

Abstract
The worldwide incidence of breast cancer affects 1.2 million women each year. In contrast to the high occurrence of this malady, a decline in mortality is reported among industrialized countries. In this respect, both awareness campaigns and substantial progress achieved in therapy and diagnosis allowed for the enhancement of the survival rate in patients with breast cancer. Undoubtedly, oncology research programs played a relevant role in the improvement of therapeutics and diagnostics for breast cancer. Major strides were reported, especially over the last decade and a half, in better understanding molecular and cellular biology events involved in breast cancer pathogenesis and progression of the disease. However, therapeutic approaches for the treatment of patients with breast cancer need further improvement. Therapeutic interventions can chronically compromise both the state of health and quality of life of breast cancer survivors. In addition, current therapeutic approaches have not significantly improved the survival rate in patients with metastatic disease. On these grounds, it is necessary to develop more efficient therapeutics and diagnostic tools, which can improve the health and quality of life of breast cancer survivors and increase the survival rate in patients with metastatic disease. In this respect, the field of cancer research has placed a particular emphasis on the elucidation of genetic and epigenetic alterations that may lead to the pathogenesis of breast cancer and contribute to its progression.
Keywords: breast cancer, genetic, epigenetic, genomic medicine, targeted therapy
1. Introduction
Breast cancer is recognized as the most common malignancy among women. The worldwide incidence of this pathological condition is reported to affect 1.2 million individuals each year (Cheema et al., 2007). In the United States of America, more than 200,000 women are diagnosed with breast cancer every year, which accounts for the 30.4% of all newly diagnosed neoplasms in that country (Jemal et al., 2006).
Substantial advances in therapy and diagnosis have enhanced the survival rate of patients with breast cancer among industrialized countries (Cheema et al., 2007). In regard with the decline of the mortality rate among patients, important achievements in the fields of molecular and cellular biology contributed considerably in improving the efficacy of therapeutic interventions for breast cancer. Research programs led to the identification of a variety of cellular factors that are involved both in breast cancer pathogenesis and progression of the disease. These factors include: HER-2 over-expression (Widakowich et al., 2007), mechanisms leading to the inactivation of the putative tumor suppressor p53 gene (Bradbury & Olopade, 2007), high levels of receptor for the insulin-like growth factor 1 (IGF-1R) (Cascio et al., 2007; Romano, 2003), CXCR4-CXCL12 axis (Smith et al., 2004), loss of estrogen receptor (ER)- α expression (Chen & Colditz, 2007; Macaluso et al., 2003), interactions between pRb2/p130 gene and ER-α promoter (Fig. 1) (Macaluso et al., 2007) and mutations in BRCA1 and BRCA2 genes (Rennert, 2007; Russo et al., 2007).
Fig. 1.
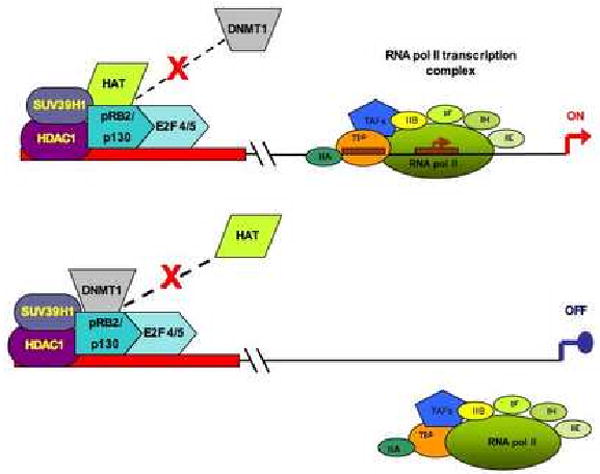
A plausible interpretation of pRb2/P130-mediated ER-α transcription regulation in human breast cancer MCF-7 cell line. The upper part of the panel shows a permissive configuration for ER-α transcription, in which a multi-molecular complex comprising pRb2/p130, HDAC1, SUV39H1 and HAT interacts with the ER-α promoter. The lower part of the panel shows a non-permissive configuration for ER-α transcription. In this case, DNMT1 is present in the multi-molecular complex, whereas HAT is released. Abbreviations: HDAC1 = histone deacetylase 1; DNMT1 = DNA (cytosine-5) methyltransferase 1; HAT = histone acetyltransferase; SUV39H1 = histone methyltransferase; TAFs = transcription activating factors; RNA pol II = RNA polymerase II; TBP = TATA binding protein. Transcription factors are reported as: IIA, IIB, IIF, IIH and IIE.
The decline of mortality rate for breast cancer is also caused by awareness campaigns and screening conducted among women in industrialized countries (Kuhl, 2002). Regrettably, therapeutic interventions for the treatment of this pathological condition are commonly associated with a variety of side effects, which can chronically compromise both the state of health and quality of life of breast cancer survivors (Widakowich et al., 2007). Typically, the symptoms of the side effects associated with the treatment may comprise: depression, chronic fatigue, dysfunction of the immune system, lymphedema, osteoporosis, neuropathy and various deficits of the upper body in terms of strength, flexibility, pain and other physiological functions.
Late recurrence of the disease sadly accounts for the death of more than 60% of patients with breast cancer (Widakowich et al., 2007). Although significant advances were reported over the years in the treatment of the four most common epithelial malignancies, such as cancers of the lung, breast, colon and prostate, the survival rate among patients with metastatic disease does not seem to be significantly affected (Chia et al., 2003; Wicha, Lin & Dontu, 2006). Indeed, further studies are needed to optimize therapeutic interventions in patients with metastatic breast cancer and diagnostic tools.
The aim of this review is to discuss the achievements reported in the field of cancer research in characterizing the genetic and epigenetic alterations that are associated with breast cancer. Indeed, these studies may lead to the design of more specific and effective therapeutics and diagnostics, which will ultimately minimize adverse effects that are related to therapy and increase the survival rate in patients with metastatic disease.
2. The role of molecular and cellular biology in the development of current therapeutics and diagnostics for breast cancer
The first impact of molecular and genetic studies on breast cancer was reported in the late 1980s. Those studies provided important insights on cellular signaling pathways involved in cell proliferation, survival and in establishing a transformed cell phenotype (Widakowich et al., 2007). A variety of important cellular regulators were identified in that period, such as: receptors for growth factors, intracellular signaling pathways, regulators of apoptosis and nuclear proteins associated with cell cycle control and deregulation. Interestingly, these findings rapidly conducted to the identification of regulatory agents for angiogenesis (Folkman, 1995). As anticipated, an over-expressed HER-2 proto-oncogene is a key component in breast cancer. HER-2 is one of the four members of the HER family of receptor tyrosine kinases, which also comprises: epidermal growth factor receptor (EGFR, or HER1), HER-3 and HER-4 (Kawaguchi et al., 2007). All of these receptors have an extra-cellular domain for the binding to the ligands, a trans-membrane moiety and a cytoplasm region (Hynes & Lane, 2005). The binding of a ligand to a specific place of the extra-cellular region of the HER family receptors induces the ATP-mediated phosphorylation in the cytoplasm domain, which, in turn, orchestrates the intracellular signaling systems. These cellular signaling systems related to the HER family plays a fundamental role in the regulation of cell proliferation, differentiation, survival and migration. In particular, HER-2 over-expression is associated with aberrant activation of intracellular mitogenic signaling and is observed in 20-25% of the global number of patients with breast cancer (Siamon et al., 1987; Siamon et al., 1989).
A decade later, the aforementioned molecular and genetic findings allowed for the first therapeutic applications of the so-called era of targeted therapy (Chabner & Roberts, 2005). These approaches aimed at targeting the biological functions of over-expressed HER-2. A recombinant humanized monoclonal antibody termed trastumab was used to displace ligands to the extra-cellular portion of HER-2, in order to silence HER-2-mediated mitogenic signaling system. Another class of therapeutic agents against HER-2 is based on small molecules that suppress ATP-mediated phosphorylation of HER-2 intracellular domain (Imay & Tanaka, 2006). This second class of therapeutics was termed small molecule tyrosine kinase inhibitors (TKIs) (Imay & Tanaka, 2006). Interestingly, TKIs can also target the intracellular signaling pathway of an over-expressed and activated IGF-1R (Romano, 2003). Trastumab and TKIs increased the arsenal of therapeutic options for breast cancer, which, until the advent of the molecular and genetic era, consisted of surgical interventions, radiotherapy and tamoxifen-based chemotherapy.
The inhibition of angiogenesis was another important strategy adopted in cancer therapy (Folkman, 1995; Huh et al., 2005). The vascular endothelial growth factor (VEGF) receptor family is an important mediator of angiogenesis (Folkman, 1995). Also in this case, a monoclonal antibody termed bevacizumab was used to displace the ligands to the VEGF receptor, in order to inhibit angiogenesis (Kim et al., 1993). Interestingly, bevacizumab was used in phase I/II clinical trials that dealt with the treatment of patients with refractory metastatic breast cancer either as a single agent (Cobleigh et al., 2003; Gordon et al., 2001), or in combination with other chemotherapeutics (Burstein, Parker, & Savoie, 2002; Margolin et al., 2001). In addition, bevacizumab was used in combination with capecitabine in 462 patients with metastatic breast cancer, who participated in a phase III clinical trial (Miller, 2003).
As anticipated, molecular biology and genetic studies led to the identification of two important prognostic markers for breast and ovarian cancer susceptibility, which were termed BRCA1 and BRCA2 (Foulkes et al., 2004; Radice, 2002; Rennert, 2007; Russo et al., 2007). Genotyping of individuals with familial history of breast and ovarian cancer revealed mutations in BRCA1 and/or BRCA2. Both factors were identified in the mid-1990s and are involved in DNA damage repair, homologous recombination, regulation of transcription and embryonic proliferation (Welcsh, Owens & King, 2000). Additionally, BRCA1 contributes to ubiquitination (Welcsh, Owens & King, 2000). Familial breast cancer is also associated with mutations of p53, PTEN, ATM, NBS1, RAD50, PALB2 and CHEK2 (Bradbury & Olopade, 2007; Walsh & King, 2007). Other gene mutations that confer breast cancer susceptibility are emerging and include the following factors: CASP8, PGR, PBRL and TGFB1 (Bradbury & Olopade, 2007; Cardillo, Yap & Castagna, 1997). Overall, the incidence of hereditary breast cancer ranges from 5 to 10% of the global number of cases (Bradbury & Olopade, 2007).
The field of oncology is continuously searching for novel diagnostics for the prognosis of sporadic and familial breast cancer. In this respect, recent clinical studies demonstrated that the presence of breast cancer micro-metastasis in the bone marrow is associated with poor prognosis (Braun et al., 2005). Interestingly, in addition to the clinical outcome, the evaluation of bone marrow in patients with breast cancer might predict their response to therapy (Braun et al., 2003).
4. From genetic to epigenetic and genomic medicine: new frontlines in the fight against breast cancer
As already mentioned, studies have identified a number of genetic alterations that are associated with hereditary breast cancer. Research programs are currently ongoing to find the remaining genetic factors that may be involved in the transmission of breast cancer hereditary traits to offspring. However, the incidence of sporadic cases of breast cancer is in the range of 90-95% (Bradbury & Olopade, 2007). Undeniably, the environment and life style also play a fundamental role in the pathogenesis and progression of this malignancy (Chen & Colditz, 2007; Foulkes et al., 2004). Some of the identified risk factors for breast cancer include: early-age menarche, postmenopausal obesity, late-age menopause, use of hormone therapy, ethnicity, nulliparity or late onset of first childbirth, never having breastfed, alcohol consumption and cigarette smoking (Albrektsen, Heuch, Thoresen, & Kvale 2006; Andrieu et al., 2006; Arsian et al., 2007; Ha et al., 2007; Maskarinec et al., 2006; Menes, Ozao & Kim, 2007; Shema et al., 2007; Vogel & Taioli, 2006). Other epidemiological studies are evaluating the possible influence of viral etiology in breast cancer (Gumus et al., 2006; Lawson, Gunzburg & Whittaker, 2006).
Evidently, sporadic and familial breast cancers have in common aberrant patterns of gene expression, which may involve the activation of cellular oncogenes and/or silencing of tumor suppressor genes. These events are ultimately responsible for the establishment of a transformed cell phenotype and may contribute to the progression of the illness. On these grounds, cancer can be considered a genetic disease that is originated either by inherited or acquired genetic mutations (Gan et al., 2003; Giordano, Fucito, Romano & Marino, 2007; Karakosta et al., 2005; Romano, Micheli, Pacilio & Giordano, 2000; Romano, 2005). In this regard, as previously discussed, genetic research revealed very important clues on breast cancer pathogenesis, which had a profound impact in medical practice in terms of development of therapeutics and diagnostics. However, genetics, per se, cannot provide the complete interpretation of various biological processes, including pathogenesis and progression of a certain illness. This became apparent after the completion of the human genome sequencing at the beginning of this decade (Guttmacher & Collins, 2002; Varmus, 2002). On one hand, altered patterns of gene expression do not necessarily involve changes in primary DNA sequence, as indicated by studies on epigenetic modifications (Jones & Baylin, 2002; Sparmann & van Lohuizen, 2006). On the other hand, besides the functional characterization of a single gene, it is important to establish how various genes interact with one another and in what succession. This notion was termed genomics and was conceived less than two decades ago (Guttmacher & Collins, 2002).
Epigenetic modifications affect the arrangement in gene expression by two main mechanisms: methylation of CpG rich islands present in the genomic DNA (Romano, Micheli, Pacilio & Giordano, 2000; Sigalotti et al., 2007) and methylation and/or acetylation of histones H3 and H4 (Jones & Baylin, 2002; Sparmann & van Lohuizen, 2006). Both events do not affect the DNA sequence at all and, yet, they have important implications on the regulation of transcriptional activity.
The methylation of cytosine residues in CpG motifs can silence a promoter by displacing transcription factors and, consequently, RNA polymerase II complexes (Romano, Micheli, Pacilio & Giordano, 2000). So far, four types of DNA methyltransferases (DNMT) that mediate the methylation of CpG islands have been identified: DNMT1, DNMT2, DNMT3a and DNMT3b (Sigalotti et al., 2007).
Histones H3 and H4 methylation and/or acetylation occur in lysine residues in the amino terminus of histonic tails (Fig. 2) (Kass, Pruss & Wolffe, 1997; Wade, Pruss & Wolffe, 1997). Methylation can also take place in arginine residues present in histone H3 and H4 tails (Stallcup, 2001).
Fig. 2.
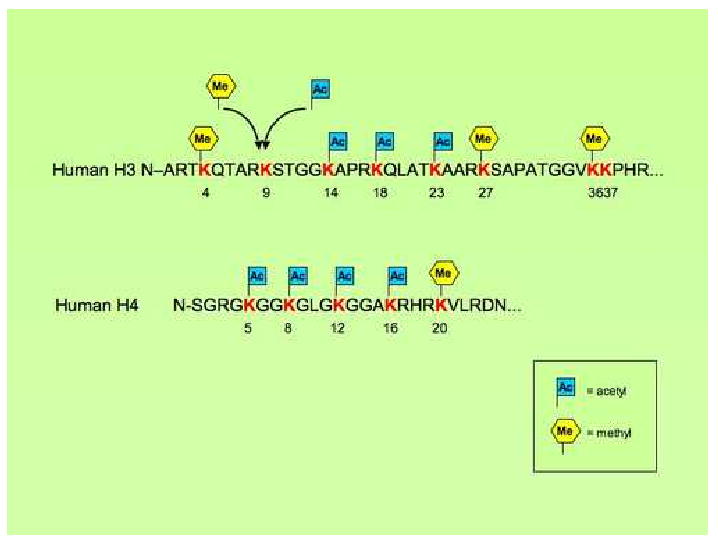
This figure shows the lysine residues in the amino terminus of the external tail of human histones H3 and H4 that can be susceptible to methylation and/or acetylation. Amino acids are reported in a single letter symbol. Numbers depict the lysines residues, which can either be methylated, or acetylated, with the only exception of lysine residue 9 in the tail of histone H3, where methylation and acetylation can both occur.
DNA methylation and histone H3 and H4 methylation and/or acetylation induce chromatin remodeling. Eukaryotic chromosomal DNA is packaged within the cellular nucleus with a roughly equal amount of proteins. Such macromolecular complexes are termed chromatin (Luger, 2006). Histones form nucleosomes, which are wrapped around by chromosomal DNA. The so-called nucleosome core particle (NCP) is the basic unit in chromatin structure and comprises an octameric histone core embraced by 147 base pairs of DNA organized in 1.65 superhelical turns. Each octameric histone core contains two copies of the following four histones: H2A, H2B, H3 and H4 (Fig. 3) (Park & Luger, 2006). The association between nucleosomes and DNA is very important for the regulation of gene expression (Luger, 2006). A tight association between nucleosomes and DNA results in chromatin condensation, with consequent suppression of gene expression (Lee, Hayes, Pruss & Wolffe, 1993; Luger, 2006). The condensation of chromatin confers a non-permissive configuration for gene expression, as various transcription factors cannot access the DNA template (Lee, Hayes, Pruss & Wolffe, 1993; Luger, 2006).
Fig. 3.
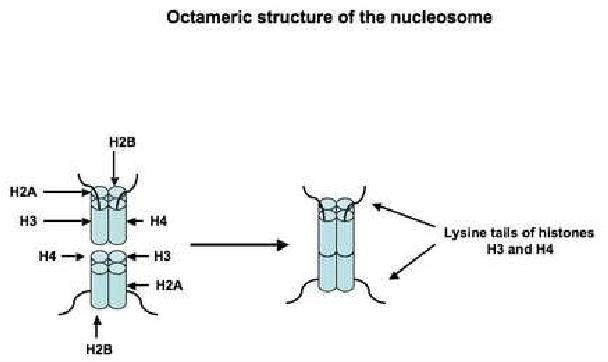
Schematic representation of the octameric structure of the nucleosome. The DNA 1.65 superhelical turns embracing the nucleosome are not shown.
Acetylation of lysine residues in external tails of histones H3 and/or H4 tends to reduce the interaction between DNA and nucleosomes, which, in turn, allows for the access of various transcription factors to the DNA (Herrera et al., 2000; Herrera, Schiltz & Bustin, 2000; Litt et al., 2001b; Marvin, Yau & Bradbury, 1990; Skiniotis, Moazed & Waltz, 2007; Wang, Moore, Laszckzak & Ausio, 2000). There are at least three possible mechanisms to interpret such a phenomenon. Acetylation, per se, can neutralize electrostatic interactions between basic lysine residues in histones tails and the DNA phosphodiester backbone (Lee, Hayes, Pruss & Wolffe, 1993; Walker, 1984). Additionally, histone tails acetylation induces conformational changes in the nucleosome, which tends to unfold by assuming an alpha-helical configuration (Herrera et al., 2000; Herrera, Schiltz & Bustin, 2000; Litt et al., 2001b; Marvin, Yau & Bradbury, 1990; Skiniotis, Moazed & Waltz, 2007; Wang, Moore, Laszckzak & Ausio, 2000). These two mechanisms involve conformational changes in nucleosome organization. Instead, the third possible mechanism envisions acetylated histone tails as mediators for the contact between transcription factors and nucleosomal DNA (Lee, Hayes, Pruss & Wolffe, 1993). Histone tail acetylation of lysine residues is carried out by histone acetyltransferases (HATs) (Rice & Allis, 2001). Interestingly, histone tails acetylation is a reversible process, as cellular enzymes termed histone deacetylases (HDACs) have the ability to remove the acetyl group from lysine residues (Rice & Allis, 2001).
In contrast to acetylation, lysine residues methylation does not affect the global charge of the histone H3 and H4 tails, which, however, tend to become more basic and hydrophobic, especially if they are bi- and tri-methylayed. On these grounds, the effect of histone tails methylation enhances the binding affinity of the nucleosome for DNA, which is an anionic macromolecule (Baxter & Byvoet, 1975; Byvoet, Shepherd, Hardin & Noland, 1972). This results in a tighter association between nucleosomes and genomic DNA, which has a propensity in conferring a non-permissive configuration for gene expression (Nielsen et al., 2001; Schotta et al., 2004; Stewart, Li & Wong, 2005). However, histone tails methylation does not always lead to suppression of transcriptional activity. In fact, histone tails methylation can have differential effects on gene expression, depending on the position of the methylated lysine residue (Rice & Allis, 2001). For instance, methylation of lysine 9 in histone H3 (H3-K9) and/or lysine 27 in histone H3 (H3-K27) is usually associated with a suppression of transcriptional activity, whereas methylation of H3-K4, H3-K36 and H3-K79 has a correlation with activation of transcription (Stewart, Li & Wong, 2005). In this respect, also the methylation of arginine residues in histones H3 and H4 tails is associated with active state of transcription (Stallcup, 2001). Mammalian arginine methyltransferases comprise: PRMT1, PRMT2/HRMT1L1, PRMT3, CARM1 and JBP1, whereas several proteins containing SET domains exhibit H3 lysine methyltransferases properties (Stallcup, 2001).
Histone tails methylation was considered an irreversible mechanism, although evidence accounting for the existence of an enzyme with histone demethylase activity was already reported in the early 1970s (Paik & Kim, 1973). With the rather recent production of antibodies against various forms of methylated histone tails, it was observed that under certain conditions the degree of arginine methylation was variable (Bannister & Kouzarides, 2005). This observation provided further support to the hypothesis that histone tails methylation can be reversible. In this regard, a final proof was provided following the identification of lysine-specific demethylase 1 (LSD1, alias BHC110, or p110) (Shi et al., 2004).
Studies on epigenetic modifications eventually led to the formulation of so-called epigenetic drugs for the treatment of cancer (Sigalotti et al., 2007; Xu, Parmigiani & Marks, 2007). Epigenetic drugs essentially fall in two categories: inhibitors of DNMT to remove aberrant patterns of DNA methylation (Sigalotti et al., 2007) and inhibitors of HDACs, which are termed HDACi (Xu, Parmigiani & Marks, 2007).
Inhibitors of DNMT aim at restoring the expression of cellular tumor suppressor genes and can be generically classified in nucleoside analogues and in non-nucleoside analogues. In nucleoside analogues, the cytidine was modified in order to neutralize DNMT activity. This class of DNMT inhibitors comprises: zebularine, 5-azacytidine, 5-aza-2′-deoxycytidine (5-AZA-CdR), 5,6-dihydro-5-azacytidine and 5-fluoro-2′-deoxycytidine (Yoo & Jones, 2006). Following the cellular uptake, kinases convert nucleosides analogues in nucleosides-like molecules, which are incorporated into the genomic DNA during the cell cycle S phase (Momparler, 2005). The inhibition of DNMT occurs at this stage. Conversely, non-nucleoside analogues are designed to inhibit DNMT activity without being incorporated into the DNA. This second category of DNMT inhibitors comprises: procaine (Villar-Garea, Fraga, Espada & Esteller, 2003), RG108 (Brueckner et al., 2005), (-)-epigallocatechin-3-gallate (Fang et al., 2003) and hydralazine (Arce et al., 2006).
Inhibition of HDACs by HDACi results in an increase acetylation pattern of histones and proteins that are involved in the regulation of gene expression, cell survival and cell proliferation (Xu, Parmigiani & Marks, 2007). Some of these non-histonic proteins comprise DNA binding transcriptional factors (p53, c-Myc, Bcl6, AML1, CREB, GATA-1, GATA-2, GATA-3, GATA-4, NF-kB, etc.), co-regulators of transcription (Rb, DEK, HMGI(Y), MSL-3, etc.), steroid receptors (ER-a, androgen receptor, glucocorticoid receptor), DNA repair enzymes (Ka70, TDG, WRN, NEIL2, FENI), chaperone protein HSP90, structural protein α-tubulin, inflammatory mediator HMGB1 and various viral factors (E1A, HIV Tat, SV40 T antigen, L-HDAg, S-HDAg).
So far, 18 HDACs have been identified and can be categorized into four classes, according to their degree of homology with their yeast counterparts (Bolden, Peart & Johnstone, 2006; Xu, Parmigiani & Marks, 2007). Chemically speaking, HDACi are based on aliphatic acids, hydroxamic acids, benzamides and cyclic tetrapeptides (Table 1) (Sigalotti et al., 2007; Xu, Parmigiani & Marks, 2007). The HDACi listed I Table 1 have been used either in phase I or phase II clinical trials, with the only exception of SAHA. The Food and Drug Administration, however, approved this compound for a clinical trial to treat cutaneous T-cell lymphoma (Xu, Parmigiani & Marks, 2007).
Table 1.
list of HDACi used in clinical trials (*SAHA has been approved for clinical trials).
| Chemical classification | Commercial names of the compounds |
|---|---|
| Aliphatic acids | Valproic acid |
| Phenylbutyrrate | |
| Savicol | |
| AN-9 | |
| Baceca | |
| Hydroxamate | LBH589 |
| ITF2357 | |
| FK228 | |
| PXD101 | |
| PCI24781 | |
| SAHA, or Zolinza, or vorinostat* | |
| Benzamide | MGCD0103 |
| MS-275 | |
| Cyclic tetrapeptide | Depsipeptide |
So-called epigenetic drugs have been utilized either alone, or in combination with other therapeutics in phase I or phase II clinical trials for the treatment of patients with hematological malignancies (Xu, Parmigiani & Marks, 2007) and other solid tumors, such as cancers of the lung, cervix, breast, head and neck, prostate and testis (Arce et al., 2006; Sigalotti et al., 2007; Xu, Parmigiani & Marks, 2007; Zembrano et al., 2005). Interestingly, epigenetic drugs seem to be well tolerated by patients (Xu, Parmigiani & Marks, 2007). Worryingly, a development of resistance to HDACi was reported among some patients. Naturally, such a finding poses a great deal of concern, especially because the cause of resistance to HDACi is not yet understood (Xu, Parmigiani & Marks, 2007).
Undoubtedly, genomics had important implications in the fields of developmental biology, stem cell research and experimental oncology (Giordano, Fucito, Romano & Marino, 2007; Guttmacher & Collins, 2002; Romano, 2005; Sparmann & van Lohuizen, 2006; Varmus, 2002). The development of genomic medicine programs is a highly ambitious goal, which may lead to discoveries of extreme importance in the study of disease, stem cell research and developmental biology. Microarray techniques are utilized for the profiling of altered gene expression patterns associated with a particular pathological condition. This information may elucidate the mechanism by which certain genetic elements may influence each other. The study of the interaction among multiple genetic factors has greatly contributed to better understand the various mechanisms associated with epigenetic modifications (Callinan & Feinberg, 2006; Feinberg, 2001; Sparmann & van Lohuizen, 2006; Valk-Lingbeek, Bruggeman & van Louizen, 2004). In this respect, an important finding was the identification of Polycomb Group (PcG) gene family, which orchestrates Homeobox (Hox) gene regulation in mammalians and has the function of maintaining silent specific sets of genes via chromatin modifications (Sparmann & van Lohuizen, 2006; Valk-Lingbeek, Bruggeman & van Louizen, 2004). On these grounds, PcG genes are essential factors in development, stem cell biology and cancer (Sparmann & van Lohuizen, 2006; Valk-Lingbeek, Bruggeman & van Louizen, 2004). Two distinct multifactor PcG complexes have been identified: polycomb repressive complex 1 (PRC1) and PRC2 (Valk-Lingbeek, Bruggeman & van Louizen, 2004). PRC1 contains more than 10 factors, including the proto-oncogene Bmi1, along with HPC, HPH and SCML (Pasini, Bracken & Helin, 2004). Conversely, PRC2 is a smaller complex that contains EED, SUZ12, EZH2 and the histone binding protein RpAp46. The factor EZH2 contains a SET domain, which confers histone methyl transferase (HMT) activity to PRC2 (Kuzmichev, et al. 2002). PRC2-mediated HMT is specific for histone H3 lysine residues 9 and 27 (Pasini, Bracken & Helin, 2004). More importantly, the presence of a functional PRC2 allows for the targeting of PRC1 to specific chromatin regions (Czermin et al., 2002; Moss & Wallrath, 2007). Thus, PRC1 and PRC2 contrive in repressing transcription in a number of genes. The pRb/E2F pathway, in turn, regulates the transcription of EZH2, EED and SUZ12 PcG genes of the PRC2 complex (Pasini, Bracken & Helin, 2004). Several studies have demonstrated the mechanism of EZH2 and EED gene regulation in cell cycle progression and in normal and malignant cell proliferation (Moss & Wallrath, 2007; Pasini, Bracken & Helin, 2004). In this respect, high levels of EZH2 expression are correlated with tumor proliferation and poor prognosis in breast cancer and lymphoma (Moss & Wallrath, 2007). It was also demonstrated that SET domain methyltransferase activity comes into play in breast cancer metastasis (Kleer et al., 2003). Additionally, Bmi-1 and EZH2 are both over-expressed in cancer cells (Dukers et al., 2004; Raaphorst et al., 2003). Bmi-1 over-expression decreases p16 and p14ARF transcriptional activity in the INK4A locus (Kim & Sharpless, 2006; Moss & Wallrath, 2007). Loss of INK4A/ARF/INK4B locus in the chromosome 9p21 is implicated in the carcinogenesis of several human malignancies (Kim & Sharpless, 2006).
Bmi-1 is an essential regulator in the self-renewal of adult hematopoietic stem cells and neuronal stem cells (Valk-Lingbeek, Bruggerman & van Lohuizen, 2004; Park et al., 2003). A conserved Bmi-1-driven gene expression pattern was similarly engaged in normal adult stem cells and eleven types of human tumors, such as lymphoma, mantle cell lymphoma, acute myeloid leukemia, mesothelioma, medulloblastoma, malignant glioma, and cancers of the breast, prostate, ovaries, bladder and lung (Glinsky, Berezovska & Glinskii, 2005). Such a pattern of Bmi-1-driven gene expression was termed eleven-gene signature and was found to be associated with poor prognosis and failure of therapy among oncological patients (Glinsky, Berezovska & Glinskii, 2005). A five-gene signature was also identified in non-small-cell lung cancer as a predictor of relapse-free and overall survival among patients affected by this malignancy (Chen et al., 2007). Another gene-signature for invasiveness was found to be associated with metastasis-free survival and overall survival in patients with breast cancer (Liu et al., 2007). Interestingly, this invasiveness gene-signature was also associated with the prognosis of lung cancer, prostate cancer and medulloblastoma (Liu et al., 2007).
Genomic medicine has the potential of developing a powerful technology leading towards personalized prognosis and therapy for patients with cancer (Herbst & Lippman, 2007). In this respect, new algorithms must be designed and should comprise genomic, proteomic, molecular imaging and clinical features (Herbst & Lippman, 2007).
Genomic can also be used to better understand the role and the characteristics of cancer stem cells, which play a primary role in tumorigenesis, maintenance of the tumor mass and in relapse of the disease by regenerating neoplastic cells with enhanced resistance to chemo- and radio-therapy (Bao et al., 2006; Giordano, Fucito, Romano & Marino, 2007; Romano, 2005). Malignant and normal stem cells share extensive phenotypic and functional features. In order to develop novel therapeutics for the selective destruction of cancer stem cells, it is essential to determine differential gene expression patterns between normal and malignant stem cells (Giordano, Fucito, Romano & Marino, 2007; Romano, 2005). For instance, a group of investigators demonstrated that dependence on tumor suppressor Pten gene allows for a distinction between normal hematopoietic stem cells and leukemia stem cells in a mouse model (Yilmaz et al., 2006). On one hand, it was observed that depletion of Pten in somatic hematopoietic stem cells resulted in myeloproliferative malady in mice within days and to transplantable leukemia within weeks. On the other hand, Pten depletion primed a proliferation of hematopoietic stem cells, which produced a high number of progeny cells lacking parental hematopoietic stem cells and, therefore, failed in regenerating the bone marrow in irradiated recipient mice. The main mediator of the effects related to Pten depletion was mTOR. Rapamycin targeting of mTOR was able to selectively destroy leukemia stem cells, without affecting normal hematopoietic stem cells. Moreover, rapamycin-mediated targeting of mTOR could restore the functions of normal hematopoietic stem cells in irradiated recipient mice (Yilmaz et al., 2006). This is one of the first reports accounting for mechanistic diversity between normal stem cells and their malignant counterparts. These differential mechanisms allowed for the selective elimination of cancer stem cells in the mouse model.
The study of the interaction among the factors encoded by multiple genes can provide a substantial contribution in interpreting biological processes that are apparently paradoxical. For instance, the already mentioned decreased expression of ER-α mediated by the tumor suppressor pRb2/p130 gene (Fig. 1) (Macaluso et al., 2003; Macaluso, Montanari & Giordano, 2006; Macaluso et al., 2007). The function of a tumor suppressor gene, such as pRb2/p130, consists in limiting cell proliferation (Giordano, Rossi, Romano & Bagella, 2007). In case of abnormal cell proliferation rate, tumor suppressor genes tend to arrest cell cycle progression by targeting the promoters driving the expression of genes encoding for factors that can be beneficial for cell proliferation. In this context, pRb2/p130 may be associated with a variety of factors that produce a non-permissive configuration for ER-α transcription, such as the pRb2/p130-E2F4-HDAC1-SUV39-H1-DNMT1 multi-molecular complex (Fig. 1) (Macaluso et al., 2003; Macaluso, Montanari & Giordano, 2006; Macaluso et al., 2007). The pRb2/p130-mediated suppression of ER-α expression is intended to contrast malignant cell proliferation. However, the loss of ER-α is one of the key events in development and progression of breast cancer, as it imparts resistance to hormone therapy to neoplastic cells (Macaluso et al., 2003; Macaluso, Montanari & Giordano, 2006; Macaluso et al., 2007).
4. Conclusion
The field of experimental oncology is frantically striving to identify novel genetic and cellular markers to improve both the efficacy of therapeutic interventions and preventive diagnosis for breast cancer, with a particular emphasis on patients with metastatic disease. Undeniably, the study of genetic and epigenetic alterations is already shedding very useful insights into a better understanding of breast cancer pathogenesis and its progression. Further advances in breast cancer research may ultimately result in the development of novel therapeutics and diagnostics to better challenge this malignancy, without affecting the health and quality of life of breast cancer survivors.
Acknowledgments
This manuscript is dedicated to the memory of Dr. Bice Perussia. The authors are grateful to Miss. Jasmine Rupert for her useful comments. This work was supported by the Sbarro Health Research Organization (SHRO) and by NIH grants (AG).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Albrektsen G, Heuch I, Thoresen S, Kvale G. Clinical stage of breast cancer by parity, age at birth, and time since birth: a progressive effect of pregnancy hormones? Cancer Epidemiol Biomarkers Prev. 2006;13(1):65–69. doi: 10.1158/1055-9965.EPI-05-0634. [DOI] [PubMed] [Google Scholar]
- Andrieu N, Goldgar DE, Easton DF, Rookus M, Brohet R, Antoniou AC, et al. Pregnancies, breast-feeding, and breast cancer rosk in the international BRCA1/2 carrier cohort study (BCCS) J Natl Cancer Inst. 2006;98(8):535–544. doi: 10.1093/jnci/djj132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arce C, Segura-Pacheco B, Perez-Cardenas E, Taia-Chaveb L, Candelaria M, Duennas-Gonzalez A. Hydralazine target: from blood to the epigenome. J Transl Med. 2006;4:10. doi: 10.1186/1479-5876-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsian AA, Zaleniuch-Jacquotte A, Lukanova A, Afanasveva Y, Katz J, Levitz M, Del Priore G, Toniolo P. Effects of parity on pregnancy hormonal profiles across ethnic groups with a diverse incidence of breast cancer. Cancer Epidemiol Biomarkers Prev. 2006;15(11):2123–2130. doi: 10.1158/1055-9965.EPI-06-0470. [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T. Reversing histone methylation. Nature. 2005;436(7054):1103–1106. doi: 10.1038/nature04048. [DOI] [PubMed] [Google Scholar]
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stm cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- Baxter CS, Byvoet P. Intercalating agents as probes of the spatial relationship between chromatin components. Biochem Biophys Res Commun. 1975;63(1):286–291. doi: 10.1016/s0006-291x(75)80041-6. [DOI] [PubMed] [Google Scholar]
- Bolden JE, Peart MJ, Johnstone RW. Anticancer activites of histone deactylase inhibitors. Nat Rev Drug Discov. 2006;5(9):769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- Bradbury AR, Olopade OI. Genetic susceptibility to breast cancer. 2007 doi: 10.1007/s11154-007-9038-0. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Braun S, Vogl FD, Naume B, Janni W, Osborne MP, Coombes RC, et al. A pooled analysis of bone marrow micrometastasis in Breast cancer. N Engl J Med. 353(8):793–802. doi: 10.1056/NEJMoa050434. [DOI] [PubMed] [Google Scholar]
- Braun S, Vogl FD, Janni W, Marth C, Schlimok G, Pantel K. Evaluation of bone marrow in breast cancer patients: prediction of clinical outcome and response to therapy. Breast. 2003;12(6):397–404. doi: 10.1016/s0960-9776(03)00143-7. [DOI] [PubMed] [Google Scholar]
- Brueckner B, Boy RG, Siedlecki P, Musch T, Kliem HC, Zielenkiewicz P, Suhai S, Wiessler M, Lyko F. Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferase. Cancer Res. 2005;65(14):6305–6311. doi: 10.1158/0008-5472.CAN-04-2957. [DOI] [PubMed] [Google Scholar]
- Burstein HJ, Parker LM, Savoie J. Phase II trial of the anti-VEGF antibody bevacizumab in combination with vinorelbine for refractory advanced breast cancer. Breast Cancer Res Treat. 2002;76(Suppl1):S115. abstract 446. [Google Scholar]
- Byvoet P, Shepherd GR, Hardin JM, Noland BJ. The distribution and turnover of labeled methyl groups in histone fractions of cultured mammalian cells. Arch Biochem Biophys. 1972;148(2):558–567. doi: 10.1016/0003-9861(72)90174-9. [DOI] [PubMed] [Google Scholar]
- Callinan PA, Feinberg AP. The emrging science of epigenomics. Hum Mol Genet. 2006;15(Spec 1):R95–101. doi: 10.1093/hmg/ddl095. [DOI] [PubMed] [Google Scholar]
- Cardillo MR, Yap E, Castagna G. Molecular genetic analysis of TGF beta1 in breast cancer. J Clin Cancer Res. 1997;16(1):57–63. [PubMed] [Google Scholar]
- Cascio S, Bartella V, Garofalo C, Russo A, Giordano A, Surmacz E. Insulin-like growth factor 1 differentially regulates estrogen receptor-dependent transcription at estrogen response element and AP-1 sites in breast cancer cells. J Biol Chem. 2007;282(6):3498–3506. doi: 10.1074/jbc.M606244200. [DOI] [PubMed] [Google Scholar]
- Chabner BA, Roberts TG., Jr Chemotherapy and the war on cancer. Nat Rev Cancer. 2005;5:65–72. doi: 10.1038/nrc1529. [DOI] [PubMed] [Google Scholar]
- Cheema B, Gaul CA, Lane K, Fiatarone Singh MA. Progressive resistance training in beast cancer: a systematic review of clinical trials. Breast Cancer Res Treat. 2007 doi: 10.1007/s10549-007-9638-0. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Chen HY, Yu SL, Chen CH, Chang GC, Chen CY, Yuan A, Cheng CL, Wang CH, et al. A five-gene signature and clinical outcome in non-small-cell lung cancer. N Engl J Med. 2007;356(1):11–20. doi: 10.1056/NEJMoa060096. [DOI] [PubMed] [Google Scholar]
- Chen WY, Colditz GA. Risk factors and hormone-receptor status: epidemiology, risk-prediction models and treatment implications for breast cancer. Nat Clin Pract Oncol. 2007;4(7):415–423. doi: 10.1038/ncponc0851. [DOI] [PubMed] [Google Scholar]
- Chia SKL, Speers C, Kang D, Yachlova Y, Malfair, Taylor S, Barnett J, et al. The impact of new chemotherapeutic and hormonal agents on the survival of women with metastatic breast cancer (MBC) in a population based cohort. Proc Am Soc Clin Oncol. 2003;22:6. abstract 22. [Google Scholar]
- Cobleigh MA, Langmuir VK, Sledge GW, Miller KD, Haney L, Novotny WF, et al. A phase I-II dose-escalation trial of bevacizumab in previously treated metastatic breast cancer. Semin Oncol. 2003;30(Suppl16):117–124. doi: 10.1053/j.seminoncol.2003.08.013. [DOI] [PubMed] [Google Scholar]
- Czermin B, Melfi R, McCabe D, Seitz V, Imhof A, Pirrotta V. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that mraks chromosomal Polycomb sites. Cell. 2002;111(2):185–196. doi: 10.1016/s0092-8674(02)00975-3. [DOI] [PubMed] [Google Scholar]
- Dukers DF, van Galen JC, Giroth C, Jansen P, Sewalt RG, Otte AP, Kluin-Nelemans HC, Meijer CJ, Raaphorst FM. Unique polycomb gene expression pattern in Hodgkin's lymphoma and Hodgkin's lymphoma-derived cell lines. Am J Pathol. 2004;164(7):873–881. doi: 10.1016/S0002-9440(10)63175-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang MZ, Wang Y, Ai N, Hou Z, Sun Y, Lu H, Welsh W, Yang CS. Tea polyphenol (-)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res. 2003;63(22):7563–7570. [PubMed] [Google Scholar]
- Feinberg AP. Methylation meets genomics. Nat Genet. 2001;27(1):9–10. doi: 10.1038/83825. [DOI] [PubMed] [Google Scholar]
- Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- Foulkes WD, Metcalfe K, Sun P, Hanna WM, Lynch HT, Ghadirian P, Tung N, et al. Estrogen receptor status in BRCA1- and BRCA2-related breast cancer: the influence of age, grade, and histological type. Clin Cancer Res. 10:2029–2034. doi: 10.1158/1078-0432.ccr-03-1061. [DOI] [PubMed] [Google Scholar]
- Gan DD, Macaluso M, Cinti C, Khalili K, Giordano A. How does a normal cell become a cancer cell? J Exp Clin Cancer Res. 2003;22(4):509–516. [PubMed] [Google Scholar]
- Giordano A, Fucito A, Romano G, Marino IR. Carcinogenesis and environment: the cancer stem cell hypothesis and implications for the development of novel therapeutics and diagnostics. Front Biosci. 2007;12:3475–3482. doi: 10.2741/2328. [DOI] [PubMed] [Google Scholar]
- Giordano A, Rossi A, Romano G, Bagella L. Tumor suppressor pRb2/p130 gene and its derived product Spa310 spacer domain as perspective candidates for cancer therapy. J Cell Physiol. 2007;213(2):403–406. doi: 10.1002/jcp.21225. [DOI] [PubMed] [Google Scholar]
- Glinsky GV, Berezovska O, Glinskii AB. Micorarray analysis reveals a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J Clin Invest. 2005;115(6):1503–1521. doi: 10.1172/JCI23412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon MS, Margolin K, Talpaz M, Sledge GW, Jr, Holmgren E, Benjamin R, et al. Phase I safety and pharmacokinetic study of recombinant human anti-vascular endothelial growth factor in patients with advanced cancer. J Clin Oncol. 2001;19:843–840. doi: 10.1200/JCO.2001.19.3.843. [DOI] [PubMed] [Google Scholar]
- Gumus M, Yumuk PF, Salepci T, Allustaoglu M, Dane F, Ekenel M, Basaran G, Kaya H, Barisik N, Turhal NS. HPV DNA frequency and subset in human breast cancer patients' normal and tumoral tissue samples. J Exp Clin Cancer Res. 2006;25(4):515–521. [PubMed] [Google Scholar]
- Guttmacher AE, Collins FS. Genomic medicine - a primer. N Engl J Med. 2002;347(19):1512–1520. doi: 10.1056/NEJMra012240. [DOI] [PubMed] [Google Scholar]
- Ha M, Mabuchi K, Sigurdson AJ, Freedman DM, Linet MS, Doody MM, Hauptmann M. Smocking cigarettes before first childbirth and risk of breast cancer. Am J Epidemiol. 2007;166(1):55–61. doi: 10.1093/aje/kwm045. [DOI] [PubMed] [Google Scholar]
- Herbst RS, Lippman SM. Molecular signatures of lung cancer - toward personalized therapy. N Engl J Med. 2007;356(1):76–78. doi: 10.1056/NEJMe068218. [DOI] [PubMed] [Google Scholar]
- Herrera JE, Schiltz RL, Bustin M. The accessibility of histone H3 tails in chromatin modulates their acetylation by P300/CBP-associated factor. J Biol Che. 2000;275(17):12994–12999. doi: 10.1074/jbc.275.17.12994. [DOI] [PubMed] [Google Scholar]
- Herrera JE, West KL, Schiltz RL, Nakatani Y, Bustin M. Histone H1 is a specific repressor of core histone acetylation in chromatin. Mol Cell Biol. 2000;20(2):523–529. doi: 10.1128/mcb.20.2.523-529.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh JY, Calvo A, Stafford J, Cheung M, Kaman R, Philip D, et al. Inhibitors of VEGF receptors significantly impairs mammary cancer growth in C3(1)/Tag transgenic mice through antiangiogenic and non-antiangiogenic mechanisms. Oncogene. 24:790–800. doi: 10.1038/sj.onc.1208221. [DOI] [PubMed] [Google Scholar]
- Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- Imay K, Tanaka A. Comparing antibody and small molecule therapy for cancer. Nat Rev Cancer. 2006;6:714–727. doi: 10.1038/nrc1913. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Want E, Murray T, Xu J, Snigal C, et al. Cancer Statistics. CA Cancer J Clin. 2006;56(2):106–130. doi: 10.3322/canjclin.56.2.106. [DOI] [PubMed] [Google Scholar]
- Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- Karakosta A, Golias C, Charalabopoulos A, Peschos D, Batistatou A, Charalabopoulos K. Genetic model of human cancer as a multistep process, Paradigm models of colorectal cancer, breast cancer, and chronic myelogenous and acute lymphoblastic leukemia. J Exp Clin Cancer Res. 2005;24(4):505–514. [PubMed] [Google Scholar]
- Kass SU, Pruss D, Wolffe AP. How does DNA methylation repress transcription? Trends Genet. 1997;13(11):444–449. doi: 10.1016/s0168-9525(97)01268-7. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Kono K, Mimura K, Mitsui F, Sugai H, Akaike H, Fujii H. Targeting EGFR and HER-2 with cetuximab and trastuzumab-mediated immunotherapy in oesophageal squamous cell carcinoma. Br J Cancer. 2007 doi: 10.1038/sj.bjc.6603885. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KJ, Li B, Winer J, Armanini M, Gillet N, Phillips HS, et al. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumor growth in vivo. Nature. 1993;362:841–844. doi: 10.1038/362841a0. [DOI] [PubMed] [Google Scholar]
- Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127(2):265–275. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, Ghosh D, Sewalt RG, Otte AP, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci USA. 2003;100(20):11606–11611. doi: 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhl CK. High-risk screening: multi-modality surveillance of women at high risk for breast cancer (proven or suspected carriers of a breast cancer susceptibility gene) J Exp Clin Cancer Res. 2002;21(Suppl 3):103–106. [PubMed] [Google Scholar]
- Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002;16(22):2893–2905. doi: 10.1101/gad.1035902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson JS, Gunzburg WH, Whittaker NJ. Viruses and human breast cancer. Future Microbiol. 2006;1:33–51. doi: 10.2217/17460913.1.1.33. [DOI] [PubMed] [Google Scholar]
- Lee DY, Hayes JJ, Pruss D, Wolffe AP. A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell. 1993;72(1):73–84. doi: 10.1016/0092-8674(93)90051-q. [DOI] [PubMed] [Google Scholar]
- Litt MD, Simpson M, Gaszner M, Allis CD, Felsenfeld G. Correlation between histone lysine methylation and developmental changes at the chicken β-globin locus. Science. 2001;293(5539):2453–2455. doi: 10.1126/science.1064413. [DOI] [PubMed] [Google Scholar]
- Litt MD, Simpson M, Recillas-Targa F, Prioleau MN, Felsenfeld G. Transitions in histone acetylation reveal boundaries of three separately regulated neighboring loci. EMBO J. 2001b;20(9):2224–2235. doi: 10.1093/emboj/20.9.2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Wang X, Chen GY, Dalerba P, Gurney A, Hoey T, Sherlock G, Lewicki J, Shedden K, Clarke MF. The prognostic role of a gene signature from tumorigenic breast-cancer cells. N Engl J Med. 2007;356(3):217–226. doi: 10.1056/NEJMoa063994. [DOI] [PubMed] [Google Scholar]
- Luger K. Dynamic nucleosomes. Chromosome Res. 2006;14:5–16. doi: 10.1007/s10577-005-1026-1. [DOI] [PubMed] [Google Scholar]
- Macaluso M, Montanari M, Giordano A. Rb family proteins as modulators of gene expression and new aspects regarding the interactions with chromatin remodeling enzymes. Oncogene. 2006;25(38):5263–5267. doi: 10.1038/sj.onc.1209680. [DOI] [PubMed] [Google Scholar]
- Macaluso M, Paggi M, Giordano A. Genetic and epigenetic alterations as hallmarks of the intricate road to cancer. Oncogene. 2003;22(42):6472–6478. doi: 10.1038/sj.onc.1206955. [DOI] [PubMed] [Google Scholar]
- Macaluso M, Cinti C, Russo G, Russo A, Giordano A. pRb2/p130-E2F4/5-HDAC1-SUV39H1-p300 and pRb2/p130-E2F4/5-HDAC1-SUV39H1-DNMT1 multimolecular complexes mediate the transcription of estrogen receptor-α in breast cancer. Oncogene. 2003;22(23):3511–3517. doi: 10.1038/sj.onc.1206578. [DOI] [PubMed] [Google Scholar]
- Macaluso M, Montanari M, Noto PB, Gregorio V, Bronner C, Giordano A. Epigenetic modulation of estrogen receptor-α by pRb family proteins: a novel mechanism in breast cancer. Cancer Res. 2007;67(16):7731–7737. doi: 10.1158/0008-5472.CAN-07-1476. [DOI] [PubMed] [Google Scholar]
- Margolin K, Gordon MS, Holigren E, Gandreault J, Novotny W, Fyfe G, et al. Phase Ib trial of intravenous recombinant humanized monoclonal antibody to vascular endothelial growth factor in combination with chremotherapy in patients with advanced cancer: pharmacologic and long-term safety data. J Clin Oncol. 2001;19:851–856. doi: 10.1200/JCO.2001.19.3.851. [DOI] [PubMed] [Google Scholar]
- Marvin KW, Yau P, Bradbury EM. Isolation and characterization of acetylated histones H3 and H4 and their assembly into nucleosomes. J Biol Chem. 1990;265(32):19839–19847. [PubMed] [Google Scholar]
- Maskarinec G, Zhang Y, Takata Y, Pagano I, Shumav DM, Goodman MT, Le Marchand L, et al. Trends of breast cancer incidence and risk factor prevalence over 25 years. Breast Cancer Res Treat. 2006;98(1):45–55. doi: 10.1007/s10549-005-9129-0. [DOI] [PubMed] [Google Scholar]
- Menes TS, Ozao J, Kim U. Breast cancer and ethnicity: strong association between reproductive risk factors and estrogen receptor status in Asian patients - a retrospective study. Breast J. 2007;13(4):352–358. doi: 10.1111/j.1524-4741.2007.00442.x. [DOI] [PubMed] [Google Scholar]
- Miller KD. E2100: a phase III trial of paclitaxel versus paclitael/bevacizumab for metastatic breast cancer. Clin Breast Cancer. 2003;3:421–422. doi: 10.3816/CBC.2003.n.007. [DOI] [PubMed] [Google Scholar]
- Momparler RL. Epigenetic therapy of cancer with 5-aza-2′-deoxycytidine (decitabine) Senim Oncol. 2005;42(3 Suppl 2):S9–16. doi: 10.1053/j.seminoncol.2005.07.008. [DOI] [PubMed] [Google Scholar]
- Moss TJ, Wallrath LL. Connections between epigenetic gene silencing and human disease. Mutat Res. 2007;618(12):163–174. doi: 10.1016/j.mrfmmm.2006.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen SJ, Schneider R, Bauer UM, Bannister AJ, Morrison A, O'Carrol D, Firestein R, Cleary M, Jenuwein T, Herrera RE, Kouzarides T. Rb targets histone H3 methylation and HP1 to promoters. Nature. 2001;412(6845):561–565. doi: 10.1038/35087620. [DOI] [PubMed] [Google Scholar]
- Paik WK, Kim S. Enzymatic demethylation of calf thymus histones. Biochem Biophys Res Commun. 1973;51(3):781–788. doi: 10.1016/0006-291x(73)91383-1. [DOI] [PubMed] [Google Scholar]
- Park IK, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL, Morrison SJ, Clarke MF. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423(6937):302–305. doi: 10.1038/nature01587. [DOI] [PubMed] [Google Scholar]
- Park YJ, Luger K. Structure and function of nucleosome assembly proteins. Biochem Cell Biol. 2006;84(4):549–558. doi: 10.1139/o06-088. [DOI] [PubMed] [Google Scholar]
- Pasini D, Bracken AP, Helin K. Polycomb group proteins in cell cycle progression and cancer. Cell Cycle. 2004;3(4):396–400. [PubMed] [Google Scholar]
- Raaphorst FM, Meijer CJ, Fieret E, Blokzijl T, Mommers E, Buerger H, Packeisen J, Sewalt RA, Otte AP, van Diest PJ. Poorly differentiated breast carcinoma is associated with increased expression of the human polycomb group EZH2 gene. Neoplasia. 2003;5(6):481–488. doi: 10.1016/s1476-5586(03)80032-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radice P. Mutations of BRCA genes in hereditary breast and ovarian cancers. J Exp Clin Cancer Res. 2002;21 3:9–12. [PubMed] [Google Scholar]
- Rennert G, Bisland-Naggan S, Barnett-Griness O, Bar-Joseph N, Zhang S, Rennert HS, Narod SA. Clinical outcomes of breast cancer in carriers of BRCA1 and BRCA2 mutations. N Engl J Med. 2007;357(2):119–123. doi: 10.1056/NEJMoa070608. [DOI] [PubMed] [Google Scholar]
- Rice JC, Allis CD. Histone methylation versus histone acetylation: new insights into epigenetic regulation. Curr Opin Cell Biol. 2001;13(3):263–273. doi: 10.1016/s0955-0674(00)00208-8. [DOI] [PubMed] [Google Scholar]
- Romano G. The role of adult stem cells in carcinogenesis. Drug News Perspect. 2005;18(9):555–559. doi: 10.1358/dnp.2005.18.9.953667. [DOI] [PubMed] [Google Scholar]
- Romano G. The complex biology of the receptor for the insulin-like growth factor-1. Drug News Perspect. 2003;16(8):525–531. doi: 10.1358/dnp.2003.16.8.829351. [DOI] [PubMed] [Google Scholar]
- Romano G, Micheli P, Pacilio C, Giordano A. Latest development in gene transfer technology: achievements, perspectives, and controversies over therapeutic applications. Stem Cells. 2000;18(1):19–39. doi: 10.1634/stemcells.18-1-19. [DOI] [PubMed] [Google Scholar]
- Russo A, Calo V, Agnese V, Bruno I, Corsale S, Augello C, Gargano G, Barbera F, Cascio S, Intrivici C, et al. BRCA1 genetic testing in 106 breast and ovarian cancer families from southern Italy (Sicily): a mutational analyses. Breast Cancer Res Treat. 2007 doi: 10.1007/s10549-006-9456-9. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Schotta G, Lachner M, Sarma K, Ebert A, Sengupta R, Reader G, Reinberg D, Jenuwein T. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 2004;18(11):1251–1262. doi: 10.1101/gad.300704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shema L, Ore L, Ben-Shakar M, Hai M, Linn S. The association between breastfeeding and breast cancer occurrence among Israeli Jewish women: a case control study. J Cancer Res, Oncol. 2007;133(8):539–546. doi: 10.1007/s00432-007-0199-8. [DOI] [PubMed] [Google Scholar]
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119(7):941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Siamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu proto-oncogene. Science. 1987;235:177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- Siamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, et al. Use of chemotherapy plus a monocloncal antibody against HER2 for metastatic breast cancer that overexpresses HER-2. N Engl J Med. 1989;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- Sigalotti L, Fratta E, Coral S, Cortini E, Covre A, Nicolay HJM, Anzalone L, et al. Epigenetic drugs as pleiotropic agents in cancer treatment: biomolecular aspects and clinical applications. J Cell Physiol. 2007;212(2):330–344. doi: 10.1002/jcp.21066. [DOI] [PubMed] [Google Scholar]
- Skiniotis G, Moazed D, Waltz T. Acetylated histone tail peptides induce structural rearrangements in the RSC chromatin remodeling complex. J Biol Chem. 2007;282(29):20804–20808. doi: 10.1074/jbc.C700081200. [DOI] [PubMed] [Google Scholar]
- Smith MCP, Luker KE, Garbow JR, Prior JL, Jackson E, Piwnica-Worms D, Luker GD. CXCR4 regulates growth of both primary and metastatic breast cancer. Cancer Res. 2004;64(23):8604–8612. doi: 10.1158/0008-5472.CAN-04-1844. [DOI] [PubMed] [Google Scholar]
- Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer. 2006;6(11):846–856. doi: 10.1038/nrc1991. [DOI] [PubMed] [Google Scholar]
- Stallcup MR. Role of protein methylation in chromatin remodeling and transcriptional regulation. Oncogene. 2001;20(24):3014–3020. doi: 10.1038/sj.onc.1204325. [DOI] [PubMed] [Google Scholar]
- Stewart MD, Li J, Wong J. Relationship between histone H3 lysine 9 methylation, transcription repression, and heterochromatin protein 1 recruitment. Mol Cell Biol. 2005;25(7):2525–2538. doi: 10.1128/MCB.25.7.2525-2538.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valk-Lingbeek ME, Bruggeman SWM, van Louizen M. Stem cells and cancer: the polycomb connection. Cell. 2004;118(4):409–418. doi: 10.1016/j.cell.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Varmus H. Getting ready for gene-based medicine. N Engl J Med. 2002;347(19):1526–1527. doi: 10.1056/NEJMe020119. [DOI] [PubMed] [Google Scholar]
- Villar-Garea A, Fraga MF, Espada J, Esteller M. Procaine is a DNA-demethylating agent with growth-inhibitory effects in human cancer cells. Cancer Res. 2003;63(16):4984–4989. [PubMed] [Google Scholar]
- Vogel VG, Taioli E. Have we found the ultimate risk factor for breast cancer? J Clin Oncol. 2006;24(12):1791–1794. doi: 10.1200/JCO.2005.05.4122. [DOI] [PubMed] [Google Scholar]
- Wade PA, Pruss D, Wolffe AP. Histone acetylation: chromatin in action. Trends Biochem Sci. 1997;22(4):128–132. doi: 10.1016/s0968-0004(97)01016-5. [DOI] [PubMed] [Google Scholar]
- Walker IO. Differential dissociation of histone tails from core chromatin. Biochemistry. 1984;23(23):5622–5628. doi: 10.1021/bi00318a037. [DOI] [PubMed] [Google Scholar]
- Wang X, Moore SC, Laszckzak M, Ausio J. Acetylation increases the alpha-helical content of the histone tails of the nucleosome. J Biol Chem. 2000;275(45):35013–35020. doi: 10.1074/jbc.M004998200. [DOI] [PubMed] [Google Scholar]
- Walsh T, King MC. Ten genes for inherited breast cancer. Cancer Cell. 2007;11(2):103–105. doi: 10.1016/j.ccr.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Welcsh PL, Owens KN, King MC. Insights into the functions of BRCA1 and BRCA2. Trends Genet. 2000;16(2):69–74. doi: 10.1016/s0168-9525(99)01930-7. [DOI] [PubMed] [Google Scholar]
- Wicha MS, Lin S, Dontu G. Cancer stem cells: an old idea-a paradigm shift. Cancer Res. 2006;66(4):1883–1890. doi: 10.1158/0008-5472.CAN-05-3153. [DOI] [PubMed] [Google Scholar]
- Widakowich C, de Azambula E, Gil T, Dinh P, Awada A, Piccart-Gebhart M. Molecular targeted therapies in breast cancer: Where are we now? Int J Biochem Cell Biol. 2007;39(78):1375–1387. doi: 10.1016/j.biocel.2007.04.015. [DOI] [PubMed] [Google Scholar]
- Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26(37):5541–5552. doi: 10.1038/sj.onc.1210620. [DOI] [PubMed] [Google Scholar]
- Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, Morrison SJ. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441(7092):475–482. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- Yoo CB, Jones PA. Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov. 2006;5(1):37–50. doi: 10.1038/nrd1930. [DOI] [PubMed] [Google Scholar]
- Zembrano P, Segura-Pacheco B, Perez-Cardenaz E, Cetina L, Revilla-Vasquez A, Taia-Chavez L, et al. A phase I study of hydralazine to demethylate and reactvate the expression of tumor suppressor genes. BMC Cancer. 2005;5(1):44. doi: 10.1186/1471-2407-5-44. [DOI] [PMC free article] [PubMed] [Google Scholar]