-Theotokis A, Rudnick G. Accessibility and conformational coupling in serotonin transporter predicted internal domains. J Neurosci. 2002;22:8370–8. doi: 10.1523/JNEUROSCI.22-19-08370.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Granas C, Ferrer J, Loland CJ, Javitch JA, Gether U. N-terminal truncation of the dopamine transporter abolishes phorbol ester- and substance P receptor-stimulated phosphorylation without impairing transporter internalization. J Biol Chem. 2003;278:4990–5000. doi: 10.1074/jbc.M205058200. [DOI] [PubMed] [Google Scholar]
- 25.Khoshbouei H, et al. N-terminal phosphorylation of the dopamine transporter is required for amphetamine-induced efflux. PLoS Biol. 2004;2:E78. doi: 10.1371/journal.pbio.0020078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fog JU, et al. Calmodulin Kinase II Interacts with the Dopamine Transporter C Terminus to Regulate Amphetamine-Induced Reverse Transport. Neuron. 2006;51:417–29. doi: 10.1016/j.neuron.2006.06.028. [DOI] [PubMed] [Google Scholar]
- 27.Li LB, et al. The role of N-glycosylation in function and surface trafficking of the human dopamine transporter. J Biol Chem. 2004;279:21012–20. doi: 10.1074/jbc.M311972200. [DOI] [PubMed] [Google Scholar]
- 28.Schenk JO. The functioning neuronal transporter for dopamine: kinetic mechanisms and effects of amphetamines, cocaine and methylphenidate. Prog Drug Res. 2002;59:111–31. doi: 10.1007/978-3-0348-8171-5_4. [DOI] [PubMed] [Google Scholar]
- 29.Seiden LS, Sabol KE, Ricaurte GA. Amphetamine: effects on catecholamine systems and behavior. Annu Rev Pharmacol Toxicol. 1993;33:639–77. doi: 10.1146/annurev.pa.33.040193.003231. [DOI] [PubMed] [Google Scholar]
- 30.Raiteri M, Cerrito F, Cervoni AM, Levi G. Dopamine can be released by two mechanisms differentially affected by the dopamine transport inhibitor nomifensine. Journal of Pharmacology & Experimental Therapeutics. 1979;208:195–202. [PubMed] [Google Scholar]
- 31.Heikkila RE, Orlansky H, Mytilineou C, Cohen G. Amphetamine: evaluation of d- and l-isomers as releasing agents and uptake inhibitors for 3H-dopamine and 3H-norepinephrine in slices of rat neostriatum and cerebral cortex. J Pharmacol Exp Ther. 1975;194:47–56. [PubMed] [Google Scholar]
- 32.Parker EM, Cubeddu LX. Comparative effects of amphetamine, phenylethylamine and related drugs on dopamine efflux, dopamine uptake and mazindol binding. J Pharmacol Exp Ther. 1988;245:199–210. [PubMed] [Google Scholar]
- 33.Fischer JF, Cho AK. Chemical release of dopamine from striatal homogenates: evidence for an exchange diffusion model. Journal of Pharmacology & Experimental Therapeutics. 1979;208:203–9. [PubMed] [Google Scholar]
- 34.Burnette WB, et al. Human norepinephrine transporter kinetics using rotating disk electrode voltammetry. Analytical Chemistry. 1996;68:2932–8. doi: 10.1021/ac960022x. [DOI] [PubMed] [Google Scholar]
- 35.Zaczek R, Culp S, De Souza EB. Interactions of [3H]amphetamine with rat brain synaptosomes. II. Active transport. Journal of Pharmacology & Experimental Therapeutics. 1991;257:830–5. [PubMed] [Google Scholar]
- 36.Sonders MS, Zhu SJ, Zahniser NR, Kavanaugh MP, Amara SG. Multiple ionic conductances of the human dopamine transporter: the actions of dopamine and psychostimulants. Journal of Neuroscience. 1997;17:960–74. doi: 10.1523/JNEUROSCI.17-03-00960.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sitte HH, et al. Carrier-mediated release, transport rates, and charge transfer induced by amphetamine, tyramine, and dopamine in mammalian cells transfected with the human dopamine transporter. Journal of Neurochemistry. 1998;71:1289–97. doi: 10.1046/j.1471-4159.1998.71031289.x. [DOI] [PubMed] [Google Scholar]
- 38.Khoshbouei H, Wang H, Lechleiter JD, Javitch JA, Galli A. Amphetamine-induced dopamine efflux. A voltage-sensitive and intracellular Na+-dependent mechanism. J Biol Chem. 2003;278:12070–7. doi: 10.1074/jbc.M212815200. [DOI] [PubMed] [Google Scholar]
- 39.Pifl C, Singer EA. Ion dependence of carrier-mediated release in dopamine or norepinephrine transporter-transfected cells questions the hypothesis of facilitated exchange diffusion. Molecular Pharmacology. 1999;56:1047–54. doi: 10.1124/mol.56.5.1047. [DOI] [PubMed] [Google Scholar]
- 40.Pifl C, Rebernik P, Kattinger A, Reither H. Zn(2+) modulates currents generated by the dopamine transporter: parallel effects on amphetamine-induced charge transfer and release. Neuropharmacology. 2004;46:223–31. doi: 10.1016/j.neuropharm.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 41.Pifl C, Drobny H, Reither H, Hornykiewicz O, Singer EA. Mechanism of the dopamine-releasing actions of amphetamine and cocaine: plasmalemmal dopamine transporter versus vesicular monoamine transporter. Molecular Pharmacology. 1995;47:368–73. [PubMed] [Google Scholar]
- 42.Pifl C, Agneter E, Drobny H, Reither H, Singer EA. Induction by low Na+ or Cl− of cocaine sensitive carrier-mediated efflux of amines from cells transfected with the cloned human catecholamine transporters. British Journal of Pharmacology. 1997;121:205–12. doi: 10.1038/sj.bjp.0701137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seidel S, et al. Amphetamines take two to tango: an oligomer-based counter-transport model of neurotransmitter transport explores the amphetamine action. Mol Pharmacol. 2005;67:140–51. doi: 10.1124/mol.67.1.. [DOI] [PubMed] [Google Scholar]
- 44.Kahlig KM, et al. Amphetamine induces dopamine efflux through a dopamine transporter channel. Proc Natl Acad Sci U S A. 2005;102:3495–500. doi: 10.1073/pnas.0407737102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Melikian HE, Buckley KM. Membrane trafficking regulates the activity of the human dopamine transporter. Journal of Neuroscience. 1999;19:7699–710. doi: 10.1523/JNEUROSCI.19-18-07699.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carvelli L, et al. PI 3-kinase regulation of dopamine uptake. J Neurochem. 2002;81:859–69. doi: 10.1046/j.1471-4159.2002.00892.x. [DOI] [PubMed] [Google Scholar]
- 47.Loder MK, Melikian HE. The Dopamine Transporter Constitutively Internalizes and Recycles in a Protein Kinase C-regulated Manner in Stably Transfected PC12 Cell Lines. J Biol Chem. 2003;278:22168–74. doi: 10.1074/jbc.M301845200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Giambalvo CT. Protein kinase C and dopamine transport--2. Effects of amphetamine in vitro. Neuropharmacology. 1992;31:1211–22. doi: 10.1016/0028-3908(92)90049-u. [DOI] [PubMed] [Google Scholar]
- 49.Kantor L, Gnegy ME. Protein kinase C inhibitors block amphetamine-mediated dopamine release in rat striatal slices. Journal of Pharmacology & Experimental Therapeutics. 1998;284:592–8. [PubMed] [Google Scholar]
- 50.Cowell RM, Kantor L, Hewlett GH, Frey KA, Gnegy ME. Dopamine transporter antagonists block phorbol ester-induced dopamine release and dopamine transporter phosphorylation in striatal synaptosomes. European Journal of Pharmacology. 2000;389:59–65. doi: 10.1016/s0014-2999(99)00828-6. [DOI] [PubMed] [Google Scholar]
- 51.Kantor L, et al. Protein kinase C and intracellular calcium are required for amphetamine- mediated dopamine release via the norepinephrine transporter in undifferentiated PC12 cells. J Pharmacol Exp Ther. 2001;297:1016–24. [PubMed] [Google Scholar]
- 52.Kantor L, Zhang M, Guptaroy B, Park YH, Gnegy ME. Repeated amphetamine couples norepinephrine transporter and calcium channel activities in PC12 cells. J Pharmacol Exp Ther. 2004;311:1044–51. doi: 10.1124/jpet.104.071068. [DOI] [PubMed] [Google Scholar]
- 53.Gnegy ME, et al. Intracellular Ca2+ regulates amphetamine-induced dopamine efflux and currents mediated by the human dopamine transporter. Mol Pharmacol. 2004;66:137–43. doi: 10.1124/mol.66.1.137. [DOI] [PubMed] [Google Scholar]
- 54.Johnson LA, Guptaroy B, Lund D, Shamban S, Gnegy ME. Regulation of amphetamine-stimulated dopamine efflux by protein kinase C beta. J Biol Chem. 2005;280:10914–9. doi: 10.1074/jbc.M413887200. [DOI] [PubMed] [Google Scholar]
- 55.Foster JD, Pananusorn B, Vaughan RA. Dopamine transporters are phosphorylated on N-terminal serines in rat striatum. J Biol Chem. 2002;277:25178–86. doi: 10.1074/jbc.M200294200. [DOI] [PubMed] [Google Scholar]
- 56.Kantor L, Hewlett GH, Gnegy ME. Enhanced amphetamine- and K+-mediated dopamine release in rat striatum after repeated amphetamine: differential requirements for Ca2+- and calmodulin-dependent phosphorylation and synaptic vesicles. Journal of Neuroscience. 1999;19:3801–8. doi: 10.1523/JNEUROSCI.19-10-03801.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pierce RC, Kalivas PW. Repeated cocaine modifies the mechanism by which amphetamine releases dopamine. Journal of Neuroscience. 1997;17:3254–61. doi: 10.1523/JNEUROSCI.17-09-03254.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Binda F, et al. Syntaxin1A Interaction with the Dopamine Transporter Promotes Amphetamine-Induced Dopamine Efflux. Mol Pharmacol. 2008 doi: 10.1124/mol.108.048447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dipace C, Sung U, Binda F, Blakely RD, Galli A. Amphetamine induces a calcium/calmodulin-dependent protein kinase II-dependent reduction in norepinephrine transporter surface expression linked to changes in syntaxin 1A/transporter complexes. Mol Pharmacol. 2007;71:230–9. doi: 10.1124/mol.106.026690. [DOI] [PubMed] [Google Scholar]
- 60.Sung U, et al. A regulated interaction of syntaxin 1A with the antidepressant- sensitive norepinephrine transporter establishes catecholamine clearance capacity. J Neurosci. 2003;23:1697–709. doi: 10.1523/JNEUROSCI.23-05-01697.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carvelli L, Blakely RD, DeFelice LJ. Dopamine transporter/syntaxin 1A interactions regulate transporter channel activity and dopaminergic synaptic transmission. Proc Natl Acad Sci U S A. 2008;105:14192–7. doi: 10.1073/pnas.0802214105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fleckenstein AE, Metzger RR, Wilkins DG, Gibb JW, Hanson GR. Rapid and reversible effects of methamphetamine on dopamine transporters. Journal of Pharmacology & Experimental Therapeutics. 1997;282:834–8. [PubMed] [Google Scholar]
- 63.Saunders C, et al. Amphetamine-induced loss of human dopamine transporter activity: an internalization-dependent and cocaine-sensitive mechanism. Proc Natl Acad Sci U S A. 2000;97:6850–5. doi: 10.1073/pnas.110035297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kahlig KM, Galli A. Regulation of dopamine transporter function and plasma membrane expression by dopamine, amphetamine, and cocaine. Eur J Pharmacol. 2003;479:153–8. doi: 10.1016/j.ejphar.2003.08.065. [DOI] [PubMed] [Google Scholar]
- 65.Kahlig KM, Javitch JA, Galli A. Amphetamine regulation of dopamine transport. Combined measurements of transporter currents and transporter imaging support the endocytosis of an active carrier. J Biol Chem. 2004;279:8966–75. doi: 10.1074/jbc.M303976200. [DOI] [PubMed] [Google Scholar]
- 66.Kahlig KM, et al. Regulation of dopamine transporter trafficking by intracellular amphetamine. Mol Pharmacol. 2006;70:542–8. doi: 10.1124/mol.106.023952. [DOI] [PubMed] [Google Scholar]
- 67.Chi L, Reith ME. Substrate-induced trafficking of the dopamine transporter in heterologously expressing cells and in rat striatal synaptosomal preparations. J Pharmacol Exp Ther. 2003;307:729–36. doi: 10.1124/jpet.103.055095. [DOI] [PubMed] [Google Scholar]
- 68.Gulley JM, Doolen S, Zahniser NR. Brief, repeated exposure to substrates down-regulates dopamine transporter function in Xenopus oocytes in vitro and rat dorsal striatum in vivo. J Neurochem. 2002;83:400–11. doi: 10.1046/j.1471-4159.2002.01133.x. [DOI] [PubMed] [Google Scholar]
- 69.Owens WA, et al. Deficits in dopamine clearance and locomotion in hypoinsulinemic rats unmask novel modulation of dopamine transporters by amphetamine. J Neurochem. 2005 doi: 10.1111/j.1471-4159.2005.03289.x. [DOI] [PubMed] [Google Scholar]
- 70.Williams JM, et al. Hypoinsulinemia regulates amphetamine-induced reverse transport of dopamine. PLoS Biol. 2007;5:2369–78. doi: 10.1371/journal.pbio.0050274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Johnson LA, Furman CA, Zhang M, Guptaroy B, Gnegy ME. Rapid delivery of the dopamine transporter to the plasmalemmal membrane upon amphetamine stimulation. Neuropharmacology. 2005;49:750–8. doi: 10.1016/j.neuropharm.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 72.Pristupa ZB, et al. Protein kinase-mediated bidirectional trafficking and functional regulation of the human dopamine transporter. Synapse. 1998;30:79–87. doi: 10.1002/(SICI)1098-2396(199809)30:1<79::AID-SYN10>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 73.Daniels GM, Amara SG. Regulated trafficking of the human dopamine transporter. Clathrin-mediated internalization and lysosomal degradation in response to phorbol esters. Journal of Biological Chemistry. 1999;274:35794–801. doi: 10.1074/jbc.274.50.35794. [DOI] [PubMed] [Google Scholar]
- 74.Sorkina T, Doolen S, Galperin E, Zahniser NR, Sorkin A. Oligomerization of dopamine transporters visualized in living cells by fluorescence resonance energy transfer microscopy. J Biol Chem. 2003;278:28274–83. doi: 10.1074/jbc.M210652200. [DOI] [PubMed] [Google Scholar]
- 75.Cervinski MA, Foster JD, Vaughan RA. Psychoactive substrates stimulate dopamine transporter phosphorylation and down regulation by cocaine sensitive and protein kinase C dependent mechanisms. J Biol Chem. 2005 doi: 10.1074/jbc.M501969200. [DOI] [PubMed] [Google Scholar]
- 76.Boudanova E, Navaroli DM, Stevens Z, Melikian HE. Dopamine transporter endocytic determinants: Carboxy terminal residues critical for basal and PKC-stimulated internalization. Mol Cell Neurosci. 2008 doi: 10.1016/j.mcn.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Boudanova E, Navaroli DM, Melikian HE. Amphetamine-induced decreases in dopamine transporter surface expression are protein kinase C-independent. Neuropharmacology. 2008;54:605–12. doi: 10.1016/j.neuropharm.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Foster JD, Adkins SD, Lever JR, Vaughan RA. Phorbol ester induced trafficking-independent regulation and enhanced phosphorylation of the dopamine transporter associated with membrane rafts and cholesterol. J Neurochem. 2008;105:1683–99. doi: 10.1111/j.1471-4159.2008.05262.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wei Y, et al. Dopamine transporter activity mediates amphetamine-induced inhibition of Akt through a Ca2+/calmodulin-dependent kinase II-dependent mechanism. Mol Pharmacol. 2007;71:835–42. doi: 10.1124/mol.106.026351. [DOI] [PubMed] [Google Scholar]
- 80.Chu PC, Lin MT, Shian LR, Leu SY. Alterations in physiologic functions and in brain monoamine content in streptozocin-diabetic rats. Diabetes. 1986;35:481–5. doi: 10.2337/diab.35.4.481. [DOI] [PubMed] [Google Scholar]
- 81.Saitoh A, Morita K, Sodeyama M, Kamei J. Effects of the experimental diabetes on dopamine D1 receptor-mediated locomotor-enhancing activity in mice. Pharmacology, Biochemistry & Behavior. 1998;60:161–6. doi: 10.1016/s0091-3057(97)00588-1. [DOI] [PubMed] [Google Scholar]
- 82.Karkanias GB, Morales JC, Li CS. Deficits in reproductive behavior in diabetic female rats are due to hypoinsulinemia rather than hyperglycemia. Horm Behav. 1997;32:19–29. doi: 10.1006/hbeh.1997.1401. [DOI] [PubMed] [Google Scholar]
- 83.Doolen S, Zahniser NR. Protein tyrosine kinase inhibitors alter human dopamine transporter activity in Xenopus oocytes. Journal of Pharmacology & Experimental Therapeutics. 2001;296:931–8. [PubMed] [Google Scholar]
- 84.Garcia BG, et al. Akt is essential for insulin modulation of amphetamine-induced human dopamine transporter cell-surface redistribution. Mol Pharmacol. 2005;68:102–9. doi: 10.1124/mol.104.009092. [DOI] [PubMed] [Google Scholar]
- 85.Galici R, et al. Selective decreases in amphetamine self-administration and regulation of dopamine transporter function in diabetic rats. Neuroendocrinology. 2003;77:132–40. doi: 10.1159/000068650. [DOI] [PubMed] [Google Scholar]
- 86.Lute BJ, et al. PI3K signaling supports amphetamine-induced dopamine efflux. Biochem Biophys Res Commun. 2008;372:656–61. doi: 10.1016/j.bbrc.2008.05.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhu MY, Shamburger S, Li J, Ordway GA. Regulation of the human norepinephrine transporter by cocaine and amphetamine. J Pharmacol Exp Ther. 2000;295:951–9. [PubMed] [Google Scholar]
- 88.Jayanthi LD, Annamalai B, Samuvel DJ, Gether U, Ramamoorthy S. Phosphorylation of the norepinephrine transporter at threonine 258 and serine 259 is linked to protein kinase C-mediated transporter internalization. J Biol Chem. 2006;281:23326–40. doi: 10.1074/jbc.M601156200. [DOI] [PubMed] [Google Scholar]
- 89.Jayanthi LD, Samuvel DJ, Ramamoorthy S. Regulated internalization and phosphorylation of the native norepinephrine transporter in response to phorbol esters. Evidence for localization in lipid rafts and lipid raft-mediated internalization. J Biol Chem. 2004;279:19315–26. doi: 10.1074/jbc.M311172200. [DOI] [PubMed] [Google Scholar]
- 90.Mazei-Robison MS, et al. Anomalous dopamine release associated with a human dopamine transporter coding variant. J Neurosci. 2008;28:7040–6. doi: 10.1523/JNEUROSCI.0473-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]