

Abstract
Although estrogens exert a pronounced protective effect on multiple sclerosis (MS) and its animal model, experimental autoimmune encephalomyelitis (EAE), their therapeutic application has been limited by undesirable side effects thought to be mediated primarily through estradiol binding to intracellular estrogen receptor alpha (iERα). In this study, we found that signaling through the putative membrane estrogen receptor, GPR30, was sufficient to mediate protection against EAE, which was significantly impaired in GPR30 gene-deficient mice. Treatment with G-1, an agonist that selectively activates GPR30 without engagement of the iERs, retained estradiol's ability to protect against clinical and histological EAE without estradiol-associated side effects, deviated cytokine profiles and enhanced suppressive activity of CD4+Foxp3+ Treg cells through a GPR30- and programmed death 1 (PD-1)-dependent mechanism. This study is the first to evaluate the protective effect of GPR30 activation on EAE, and provides a strong foundation for the clinical application of GPR30 agonists such as G-1 in MS.
Keywords: GPR30, G-1, Estrogen, Multiple sclerosis, Experimental Autoimmune Encephalomyelitis, Programmed death 1, T-lymphocyte
INTRODUCTION
Sex steroids and glucocorticoids have long been utilized in the clinic to control allergy, asthma and autoimmune diseases due to their anti-inflammatory effects. Multiple sclerosis (MS) is a debilitating neurological autoimmune disease with higher incidence in women(1). However, MS relapse rates are decreased during late pregnancy(2,3) and treatment with pregnancy levels of estriol reduces CNS lesions(4,5). We demonstrated previously(6,7) that relatively low doses of 17β-estradiol (E2) and estriol (E3) confer potent protection against clinical and histological signs of experimental autoimmune encephalomyelitis (EAE), an animal model of MS. However, clinical application of estrogens, particularly estradiol, in MS is limited by undesirable side effects ranging from the triggering of breast and uterine cancer to the loss of appetite, rapid weight gain and fluid retention. Most “estrogenic” side effects are believed to be mediated by estradiol through classic intracellular estrogen receptors (iERs), particularly ERα (Esr1) (8). Consequently, there is much interest in identifying estrogen-like molecules that exhibit neuroprotective effects in MS without activating classic iERs.
Several studies have demonstrated rapid estrogen signaling that appears to be initiated at the plasma membrane and affects genes that lack the putative estrogen response element(9). Recently, GPR30(10), a G protein-coupled receptor located on the plasma and endoplasmic reticulum membrane that binds estradiol with high affinity, has been recognized as a putative membrane estrogen receptor (mER)(11-13). GPR30 signaling can account for several E2-induced effects such as inhibition of oxidative stress-induced apoptosis and upregulation of NGF in macrophages(12). Recently, we reported the generation of GPR30-deficient mice (GPR30KO)(14) that possesses many similarities to the mouse strain generated independently by Mårtensson et al.(15). Mice homozygous for GPR30KO are viable, fertile, and do not display any gross physical or neurological abnormalities; however, E2-induced thymocyte apoptosis and thymic atrophy were drastically mitigated. In the current study, we evaluated the role of GPR30 in E2-mediated protection against EAE.
MATERIALS AND METHODS
Mice
C57BL/6 (B6) mice were purchased from the animal service at National Cancer Institute (Frederick, MD). The generation of GPR30- (Gper−/−, GPR30KO and the respective wild-type (WT) control mice from the colony were described before(14). PD-1 gene-deficient (PD-1KO) mice were obtained from Dr. Tasuku Honjo at Kyoto University (Kyoto, Japan). GPR30KO mice were generated on the 129Sv background and backcrossed with B6 mice for 6 generations. PD-1KO strains were backcrossed with B6 mice for more than 10 generations. All mice used for experiment were age-matched (eight- to ten-week old) female, and rested for at least 7 days prior to treatment or immunization. Animals were bred (for gene-deficient strains), housed and cared for according to institutional guidelines in the animal resource facility at the Veterans Affairs Medical Center, Portland, OR.
Reagents
Mouse (m) MOG-35-55 peptide (MEVGWYRSPFSRVVHLYRNGK) was synthesized using solid phase techniques and was purified by HPLC at Beckman Institute, Stanford University (Palo Alto, CA). Heat killed Mycobacterium tuberculosis H37RA is a product of DIFCO (Detroit, MI). Pertussis toxin (PTX) was purchased from List Biological Laboratories (Campbell, CA). [3H] thymidine was purchased from Perkin-Elmer (Boston, MA). 17β-estradiol (E2) and corn oil were purchased from Sigma-Aldrich (St. Louis, MO). 2.5 mg/60 day release E2 and vehicle (placebo) pellets were purchased from Innovative Research of America (Sarasota, FL). G-1 powder was purchased from Cayman Chemicals (Ann Arbor, MI) and packaged into slow-release pellets by Innovative Research of America. All fluorescein-labeled antibodies were purchased from eBioscience (San Diego, CA). Trilogy® antigen unmasking reagent was purchased from Cell Marque (Hot Springs, AR). MicroBeads for MACS cell separation were purchased from Miltenyi Biotec (Bergisch Gladbach, Germany). Luminex Bio-Plex mouse cytokine assay kit was purchased from Bio-Rad (Hercules, CA). VECTOR® MOM™ Immunodetection peroxidase kit and Hematoxylin QS were purchased from Vector Laboratories (Burlingame, CA). Monoclonal antibodies SMI32 and SMI312 were purchased from Covance (Princeton, New Jersey). DakoCytomation liquid DAB substrate is a product of DakoCytomation (Carpinteria, CA). Cytoseal™ XYL mounting medium is from Richard-Allan Scientific (Kalamazoo, MI).
Induction of EAE
Mice were inoculated s.c. in the flanks with 0.2 ml of an emulsion containing 200 μg of mMOG-35-55 peptide and an equal volume of complete Freund's adjuvant (CFA) containing 200 μg of heat-killed Mycobacterium tuberculosis H37RA. On the same day and 2 days after the immunization, each mouse was injected i.v. with 75 and 200 ng of pertussis toxin (PTX), respectively. The mice were assessed daily for clinical signs of EAE according to the following scale: 0 = normal, 1 = limp tail or mild hindlimb weakness, 2 = moderate hindlimb weakness or mild ataxia, 3 = moderately severe hindlimb weakness, 4 = severe hindlimb weakness or mild forelimb weakness or moderate ataxia, 5 = paraplegia with no more than moderate forelimb weakness, and 6 = paraplegia with severe forelimb weakness or severe ataxia, moribund condition or dead.
Treatments
To investigate the effect of estrogen and G-1, 3-mm pellets containing various dose of E2 and G-1 were implanted subcutaneously (dorsally) into mice one week before immunization. These pellets are designed to release their contents at a constant rate over 40 or 60 days. Serum levels of E2 were monitored by radioimmunoassay as described previously(6,16). Alternatively, both reagents (0.04 mg/kg/day for E2 and 0.1 mg/kg/day for G-1) were dissolved in vehicle (10% ethanol and 90% corn oil) and administered s.c. daily to mice from one week prior to immunization to the end of the experiment.
Flow cytometry
For membrane staining, 1 million cells were stained at 4°C in the dark with appropriate Ab dilutions in staining buffer (PBS containing 0.5% BSA and 0.02% sodium azide). Intracellular staining for FoxP3 was performed following the protocol recommended by eBioscience. Briefly, 4 million cells were surface stained following standard procedures. After washing, the cells were fixed overnight and washed twice with 0.5 ml of permeabilization buffer. The cells were co-stained for 15 min with FcBlock and IgG-PerCP followed 30 min later with fluorescent-labeled antibodies to FoxP3 or isotype control. The cells were then washed twice with 2 ml of permeabilization buffer and once with 1 ml of staining buffer, and re-suspended in staining buffer. Flow cytometry data were collected on LSRII and FACSCalibur flow cytometers (BD Bioscience), and analyzed using FlowJo software (Tree Star, Ashland. OR). Data represent 50,000-100,000 events, unless otherwise noted.
Lymphocyte proliferation assay
Splenocytes or T cells were harvested and cultured in a 96-well flat-bottom tissue culture plate at 4 × 105 cells/well in stimulation medium in the presence of APC, irradiated (2500 rad) syngeneic thymocytes at a ratio of 1:10 (T: APCs), either with or without mMOG-35-55 peptide at varying concentrations. The cells were incubated for 3 days at 37°C in 7% CO2, and pulsed with 0.5 μCi of [3H]thymidine for the final 18 h of incubation. The cells were harvested onto glass fiber filters, and incorporated radioactivity was measured by a liquid scintillation counter. The cpm values (mean ± SD) were calculated from triplicate wells. Stimulation index (SI) was calculated by dividing the experimental cpm by the control cpm.
Cytokine determination by Luminex kit
Lymph node (LN) and spleen cells were cultured at 4 × 106 cells/well in a 24-well flat-bottom culture plate in stimulation medium (RPMI 1640, 1% sodium pyruvate, 1% l-glutamine, 0.4% 2-β-ME, 10% FBS) with 25 μg/ ml mMOG-35–55 peptide for 48 h. Supernatants were then harvested and stored at −80°C until tested for cytokines. Culture supernatants were assessed for cytokine levels using a Luminex Bio-Plex mouse cytokine assay kit following manufacturer's instructions. The following cytokines were determined in a single assay in three separate experiments: IL-1β, IFN-γ, TNF-α, IL-2, IL-4, IL-5, IL-6, IL-12, IL-13, and IL-17.
Pathology and Immunohistochemistry (IHC)
Intact spinal columns were removed from experimental and control groups of mice. The spinal cords were dissected after fixation in 4% paraformadehyde, dehydrated and embedded in paraffin before sectioning. To examine neuroinflammation, the sections were stained with hematoxylin and eosin (H&E). To examine demyelination, the sections were stained with Luxol fast blue plus periodic acid Schiff (LFB-PAS). IHC was performed as described(6,17). Briefly, spinal cords were fixed in 4% paraformaldehyde (mass/volume in PBS, pH 7.4) at 4°C for at least 48 h. The spinal cords were dissected out from the columns, cut into sections 1–2 mm in length from the sampled thoracic or limbic cords, dehydrated and embedded in paraffin blocks. Then, 10 μm thick sections were cut from paraffin blocks and mounted onto pre-cleaned microscope slides. The sections were dewaxed and rehydrated sequentially by xylene (2 min), gradient ethanol (100%, 95%, 85%, 2 min each) and PBS (5 min), and then cooked (120°C) in antigen unmasking agent Trilogy® for 10min in a pressure steamer. The endogenous peroxidase activity was blocked with 3% hydrogen peroxide in tap water for 5 min. The sections were incubated 1h in a working solution of Mouse IgG blocking reagent from the VECTOR® M.O.M™ Immunodetection peroxidase kit, and then incubated sequentially with primary antibody (SMI312 1:3000 or SMI32 1:1000 diluted in MOM™ diluents) for 30 min, M.O.M™ biotinylated anti-mouse IgG reagent for 10 min, VECTASTAIN® ABC reagent for 5 min, and DakoCytomation liquid DAB substrate until sections turned light brown. The slides were counterstained with hematoxylin for 30–60s to visualize nuclei, washed with tap water, dehydrated, and mounted with Cytoseal™ XYL mounting medium. The sections were analyzed by light microscopy after staining and recorded with a digital camera. The damaged areas were labeled out by hands and traced with Bioquant software (Bioquant, Nashville, TN). The numbers of injured axons were counted by Scion Image (Frederick, MD).
Statistical analysis
Mean values from each experiment were compared statistically. Differences in group daily clinical scores, peak scores and CDI were evaluated by Kruskal-Wallis test followed by Mann-Whitney (unpaired Student's t-test was employed for daily EAE score or CDI if none of the mice were sick in one of the comparison groups); incidence was evaluated by Fisher's exact test; disease onset, uterine, body and femur weights were measured by one-way ANOVA followed by Newman-Kuels test; endogenous steroid levels in serum, cytokine secretion, T cell proliferation and flow cytometric data were compared by unpaired Student's t-test. Data are represented as mean ± standard deviation of the mean (SD). All presented data represent one of 2-4 independent experiments.
RESULTS
GPR30 is sufficient for E2-induced protection against EAE
The contribution of GPR30 to E2-induced protection against EAE was evaluated in age-matched WT C57BL/6, GPR30KO and heterozygous (GPR30+/−) female mice from the same colony. Mice were implanted with E2 (2.5 mg/60 day release) or placebo pellets one week prior to immunization with mouse (m)MOG-35-55 peptide in complete Freund's adjuvant (CFA) with additional pertussis toxin (Ptx) given on days 0 and 2. Serum E2 levels were 1.5-2 ng/ml (Fig. 3). As shown in Fig. 1A and Table I, placebo-treated WT and GPR30KO mice developed severe EAE with onset on Day 11 and peak about Day 15, whereas E2-treated WT mice developed no signs of disease. In contrast, E2-treated GPR30KO and heterozygous mice had delayed onset and were only partially protected from EAE. Pathologically, all the placebo-treated mice had substantial immune cell infiltration and demyelination in the CNS (Fig. 1B), whereas no pathological signs of EAE were found in E2-treated WT mice. In E2-treated GPR30KO mice, however, cellular infiltration and demyelination were visibly present, but at a reduced level. T cell responses to MOG peptide were similarly reduced by E2 treatment in both WT and GPR30KO mice (Fig. 1C), suggesting that GPR30 is not directly linked to suppression of T cell proliferation. Taken together, these results indicate a significant, but not exclusive role for GPR30 in E2-mediated protection against EAE.
Fig. 3. G-1 treatment did not change the serum levels of E2, progesterone or testosterone and only slightly lowered corticosteroid.

Steroid levels were measured in sera collected from placebo-, 3.7 mg G-1- and 2.5 mg E2-treated mice used in clinical experiments shown in Fig. 2a. *P < 0.05 or ***P < 0.001.
Fig. 1. E2-induced protection against EAE is reduced in GPR30KO mice.

Age-matched GPR30KO, WT and heterozygous mice from the same colony were implanted with 2.5 mg/60 day release E2 or placebo pellets one week prior to immunization with mMOG-35-55 peptide plus CFA and PTX. The mice were scored for EAE development and euthanized 25 days after immunization. A. The protective effect of E2 was reduced but not completely abrogated in GPR30KO mice. B. E2 did not prevent CNS infiltration and demyelination in GPR30KO mice. For quantification, 5 mice randomly selected from each group were included, and similar differences were noted consistently in all mice from each group. C. GPR30 was not directly involved in suppression of T cell proliferation to mMOG-35-55 peptides. CPM of control = 7199.5 ± 1080.6 (Spleen) or 5617.1 ± 665.1 (LN). *P < 0.05 or **P < 0.01 as compared to placebo control. The experiment was repeated 3 times with at least 7 mice in each group.
Table I.
Gene disruption of GPR30 weakens the therapeutic effects of E2 on EAEa.
| Genotype | pellet | n | Incidence | Mortality | Onset | Peak | Daily | CDI |
|---|---|---|---|---|---|---|---|---|
| WT | Placebo | 10 | 9/10 | 0/10 | 15.3±2.1 | 4.1±0.9 | 3.4±0.7 | 33.1±16.3 |
| E2 | 10 | 0/10 | 0/10 | - | 0** | 0** | 0** | |
| GPR30KO | Placebo | 7 | 6/7 | 0/7 | 13.4±0.53 | 3.8±1.7 | 3.6±0.9 | 40.1±13.2 |
| E2 | 7 | 4/7 | 0/7 | 18.0±1.4** | 2.5±2.3# | 3.7±1.3 | 17.2±17.4*# | |
| GPR30+/− | Placebo | 10 | 9/10 | 0/10 | 13±2.8 | 3.6±1.6 | 3.5±0.6 | 38.9±31.4 |
| E2 | 10 | 5/10 | 0/10 | 20.0±1.0** | 2.1±2.3*# | 2.6±0.8 | 11.3±13.0**# |
Mice were immunized one week after implantation with 2.5 mg/60 day release E2 pellets. The experiment was concluded 25 days after immunization.
P < 0.05 or
P < 0.01 as compared to placebo control,
P < 0.05 as compared to E2-treated WT.
Activation of GPR30 confers protection against clinical EAE
Although the above results indicated an indispensible role for GPR30 in E2-mediated protection against EAE, we sought to determine whether activation of GPR30 alone could provide protection. We thus compared the efficacy of G-1, the only specific GPR30 agonist available, and E2 in protecting wild-type C57BL/6 mice against EAE. Initially, G-1 (1.8, 0.1 or 0.01 mg/40 day release pellets), E2 (2.5, 0.1 or 0.025 mg/60 day release pellets) or placebo were administered to mice 7 days prior to induction of EAE. Administration of either 1.8 mg/40 day release G-1 or the molar equivalent level of 2.5 mg/60 day release E2 pellets completely protected the mice from clinical EAE, with lower doses of G-1 being less effective than E2 (Fig. 2A and Table II). Subcutaneous injections of G-1 (20 ug/mouse/day in corn oil) and E2 (1 ug/mouse/day in corn oil) were less protective against EAE (Fig. 2B and Table II) when compared to pellets. G-1 and E2 treatment nominally lowered ex vivo T cell proliferation responses to mMOG-35-55 peptide in this particular experiment, but statistical significance was not reached (Fig. 2C). Therefore, we conclude that G-1, when administered alone at relatively high doses, was sufficient to induce complete protection against clinical EAE without observable effects on proliferation of antigen-specific T cells.
Fig. 2. Activation of GPR30 conferred substantial protection against clinical EAE in WT B6 mice.

A. Treatment with G-1 delayed and ameliorated EAE in a dose-dependent manner. Various doses of G-1, E2 or placebo pellets were imbedded underneath the skin on the back of the mice. One week following the start of treatment, the mice were immunized with mMOG-35-55 peptide plus CFA and PTX. The mice were scored for EAE development and euthanized 34 days after immunization for ex vivo studies. P < 0.05 or 0.01 for 2.5 and 0.1 mg E2- and 3.7 mg G-1-treatment groups from Day 14 to 34, and for 0.1 mg G-1-treatment group from Day 14 to 19 and from Day 25 to 34. The experiment was repeated 2 times with 5-8 mice in each group. B. G-1 injected s.c had nominally less protection than E2 against EAE. G-1 (20 ug/mouse/day in 100 ul of 10% ethanol and 90% olive oil), E2 (1 ug/mouse/day in 100 ul of 10% ethanol and 90% olive oil) or placebo (100 ul of 10% ethanol and 90% olive oil) were daily injected underneath the neck skin of the mice one week prior to immunization. The mice were monitored for changes in clinical EAE scores and euthanized 20 days after the immunization. P < 0.05 or 0.01 for E2-treatment groups from Day 11 to 20, and for G-1-treatment group from Day 11 to 17 as indicated by One-way ANOVA followed by Newman-Kuels multiple comparisons test. C. T cell proliferation to mMOG-35-55 from G-1, E2 and placebo-treated mice. Splenocytes and lymph node cells were obtained ex vivo from G-1- or E2-implanted mice as depicted in (a) and incubated with various concentration of antigenic peptides. Statistical difference was not reached among different treatment groups. D. G-1- but not E2-indcued protection against EAE was abrogated in GPR30KO mice. Neither G-1 nor E2 affected T cell proliferation to mMOG-35-55. GPR30KO mice were immunized one week after implantation with 1.8 mg/40 day release G-1 or 2.5 mg/60 day release E2 pellets for a week. The experiment was concluded 29 days after immunization for ex vivo experiments. *P < 0.05 or **P < 0.01 compared to placebo control. The experiment was repeated 2 times with 7-10 mice in each group. E. G-1 treatment did not increase the uterine weight in B6 mice. At the end of the clinical experiment, the weights of the whole mice, uteri and femurs were measured. *P < 0.05 compared to placebo control.
Table II.
The protective effects of G-1 vs. E2 in wild-type B6 miceb.
| Treatment | n | Incidence | Mortality | Onset | Peak | Daily | CDI |
|---|---|---|---|---|---|---|---|
| Placebo pellets | 7 | 7/7 | 0/7 | 13.7±1.1 | 3.9±0.3 | 2.0±0.54 | 51.9±13.5 |
| 1.8 mg G-1 | 7 | 0/7** | 0/7 | - | 0** | 0** | 0** |
| 0.1mg G-1 | 6 | 3/6 | 0/6 | 16.7±2.3* | 1.5±1.6** | 0.75±1.3* | 18.8±28.3* |
| 0.01 mg G-1 | 6 | 5/6 | 0/6 | 13.6±1.6 | 3.1±1.7 | 1.8±1.6 | 43.8±39.9 |
| 2.5 mg E2 | 7 | 0/7** | 0/7 | - | 0** | 0** | 0** |
| 0.1 mg E2 | 6 | 3/10** | 0/10 | 21.0±6.1** | 0.8±1.2** | 0.3±0.6** | 9.1±14.8** |
| 0.025 mg E2 | 5 | 4/5 | 0/5 | 14.8±0.5 | 2.8±1.6 | 1.7±1.0 | 37.0±21.6 |
| Placebo injection | 8 | 8/8 | 0/7 | 11.1±2.0 | 5.3±2.4 | 5.9±2.4 | 42.7±8.1 |
| G-1 injection | 7 | 6/8 | 1/8 | 13.7±1.3 | 3.7±1.6* | 2.7±1.3* | 19.7±11.3* |
| E2 injection | 7 | 3/8* | 1/8 | 15.3±1.5 | 2.3±2.9* | 2.3±0.3* | 10.5±13.9** |
Mice were immunized one week after implantation with G-1 or E2 pellets at different doses. The experiment was concluded 34 days after immunization.
P < 0.05 or
P < 0.01 as compared to placebo controls.
Although G-1 was reported previously to bind specifically to GPR30 (18) and not to any of the iERs in vitro, its specificity has not yet been characterized in vivo. Using GPR30KO mice, we tested critically whether GPR30 is required for G-1 induced protection against EAE. GPR30 mice were implanted with placebo, G-1 or E2 pellets 7 days prior to immunization. Although the protective effect of E2 against EAE was only partially offset by the absence of GPR30, treatment with G-1 was completely ineffective in GPR30KO mice (Fig. 2D and Table II). Therefore, G-1-induced protection against clinical EAE was a result of specific activation of GPR30.
Of importance, treatment with G-1 in vivo lacked the “estrogenic effects” of E2. In contrast to E2, G-1 treatment did not significantly change the weight of uteri, a prominent and well-known estrogenic effect (Fig. 2E). Neither G-1 nor E2 significantly changed the body and femur weights. Additionally, there were no abnormalities by H&E staining in liver, eyes, heart, mammary gland, brain, spleen, kidney, muscle or lung from 1.8 mg G-1-treated naïve mice (data not shown). To rule out the possibility that G-1 prevented EAE by regulating endogenous steroid hormones, levels of E2, progesterone, testosterone and corticosteroid were measured in sera from mice treated with placebo, 3.7mg G-1 or 2.5mg E2. The results showed that G-1 slightly lowered the level of corticosteroid but did not affect any of the other steroid hormones tested (Fig. 3), thus ruling out the possibility that upregulation of endogenous anti-inflammatory steroid hormones was responsible for the clinical improvement caused by G-1 treatment.
G-1 treatment reduced CNS inflammation, demyelination and axonal damage
Consistent with the clinical observations, both G-1 and E2 treatments markedly ameliorated immune cell infiltration and demyelination in the CNS as indicated by H&E and LFB-PAS staining (Fig. 4, upper panels). Moreover, both agents reduced the level of axonal damage in spinal cord sections (Fig. 4, lower panels). Existing axons can be visualized by immunohistochemical staining with SMI312, an antibody cocktail that stains phosphorylated (healthy) neurofilaments (NFL), and the degree of ongoing damage can be seen by staining non-phosphorylated neurofilaments (NPNFL) with antibody SMI32, which specifically detects injured and demyelinated axons(19,20). In placebo-treated WT mice, axonal staining was markedly reduced in the presence of inflammatory mononuclear cells, resulting in severe loss of SMI312 staining in the outer region of white matter where most neuroinflammation occurred (Fig 4, lower left panel). In contrast, axons in the spinal cords of G-1 and E2-treated mice were well preserved. Additionally, sections from both G-1 and E2 treated mice showed much less SMI32 staining in the white matter of the spinal cords (Fig. 4, lower right panel). Thus, G1 and E2 induced comparable neuroprotective effects in EAE.
Fig. 4. G-1 treatment reduced CNS infiltration (H&E), demyelination (LFB-PAS), axonal loss (NFLs) and ongoing axonal damage (dephosphorylated NFLs).
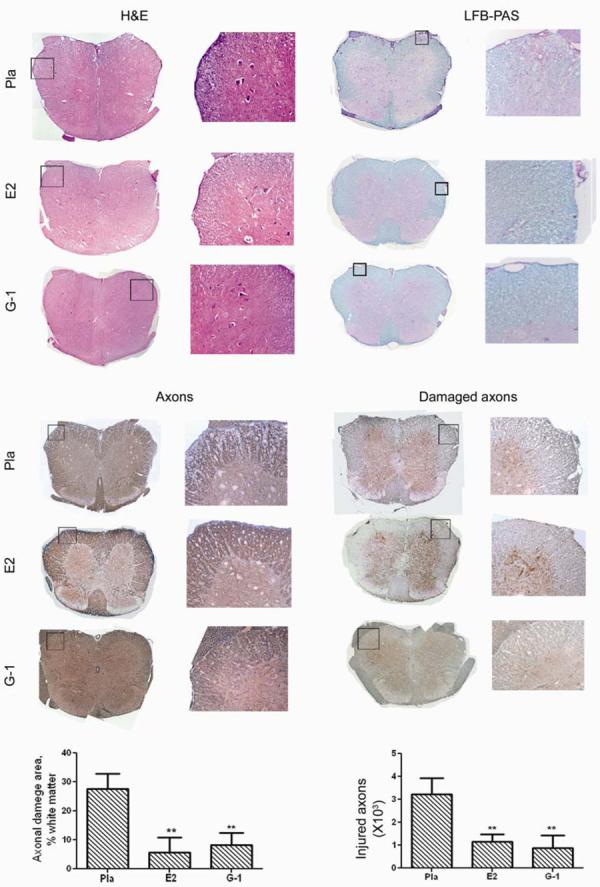
The mice from the clinical experiment shown in Fig. 2a were euthanized at the end of the experiment and spinal cords from >3 mice from each group were dissected for histology. Immune cell infiltration and demyelination of CNS were examined with H&E and LFBPAS staining. Total and damaged axons were examined with immunohistological staining for neurofilaments (NFLs) or dephosphorylated NFLs.
GPR30 upregulates PD-1 in Treg cells
Our previous results showed that E2-treatment increased the percentage of CD4+FoxP3+ Treg cells and PD-1 expression within this cell population(6,16,21). We thus studied whether GPR30 is required for E2-induced upregulation of Foxp3 or PD-1 expression in Treg cells. Splenocytes were harvested from placebo- or E2-treated WT and GPR30KO mice (from Fig. 1A) and stained for CD4, FoxP3 and PD-1. The results showed that GPR30 was not required for E2-induced upregulation of CD4+FoxP3+ T cells (data not shown). However, E2-induced upregulation of PD-1 expression in these cells was abolished in splenocytes from GPR30KO mice (Fig. 5A). E2 did not induce a significant shift in PD-1 expression in the non-regulatory CD4+FoxP3− T cells in WT or GPR30KO mice. Therefore, we conclude that GPR30 is required for E2-induced upregulation of PD-1 in Treg cells.
Fig. 5. GPR30 activation enhanced Treg cell function by upregulating PD-1 expression level.
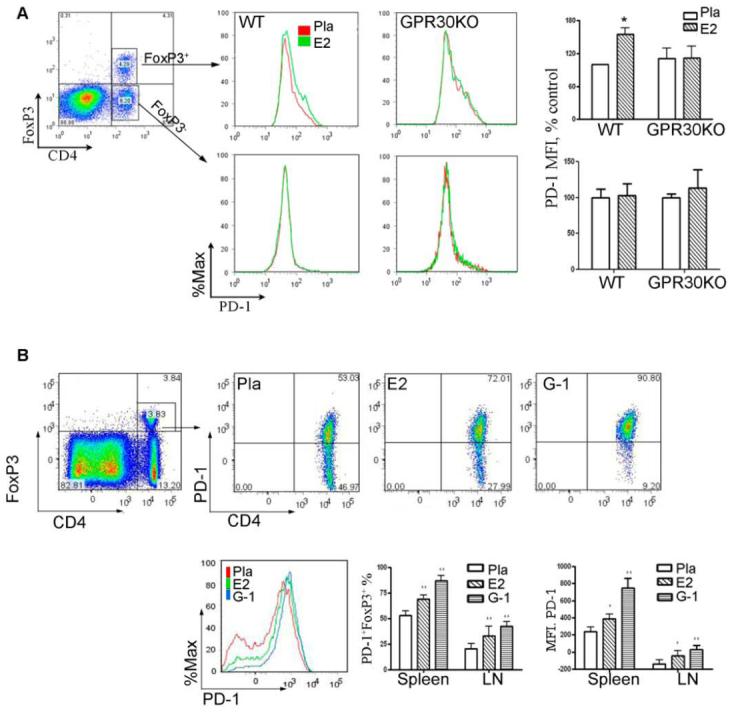
A. GPR30 was required for E2-induced upregulation of PD-1 expression levels in CD4+FoxP3+ Treg cells. E2 treatment increased the expression of PD-1 in CD4+FoxP3+, but not CD4+FoxP3− T cells. Splenocytes or lymph node cells from GPR30KO mice were obtained at the end of clinical experiment in Fig. 1A and stained for FoxP3 and PD-1, as well as cellular markers for T cells and APCs. The expression levels of PD-1 were analyzed by gating on CD4+FoxP3+ or CD4+FoxP3− T cells from WT or GPR30 mice. The bar charts show the average MFI of PD-1 in CD4+FoxP3+ and CD4+FoxP3− T cells from all mice in each group. B. G-1 treatment significantly increased PD-1 expression levels in CD4+FoxP3+ T cells. Splenocytes or lymph node cells were obtained at the end of clinical experiment in Fig. 2a and stained for FoxP3 and PD-1, as well as cellular markers for T cells and APCs. The expression levels of PD-1 were analyzed by gating on CD4+FoxP3+ T cells from placebo-, E2- or G-1-treated WT mice. The dot plot shows the marked shift of CD4+FoxP3+ T cells from PD-1− to PD-1+ after treatment with G-1 or E2. The histogram overlay compares the MFI of PD-1 in CD4+FoxP3+ T cells from different treatment groups. The bar charts show the average percentages of PD-1+ CD4+FoxP3+ T cells and the average MFI of PD-1 in CD4+FoxP3+ T cells from all mice in each group. *P < 0.05 or **P < 0.01 compared to placebo control. The experiment was repeated 2 times with a total of 7-10 mice in each group.
We then tested whether activation of GPR30 upregulates PD-1 in CD4+FoxP3+ Treg cells. Splenocytes were harvested from placebo-, E2- or G-1-treated mice after EAE induction and stained for CD4, FoxP3 and PD-1. Roughly half of the CD4+FoxP3+ cells from the spleen of placebo-treated mice were negative or low for PD-1 expression (Fig. 5B). Of critical importance, both E2 and G-1 treatments strongly enhanced the staining intensity of PD-1 and converted a majority of the CD4+FoxP3+PD-1− cells to CD4+FoxP3+PD-1+ cells. Surprisingly, G-1 was even more potent than E2 in boosting PD-1 expression in CD4+FoxP3+ cells, suggesting that the iER pathways for E2 might have opposing effects on GPR30. Functional assays using FoxP3-GFP “knock-in” mice indicated that GFP+ PD-1+ Treg cells were more suppressive than GFP+ PD-1− Treg cells (submitted for publication). Thus, activation of GPR30 may cause Treg cells to shift from PD-1− to PD-1+ with enhanced suppressive function.
PD-1 is critical to G-1-induced EAE protection and decrease of IL-17 production
To evaluate if G-1-induced upregulation of PD-1 is important for EAE protection, PD-1KO mice were implanted with 2.5 mg/60 day release E2, 1.8 mg/40 day release G-1, or placebo pellets one week prior to immunization to induce EAE. As shown in Fig. 6A and Table IV, G-1 failed to protect against clinical EAE in PD-1KO mice, whereas E2 retained partial efficacy. In addition, G-1 treatment did not significantly change T cell proliferation to mMOG-35-55 (Fig. 6B). Thus, upregulation of PD-1 is of critical importance for G-1-induced EAE protection.
Fig. 6. PD-1 is required for G-1-induced EAE protection and cytokine deviation.

PD-1KO mice (A) and WT mice (as in Fig 2 and Table II) were immunized one week after implantation with placebo, 1.8 mg/40 day release G-1 or 2.5 mg/60 day release E2 pellets. The mice were observed for EAE development for 20 days and euthanized. Splenocytes from placebo and G-1 treatment groups were evaluated for T cell proliferation (B) and cytokine secretion in 48 h supernatants (C). *P < 0.05 or **P < 0.01 compared to placebo. The experiment was repeated 2 times with a total of 7-10 mice in each group.
Table IV.
The protective effect of G-1, but not E2, was abolished in PD-1KO miced.
| Treatment | n | Incidence | Mortality | Onset | Peak | Daily | CDI |
|---|---|---|---|---|---|---|---|
| Placebo | 5 | 5/5 | 1/5 | 12.2 ± 0.8 | 5.1±0.2 | 3.5 ± 0.5 | 38.9 ± 5.1 |
| G-1 | 5 | 5/5 | 1/5 | 13.8 ± 1.3 | 4.8 ± 0.6 | 2.7 ± 0.8 | 30.6 ± 9.1 |
| E2 | 5 | 5/5 | 0/5 | 16.4 ± 0.9* | 4.1 ± 0.8 | 1.5 ± 0.6* | 16.4 ± 6.3** |
PD-1KO mice were immunized one week after implantation with 1.8 mg/40 day release G-1 or 2.5 mg/60 day release E2 pellets for a week.
P < 0.05 or
P < 0.01 as compared to placebo control.
Lastly, we evaluated the effects of G-1 treatment and PD-1 expression on cytokine profiles in splenocytes harvested from G-1- or placebo-treated WT and PD-1KO immunized mice. As shown in Fig. 6C, G-1-treatment significantly reduced secretion of IL-17 and IL-2, critical pro-inflammatory cytokines that play a vital role in EAE induction(22), and increased the secretion of IL-10, a key anti-inflammatory cytokine. Interestingly, G-1 also increased the production of IFN-γ, a hallmark Th1 cytokine, as well as IL-6, and suppressed IL-4, but did not significantly change the production of any other cytokines examined, including IL-1β, IL-5, IL-12, and TNF-α. Of importance, this cytokine profile was drastically altered in G-1 treated PD-1KO mice. The decrease of IL-17 and increase of IL-6, IL-10 and IFN-γ production were abolished, whereas the reduction in IL-2 and IL-4 remained. Thus, PD-1 was required for GPR30 downregulation of IL-17 but not IL-2 and IL-4, and upregulation of IL-6, IL-10 and IFNγ. The result that G-1 treatment reduced IL-17 production through a PD-1-dependent mechanism is of particular importance because this cytokine has been closely linked to neuroinflammation in EAE.
DISCUSSION
The results presented above demonstrate for the first time that the mER, GPR30, is both necessary and sufficient for full E2-mediated protection against EAE. Moreover, the agonist G-1, which selectively activates GPR30 without engagement of the iERs, retained E2's ability to protect against clinical and histological EAE without obvious “estrogenic side effects” including increased uterine weight. The prospect of using estrogens to treat autoimmune diseases such as MS has been complicated by considerable side effects and risk, which are believed to be associated with transcription modifying functions of iERs. Our study thus demonstrates possible clinical significance of G-1 activation of GPR30 and the selective upregulation of PD-1 by CD4+Foxp3+ Treg cells.
In this study, we employed the GPR30KO mouse model to define whether GPR30 is indispensible for E2-mediated full protection against EAE. Our result indicated that GPR30 is an active contributor but not the only player in mediating the suppressive effects of E2 on EAE. This conclusion is consistent with our previous results on iER gene-deficient mice(23), which showed that the protective effect of E2 was mainly dependent on ERα. In that study, although E2-treatment delayed the disease onset for 4 days, it did not reduce the incidence, disease peak, or CDI score. Similarly, Voskuhl et al. reported complete loss of protection by estriol treatment in ERKO mice. However, as shown in Fig. 1, there clearly is residual E2 protection in GPR30 KO mice, indicating contribution of other receptors, presumably ERα. Thus, it is likely that both GPR30 and ERα participate in E2-mediated protection in an additive manner. However, based on the difference in residual protection in ERKO vs. GPR30KO mice treated with E2, we think that there is a lesser contribution of GPR30 to protection when compared to ERα. We cannot yet conclude that E2-induced protection against EAE is mediated exclusively by ERα plus GPR30, but GPR30KO and ERKO double-gene-deficient mice are currently being constructed to address this issue.
Since our result in GPR30KO mice indicated that GPR30 contributed to E2-mediated protection against EAE, we determined whether activation of GPR30 alone could confer protection against EAE. The development of selective agonists for mER made it possible to specifically activate mER without transactivation of iERs. Qiu et al. described a synthesized compound, STX, that does not exhibit any binding affinity for the nuclear receptors ERα or ERβ, but mimics the quick effect of estrogens on neurons(24). Yet, the molecular target of STX has not been identified and it remains to be determined whether STX indeed interacts with a mER. Besides STX, the only other reported mER ligand is G-1, which we selected for further study due to its ability to bind GPR30 with high affinity and excellent specificity(25). Remarkably, none of the mice receiving the highest dose G-1 pellets showed any clinical signs of EAE. In the absence of GPR30, the effect of G-1, but not E2, completely disappeared. These results suggested that solo activation of GPR30 is sufficient to confer protection against EAE. The protective effects of G-1 in WT mice eliminated the possibility that the observed role of GPR30 could be due merely to an artifact of gene deletion or a compensation effect in the developing animal.
How could G-1 treatment at a sufficiently high dose completely protect mice against EAE while GPR30 is only one of the receptors that mediate the protective effect of E2 against EAE? Since G-1 did not protect the mice from EAE in GPR30KO mice, we do not believe that transactivation of ERα by G-1 played a significant role. Also, we did not detect any significant increase in the serum levels of estradiol, corticosterone, progesterone or testosterone in G-1-treated mice, thus ruling out the possibility that upregulation of endogenous steroid hormones accounted for the clinical and neuroprotective effects of G-1 treatment. Thus, we conclude that ERα and GPR30 work together additively to achieve optimal protection, and each receptor may adequately compensate the functional loss of the other.
Mechanistically, we observed that G-1 treatment did not directly suppress pathogenic T cell proliferation, nor did it increase the percentage of CD4+FoxP3+ Treg cells. Nevertheless, both E2 and G-1 markedly increased the level of PD-1 expression in CD4+FoxP3+ Treg cells. PD-1 has been found to play a crucial role in the development and maintenance of peripheral tolerance(26). In a separate study, we sorted PD-1+ and PD-1− Treg cells from FoxP3-GFP “knock-in” mice and found that PD-1+ Treg cells had increased suppressive function (submitted for publication). Moreover, the therapeutic effect of G-1, but not E2, disappeared completely in PD-1KO mice. Thus, it seems that G-1 is exclusively dependent on the presence of PD-1 to function, but the effects of E2 could be mediated by alternative pathways in the absence of PD-1. Taken together, these results suggest that G-1 may suppress EAE by upregulation of the PD-1 signaling pathway in CD4+FoxP3+ cells.
The result that both E2 and G-1 decrease IL-17 in vivo is remarkable since this cytokine has been closely linked to the development of EAE. That G-1 failed to reduce IL-17 production in PD-1 gene-deficient mice strongly supports the idea that G-1 regulates IL-17 production via manipulation of PD-1. Increased secretion of the prototypic Th1 cytokine, INF-γ, seemed to be in conflict with the fact that G-1 ameliorated disease severity and tissue damage. Unlike IL-17, however, IFN-γ appears to be pleiotrophic. Although administration of IFN-γ worsened symptoms of MS(27), it improved EAE in mice(28-30), and IFN-γ gene-disrupted mice were more susceptible to the induction of EAE(29). Thus, increased production of IFN-γ may actually facilitate the containment of EAE in some circumstances. However, our previous results indicated that E2 treatment reduced IFN-γ (23), raising the possibility that effects of E2 and G1 may differ with regards to IFN-γ production and its effect on protection against EAE.
Our results showed that treatment with G-1 in vivo lacked some of the prominent “estrogenic effects” of E2 and did not cause any abnormalities in liver, eyes, heart, mammary gland, brain, spleen, kidney, muscle or lung. Although we believe that the use GPR30 ligands will avoid some of the side effects mediated by iERα, it remains to be seen whether or not GPR30 agonists represent a “safer” alternative to estrogen treatment. For instance, GPR30 may play an important role in promoting breast and uterine cancer progression (12). Thus, G-1 signaling through GPR30 might be as potent as estradiol signaling through iERα with respect to carcinogenesis. Nevertheless, our study demonstrated that it is possible to avoid some of the side effects mediated through iERs by specifically targeting the membrane receptor, while retaining much of the therapeutic efficacy of estradiol, at least in EAE.
Taken together, we showed that the putative mER, GPR30, is sufficient, yet not exclusively responsible for full E2-mediated protection against EAE. Treatment with G-1 that specifically targets GPR30 suppressed clinical and histological EAE and the production of IL-17 by upregulation of PD-1 expression in regulatory T cells. This study is the first to evaluate the contribution of a membrane steroid receptor in suppression of autoimmune disease in an animal model, and may provide the necessary foundation for the clinical application of membrane steroid receptor agonists such as G-1 in human subjects.
Table III.
The protective effect of G-1 was abrogated in GPR30KO micec.
| Treatment | n | Incidence | Mortality | Onset | Peak | Daily | CDI |
|---|---|---|---|---|---|---|---|
| Placebo | 10 | 9/10 | 0/10 | 14.7 ± 1.5 | 4.0 ± 1.6 | 2.5 ± 1.1 | 49.7 ± 22.8 |
| G-1 | 8 | 8/8 | 0/8 | 13.8 ± 1.2 | 4.5 ± 0.7 | 3.0 ± 0.7 | 59.9 ± 13.4 |
| E2 | 7 | 4/7 | 0/7 | 18.0±1.4** | 2.5±2.3# | 3.7±1.3 | 17.2±17.4*# |
GPR30KO mice were immunized one week after implantation with 1.8 mg/ 40 day release G-1 or 2.5 mg/60 day release E2 pellets for a week. The experiment was concluded 29 days after immunization.
P < 0.05 or
P < 0.01 as compared to placebo control.
P < 0.05 as compared to G-1-treated mice.
ACKNOWLEDGMENTS
The authors thank Paul Bui in the Portland VA Medical Center Veterinary Medical Unit for providing excellent breeding service and animal care, Dr. Francis Pau for measuring the serum hormone levels, Elizabeth Rick and Dr. Martin Kelly for assistance in genotyping, Dr. Tasuku Honjo at Kyoto University for providing PD-1KO mice, Dr. Steve Ziegler and Alexander Rudensky for providing FoxP3-GFP mice, and Eva Niehaus for assistance in manuscript preparation.
Footnotes
This work was supported by NIH Grants NS45445 and NS49210; National Multiple Sclerosis Society Grants RG3405; The Nancy Davis Center Without Walls; and the Biomedical Laboratory R&D Service, Department of Veterans Affairs.
Correspondence should be addressed to: Halina Offner (offnerva@ohsu.edu) , Portland VA Medical Center, 3710 SW US Veterans Hospital Rd, Portland, OR; 503/721-7893 Phone; 503/721-7975 FAX; or Chunhe Wang (wangch@ohsu.edu)
References
- 1.Whitacre CC. Sex differences in autoimmune disease. Nat. Immunol. 2001;2:777–780. doi: 10.1038/ni0901-777. [DOI] [PubMed] [Google Scholar]
- 2.Abramsky O. Pregnancy and multiple sclerosis. Ann. Neurol. 1994;36(Suppl):S38–S41. doi: 10.1002/ana.410360712. [DOI] [PubMed] [Google Scholar]
- 3.Confavreux C, Hutchinson M, Hours MM, Cortinovis-Tourniaire P, Moreau T. Rate of pregnancy-related relapse in multiple sclerosis. Pregnancy in Multiple Sclerosis Group. N. Engl. J. Med. 1998;339:285–291. doi: 10.1056/NEJM199807303390501. [DOI] [PubMed] [Google Scholar]
- 4.Sicotte NL, Liva SM, Klutch R, Pfeiffer P, Bouvier S, Odesa S, Wu TC, Voskuhl RR. Treatment of multiple sclerosis with the pregnancy hormone estriol. Ann. Neurol. 2002;52:421–428. doi: 10.1002/ana.10301. [DOI] [PubMed] [Google Scholar]
- 5.Soldan SS, varez Retuerto AI, Sicotte NL, Voskuhl RR. Immune modulation in multiple sclerosis patients treated with the pregnancy hormone estriol. J. Immunol. 2003;171:6267–6274. doi: 10.4049/jimmunol.171.11.6267. [DOI] [PubMed] [Google Scholar]
- 6.Bebo BF, Jr., Fyfe-Johnson A, Adlard K, Beam AG, Vandenbark AA, Offner H. Low-dose estrogen therapy ameliorates experimental autoimmune encephalomyelitis in two different inbred mouse strains. J. Immunol. 2001;166:2080–2089. doi: 10.4049/jimmunol.166.3.2080. [DOI] [PubMed] [Google Scholar]
- 7.Ito A, Bebo BF, Jr., Matejuk A, Zamora A, Silverman M, Fyfe-Johnson A, Offner H. Estrogen treatment down-regulates TNF-alpha production and reduces the severity of experimental autoimmune encephalomyelitis in cytokine knockout mice. J. Immunol. 2001;167:542–552. doi: 10.4049/jimmunol.167.1.542. [DOI] [PubMed] [Google Scholar]
- 8.Dechering K, Boersma C, Mosselman S. Estrogen receptors alpha and beta: two receptors of a kind? Curr. Med. Chem. 2000;7:561–576. doi: 10.2174/0929867003375010. [DOI] [PubMed] [Google Scholar]
- 9.Toran-Allerand CD, Singh M, Setalo G., Jr. Novel mechanisms of estrogen action in the brain: new players in an old story. Front Neuroendocrinol. 1999;20:97–121. doi: 10.1006/frne.1999.0177. [DOI] [PubMed] [Google Scholar]
- 10.Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307:1625–1630. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- 11.Prossnitz ER, Arterburn JB, Sklar LA. GPR30: A G protein-coupled receptor for estrogen. Mol. Cell Endocrinol. 2007;265-266:138–142. doi: 10.1016/j.mce.2006.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prossnitz ER, Arterburn JB, Smith HO, Oprea TI, Sklar LA, Hathaway HJ. Estrogen signaling through the transmembrane G protein-coupled receptor GPR30. Annu. Rev. Physiol. 2008;70:165–190. doi: 10.1146/annurev.physiol.70.113006.100518. [DOI] [PubMed] [Google Scholar]
- 13.Prossnitz ER, Oprea TI, Sklar LA, Arterburn JB. The ins and outs of GPR30: A transmembrane estrogen receptor. J. Steroid Biochem. Mol. Biol. 2008;109:350–353. doi: 10.1016/j.jsbmb.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang C, Dehghani B, Magrisso IJ, Rick EA, Bonhomme E, Cody DB, Elenich LA, Subramanian S, Murphy SJ, Kelly MJ, Rosenbaum JS, Vandenbark AA, Offner H. GPR30 contributes to estrogen-induced thymic atrophy. Mol. Endocrinol. 2008;22:636–648. doi: 10.1210/me.2007-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mårtensson UE, Salehi SA, Windahl S, Gomez MF, Swärd K, Daszkiewicz-Nilsson J, Wendt A, Andersson N, Hellstrand P, Grände PO, Owman C, Rosen CJ, Adamo ML, Lundquist I, Rorsman P, Nilsson BO, Ohlsson C, Olde B, Leeb-Lundberg LM. Deletion of the G protein-coupled Receptor GPR30 Impairs Glucose Tolerance, Reduces Bone Growth, Increases Blood Pressure, and Eliminates Estradiol-stimulated Insulin Release in Female Mice. Endocrinology. 2008 Oct 9; doi: 10.1210/en.2008-0623. 2008. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 16.Polanczyk MJ, Hopke C, Vandenbark AA, Offner H. Treg suppressive activity involves estrogen-dependent expression of programmed death-1 (PD-1) Int. Immunol. 2007;19:337–343. doi: 10.1093/intimm/dxl151. [DOI] [PubMed] [Google Scholar]
- 17.Wang C, Gold BG, Kaler LJ, Yu X, Afentoulis ME, Burrows GG, Vandenbark AA, Bourdette DN, Offner H. Antigen-specific therapy promotes repair of myelin and axonal damage in established EAE. J. Neurochem. 2006;98:1817–1827. doi: 10.1111/j.1471-4159.2006.04081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bologa CG, Revankar CM, Young SM, Edwards BS, Arterburn JB, Kiselyov AS, Parker MA, Tkachenko SE, Savchuck NP, Sklar LA, Oprea TI, Prossnitz ER. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat. Chem. Biol. 2006;2:207–212. doi: 10.1038/nchembio775. [DOI] [PubMed] [Google Scholar]
- 19.Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. N. Engl. J. Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- 20.Pitt D, Werner P, Raine CS. Glutamate excitotoxicity in a model of multiple sclerosis. Nat. Med. 2000;6:67–70. doi: 10.1038/71555. [DOI] [PubMed] [Google Scholar]
- 21.Polanczyk MJ, Carson BD, Subramanian S, Afentoulis M, Vandenbark AA, Ziegler SF, Offner H. Cutting edge: estrogen drives expansion of the CD4+CD25+ regulatory T cell compartment. J. Immunol. 2004;173:2227–2230. doi: 10.4049/jimmunol.173.4.2227. [DOI] [PubMed] [Google Scholar]
- 22.Bettelli E, Oukka M, Kuchroo VK. T(H)-17 cells in the circle of immunity and autoimmunity. Nat. Immunol. 2007;8:345–350. doi: 10.1038/ni0407-345. [DOI] [PubMed] [Google Scholar]
- 23.Polanczyk M, Zamora A, Subramanian S, Matejuk A, Hess DL, Blankenhorn EP, Teuscher C, Vandenbark AA, Offner H. The protective effect of 17beta-estradiol on experimental autoimmune encephalomyelitis is mediated through estrogen receptor-alpha. Am. J. Pathol. 2003;163:1599–1605. doi: 10.1016/s0002-9440(10)63516-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qiu J, Bosch MA, Tobias SC, Krust A, Graham SM, Murphy SJ, Korach KS, Chambon P, Scanlan TS, Ronnekleiv OK, Kelly MJ. A G-protein-coupled estrogen receptor is involved in hypothalamic control of energy homeostasis. J. Neurosci. 2006;26:5649–5655. doi: 10.1523/JNEUROSCI.0327-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Revankar CM, Mitchell HD, Field AS, Burai R, Corona C, Ramesh C, Sklar LA, Arterburn JB, Prossnitz ER. Synthetic estrogen derivatives demonstrate the functionality of intracellular GPR30. ACS Chem. Biol. 2007;2:536–544. doi: 10.1021/cb700072n. [DOI] [PubMed] [Google Scholar]
- 26.Keir ME, Francisco LM, Sharpe AH. PD-1 and its ligands in T-cell immunity. Curr. Opin. Immunol. 2007;19:309–314. doi: 10.1016/j.coi.2007.04.012. [DOI] [PubMed] [Google Scholar]
- 27.Panitch HS, Hirsch RL, Schindler J, Johnson KP. Treatment of multiple sclerosis with gamma interferon: exacerbations associated with activation of the immune system. Neurology. 1987;37:1097–1102. doi: 10.1212/wnl.37.7.1097. [DOI] [PubMed] [Google Scholar]
- 28.Billiau A, Heremans H, Vandekerckhove F, Dijkmans R, Sobis H, Meulepas E, Carton H. Enhancement of experimental allergic encephalomyelitis in mice by antibodies against IFN-gamma. J. Immunol. 1988;140:1506–1510. [PubMed] [Google Scholar]
- 29.Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L, Dalton D, Fathman CG. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE) J. Immunol. 1996;156:5–7. [PubMed] [Google Scholar]
- 30.Voorthuis JA, Uitdehaag BM, De Groot CJ, Goede PH, van der Meide PH, Dijkstra CD. Suppression of experimental allergic encephalomyelitis by intraventricular administration of interferon-gamma in Lewis rats. Clin. Exp. Immunol. 1990;81:183–188. doi: 10.1111/j.1365-2249.1990.tb03315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]