

Abstract
Uracil DNA glycosylase (UNG) is a powerful DNA repair enzyme that has been shown to stabilize a glycosyl cation reaction intermediate and a related tight binding inhibitor using electrostatic interactions with the +1 and −1, but not the +2, phosphodiester group of the single-stranded DNA substrate Ap2+Ap1+Up1−ApA. These experimental results differed considerably from computational findings using duplex DNA, where the +2 phosphate was found to stabilize the transition state by ~5 kcal/mol, suggesting that UNG uses different catalytic strategies with single-stranded and double-stranded DNA substrates. In addition, the computational studies indicated that the conserved and positively charged His 148 (which hydrogen bonds to the +2 phosphate) destabilized the glycosyl cation intermediate by 6–8 kcal/mol through anticatalytic electrostatic interactions. To evaluate these interesting proposals, we measured the kinetic effects of neutral methylphosphonate (MeP) stereoisomers at the +1 and +2 positions of a 12-mer dsDNA substrate and also the catalytic contribution and ionization state of His 148. For MeP substitutions at the +1 position, single-turnover kinetic studies showed that the activation barrier was increased by 9.8 and 3.1 kcal/mol, corresponding to a stereoselectivity of nearly 40000-fold for the respective MeP isomers. Identical to the findings with ssDNA, MeP substitutions at the +2 position resulted in only small changes in the activation barrier (±0.3 kcal/mol), with little stereoselectivity (~4-fold). However, the H148A mutation destabilizes both the ground state and transition states by 2.4 and 4.3 kcal/mol, respectively. Thus, His 148 is catalytic because it stabilizes the transition state to a greater extent (1.9 kcal/mol) than the ground state. Heteronuclear NMR studies established that His 148 was neutral in the free enzyme at neutral pH, and in conformational exchange in a specific DNA complex containing uracil. We conclude that the +1 and +2 phosphate esters play identical catalytic roles in the reactions of single-stranded and double-stranded DNA substrates, and that His 148 serves a catalytic role by positioning the substrate and catalytic water, or by an environmental effect.
The diverse functional groups of substrates can provide unique opportunities for enzymes to co-opt mechanisms of catalysis that may not be possible in the absence of these groups. One example is the polymeric and negatively charged DNA phosphate ester backbone that can provide a robust scaffold for electrostatically driven binding interactions for enzymes that recognize DNA substrates (1). Quite often, such interactions may involve noncovalent bonding at one or more phosphate sites distant from the site of chemical reaction, and the favorable binding energy is used to drive the substrate into a reactive state that would be otherwise energetically inaccessible. More remarkably, the DNA repair enzyme uracil DNA glycosylase (UNG)1 has been shown to use the negatively charged phosphate backbone in specific stabilization of a high-energy glycosyl cation intermediate along the reaction coordinate for cleavage of the glycosidic bond of deoxyuridine in DNA (2–6). Despite the general consensus that substrate-assisted electrostatic catalysis is occurring with UNG, significant discrepancies between experimental and computational findings that cloud our understanding of catalysis in this system (2, 4–6) and, more generally, the very nature of electrostatic catalysis remain (7, 8).
The first study to quantify the energetic importance of phosphate interactions in UNG catalysis reconstructed the optimal substrate 5′ Ap1+Up1−Ap2−A 3′ by systematically adding nucleotides and phosphate groups onto the minimal substrate deoxyuridine (4, 9). Although deoxyuridine is 4 billion-fold less reactive than 5′ ApUpApA 3′, reconstruction determined that the simple addition of +1 and −1 phosphate ester linkages increased the cleavage rate by more than 105-fold over that of deoxyuridine (Figure 1) (4). A more targeted study later used neutral methylphosphonate (MeP) substitutions at the +2, +1, −1, and −2 phosphate ester linkages of 5′ Ap2+Ap1+Up1−Ap2−A 3′ to show that phosphate charge was important at the +1, −1, and −2 positions (a range of damaging effects of 2–5 kcal/mol was observed depending on the site of substitution), but entirely unimportant at the +2 position (2). The attribution of the deleterious MeP effects to favorable electrostatic interactions with the high-energy glycosyl cation intermediate was based on the observation that a given MeP substitution had a large and equal damaging effect on kcat/Km and on binding of a cationic 1-azadeoxyribose transition state analogue inhibitor (Ki), but a weaker effect on binding of the substrate (2). (A plot of log kcat/Km against log Ki was linear with a slope of −1; a plot of log Km against log Ki had a smaller slope of +0.5.)
Figure 1.

Interactions along the reaction coordinate of UNG. E, free UNG (PDB entry 1AKZ); EI, UNG complex with nonspecific DNA showing a partially flipped base (PDB entry 2OXM); ES, UNG complex with substrate analogue DNA with uracil fully flipped into the active site (PDB entry 1EMH); ES‡, UNG complex with DNA containing a 1-azadeoxyribose glycosyl cation mimic and uracil (PDB entry 1Q3F); EP, UNG product complex with abasic DNA and uracil (PDB entry 1EMJ). Distances indicate potentially important interactions made by the +1 and +2 phosphates, His 148, and the catalytic water in each step along the reaction coordinate.
Two computational studies have been performed to elucidate electrostatic catalysis by the DNA phosphates in duplex DNA substrates of UNG with results that differed markedly from the results of experiments with single-stranded (ss) DNA (5, 6). The primary conclusion from the first study was that phosphate electrostatic interactions with the cationic transition state accounted for 17.5–21.9 kcal/mol toward activation barrier lowering, which included a large 5 kcal/mol favorable electrostatic interaction between the +2 phosphodiester monoanion and the glycosyl cation species. In addition, computations indicated a 6–8 kcal/mol anticatalytic electrostatic interaction between the imidazolium side chain of His 148 and the cationic transition state. This result was also difficult to reconcile with previous mutagenesis experiments that showed a small 2-fold damaging effect of the H148L mutation on the steady state kcat value (Figure 1) (10). Thus, a second theoretical study was performed to explore the nature of these discrepancies with experimental work, uncovering two potential reasons for the disparate results (6). First, it was suggested that the diminished importance of charge at the +2 position in ssDNA arose from an increased level of solvent shielding of the +2 position as compared to double-stranded (ds) DNA substrates. Second, removal of the charge at the +2 phosphate was proposed to lead to neutralization of His 148, due to its direct interaction with this phosphate in the ES complex (Figure 1). Thus, the lack of an energetic effect upon removal of the +2 phosphate charge was due to compensating effects: loss of the anticatalytic effect of the His 148 imidazolium ion and the favorable interaction of the +2 phosphate monoanion.
Here we return to test these proposals using dsDNA substrates with MeP substitutions at the +1 and +2 positions and by deletion mutagenesis of His 148. We find no support for either theoretical explanation and instead conclude that the same catalytic mechanisms are used for both ssDNA and dsDNA substrates. In addition, His 148, in its neutral form, is uncovered as an important catalytic group that likely orients the catalytic water in the transition state for hydration of the glycosyl cation intermediate or plays a structural role in organizing the active site.
EXPERIMENTAL PROCEDURES
Enzymes
Both bacterial and human enzymes are used in this work. The human enzyme has properties that make it more favorable for heteronuclear NMR studies, and since all of the previous kinetic work has been performed using the Escherichia coli enzyme, we perform all of the kinetic and thermodynamic studies using the bacterial form. The H148A mutation of the human enzyme corresponds to the H67A mutation of the bacterial enzyme. Recombinant UNG from E. coli strain B (eUNG) was purified to >99% homogeneity using a T7 polymerase-based overexpression system as previously described (11). The eUNG enzyme concentration was determined using an extinction coefficient of 38500 M−1 cm−1 at 280 nm. 15N-labeled human UNG2 (hUNG) samples were prepared by growing BL21(DE3)pLysS cells (Stratagene), containing the pET21a (UNG) or pET21a (H148A) plasmid, in MOPS minimal medium (12) containing 1.0 g/L 99% 15N-labeled ammonium chloride (Cambridge Isotope, Andover, MA) as the sole nitrogen source and purified as previously described (13). The hUNG enzyme concentration was determined using an extinction coefficient of 33680 M−1 cm−1 at 280 nm. The eUNG H67A mutation (corresponding to H148A in human UNG) was generated using the QuikChange double-stranded mutagenesis kit (Stratagene, La Jolla, CA) and confirmed by sequencing both strands of the DNA. The purity of all enzymes were greater than 95%as judged by sodium dodecyl sulfide—poly aery lamide gel electrophoresis (SDS–PAGE) with visualization by Coomassie blue staining. The purified enzymes are estimated to be greater than 90% active based on the observation that 1 equiv of enzyme is required to titrate a tight binding transition state inhibitor under conditions where the inhibitor concentration was 100 times greater than its Kd (stoichiometric binding conditions) (3, 14).
DNA Substrates
The nucleoside phosphoramidites and methylphosphonamidites were purchased from Glen Research (Sterling, VA). The single strands of DNA oligonucleotides U, Z, and F, and both strands of P, were synthesized using standard phosphoramidite chemistry with an Applied Biosystems 390 synthesizer (see Table 1 for nucleotide sequences and nomenclature). Methylphosphonate (MeP) oligonucleotides were deprotected in 0.5 mL of a 45: 45:10 acetonitrile/ethanol/ammonium hydroxide mixture for 0.5 h, followed by addition of 1 equiv volume of ethylene-diamine and incubation for 6 h at room temperature. The MeP oligonucleotides were first purified by anion exchange high-performance liquid chromatography (HPLC), followed by desalting and resolving of MeP isomers using a C-18 reversed phase preparative column (Phenomenex Aqua column) with gradient elution from 23 to 28% acetonitrile in 0.1 M aqueous TEAA (pH 7.0) over the course of 3.5 h. Standard oligonucleotides were deprotected in 30% aqueous ammonium hydroxide for 10 h at 50 °C. The oligonucleotides were then purified by anion exchange HPLC and desalted by C-18 reversed phase HPLC. The complementary strands of U, Z, and F were purchased from Integrated DNA Technologies. DNA was purified by anion exchange HPLC and desalted using a C-18 reversed phase HPLC column. The purity and nucleotide composition of the DNA were assessed by analytical reversed phase HPLC and MALDI-TOF. The concentrations were determined by UV absorption measurements at 260 nm, using the pairwise extinction coefficients for the constituent nucleotides.
Table 1.
DNA Duplex Abbreviations and Respective Sequencesa
| Duplex Abbreviation | Sequencea |
|---|---|
| U | 5′-GCGCAUAGTCGG-3′ 3′-CGCGTATCAGCC-5′ |
| P | 5′-GCGGCCAAAFAAAAAAGCGC-3′ 3′-CGCCGGTTTPTTTTTTCGCG-5′ |
| F | 5′--GCGCAFAGTCG-3′ 3′-CGCGTATCAGC-5′- |
| Z | 5′-GCGCAZAGTCG-3′ 3′-CGCGTATCAGC-5′ |
|
| |
| U-19 | 5′-GCGGCCAAAUAAAAAAGCGC-3′ 3′-CGCCGGTTTPTTTTTTCGCG-5′ |
Abbreviations: U, deoxyuracil; P, 2-aminopurine; F, tetrahydrofuran abasic site analogue; Z, 1-azadeoxyribose. The following nomenclature is used to identify the +2, +1, and −1 phosphates: 5′ GCGC+2-pA+1pU−1pAGTCGG 3′.
The uracil-containing strand of oligonucleotide U (Table 1) was 5′-32P-end-labeled using [γ-32P]ATP (GE Healthcare) in the presence of T4 polynucleotide kinase (New England Biolabs, Ispwich, MA). The reaction was quenched by heating the mixture to 65 °C for 20 min in the presence of 1 mM EDTA, and the fractional incorporation of the label was determined by resolving the [γ-32P]ATP from the radiolabeled oligonucleotide on a denaturing 19% polyacrylamide gel. The gel was exposed on an imaging plate (FujiFilm) and imaged with a phosphorimager (FujiFilm BAS-2500), and the radioactivity was counted using Image Gauge (FujiFilm). The unincorporated label was separated from the oligonucleotide using a Micro Bio-Spin 6 chromatography column (Bio-Rad). The concentration of the DNA was determined by its specific activity.
Duplex hybridization reactions involved mixing 1 equiv of the labeled single strand with a 10% excess of complementary strand in 10 mM NaH2PO4 (pH 7.0) and 25 mM NaCl. The hybridization mixture was heated to 95 °C for 5 min and allowed to cool to room temperature over 4 h. Duplex formation was confirmed by analyzing reaction mixtures on a native 19% polyacrylamide gel with either ethidium bromide staining or autoradiography.
Rapid Chemical Quench Kinetic Studies using E. coli UNG
The single-turnover kinetics of uracil glycosidic bond cleavage were determined at 25 °C in TEN buffer [10 mM Tris-HCl (pH 8.0), 1 mM EDTA, and 60 mM NaCl] using a KinTek (University Park, PA) RQF-3 rapid quench instrument with excess eUNG (final concentration of 10 μM) and limiting amounts (10–20 nM) of the 5′-32P-end-labeled duplex DNA substrates U(OxP), U(+1MeP), and U(+2MeP). Reactions were initiated by mixing equal volumes (~24 μL) of the labeled substrate and enzyme solutions. At the designated time point, the reactions were quenched with 0.5 M HCl delivered from the quench syringe. The quenched reaction mixtures were collected in 1.5 mL polyethylene tubes, and immediately, 100 μL of a phenol/chloroform/isoamyl alcohol mixture (25:24:1 volume ratio) was added to denature the enzyme. The reaction mixtures were vortexed, and 20 μL of 3 M Tris base was added to adjust the pH to ~7 to prevent degradation of the DNA under acidic conditions. The samples were then centrifuged at 16100g for 10 min to separate the organic and aqueous layers. A 5 μL aliquot of the aqueous layer was mixed with 10 μL of loading buffer (95% volume ratio formamide, 0.05% mass fraction bromophenol blue, and 0.05% mass fraction xylene cyanol) and was heated to 90 °C for 3 h to thermally cleave the abasic sites (via β-elimination) generated by uracil cleavage by UNG. The samples were analyzed by electrophoresis on a 19% polyacrylamide gel containing 8 M urea. The counts in each resolved band were measured using a phosphorimager, and the fractions of the total radioactivity in the substrate and β-elimination reaction product bands at each reaction time point were quantified using Image Gauge (FujiFilm). The data were fit to a first-order exponential decay rate equation using Graphpad Prism. The reported single-turnover rate constants (kcl) were found to be independent of enzyme concentration, indicating that the measured rates are not limited by bimolecular encounter (data not shown).
Steady State Kinetic Measurements
The steady state kinetics of cleavage of uracil by 10 nM H67A at 25 °C were measured by following the fluorescence increase of a 2-aminopurine base that was incorporated opposite the excised uracil in U-19 dsDNA as previously described (Table 1) (15), and the data were fitted to eq 1, where kobsd = v/[E]tot.
| (1) |
Equilibrium Binding Measurements Using E. coli UNG
The dissociation constants for binding of F or Z to the UNG–uracil complex were determined by competition binding experiments in which a 2-aminopurine (2-AP)-labeled 19-mer duplex abasic product analogue DNA (P, Table 1) was displaced from the enzyme–uracil (EU) complex. P has previously been shown to undergo a strong fluorescence decrease upon binding to UNG, which can be used as a spectroscopic signal to monitor its binding or, alternatively, its displacement by other ligands (2, 15). All measurements were performed in TMN buffer [10 mM Tris-HCl (pH 8.0), 2.5 mM MgCl2, and 25 mM NaCl] in the presence of 1 mM uracil at 25 °C. This concentration of uracil is approximately 15-fold greater than the apparent KD of the enzyme for uracil at pH 8 and ensures that all enzyme is bound as EU (16). The KD values of P for the wild-type (wt) and “H148A” EU complexes were determined using direct binding measurements in which a decrease in 2-AP fluorescence was followed upon titration of a fixed concentration of P (200 nM) with increasing concentrations of UNG (0–740 nM for wt and 0–3 μM for H67A). All fluorescence experiments were performed on a Spex Fluoromax-3 spectrofluorimeter using an excitation wavelength of 320 nm and collecting emission from 340 to 450 nm. The fluorescence intensity (I) at 370 nm was plotted against [EU]tot to yield the KD from eq 2:
| (2) |
In the competition binding experiments for determining the affinity of the neutral abasic analogue (F) or the cationic transition state analogue (Z) for the UNG–uracil (EU) complex, increasing concentrations of F or Z were titrated into a solution containing 200 nM P, 300 nM UNG (wild-type or “H148A”), and a saturating concentration of uracil (1 mM). The KD values for binding of F and Z to the EU complex were determined using Dynafit using the known dissociation constants of P for the wild-type and mutant EU complexes and the equilibria shown in eqs 3–5 (2).
| (3) |
| (4) |
| (5) |
NMR Spectroscopy
Two-dimensional (2D) 1H–15N LR-HMQC spectra were acquired on wild-type and H148A mutant human UNG to correlate the histidine Nδ1 and Nε2 atoms with the Hδ2 and Hε1 ring protons in the imidazole side chain (16, 17). This experiment differed from a conventional HMQC experiment in that a 22 ms dephasing delay was used to optimize signals from the two- and three-bond coupled nitrogens and carbon-bound protons in the imidazole ring of histidine residues and suppress signals from one-bond coupled 15N–1H amide resonances. Spectra were acquired at 20 °C using uniformly 15N-labeled wt and H148A hUNG (0.5 mM) in buffer containing 10 mM NaH2PO4, 100 mM NaCl, and 1 mM DTT in a 300 μL volume using a low-volume NMR tube (Shigemi, Inc.). Spectra of the bound proteins were acquired using the same conditions, but with the addition of F (0.6 mM) and uracil base (0.8 mM). The 1H and 15N carriers were set at 4.82 and 205.0 ppm, respectively. The 15N dimension was collected with 452 complex points and a 123 ppm sweep width, and the 1H dimension was collected with 1024 complex points and a 13.3 ppm sweep width. All experiments were performed on a Varian Unity Plus 600 MHz spectrometer (Varian NMR Systems) equipped with a 5 mm triple-resonance CryoProbe with z-axis-pulsed magnetic field gradients. Data were processed using NMRPipe and analyzed using NMRDraw (18).
RESULTS AND DISCUSSION
Methylphosphonate Substitution Strategy
The strategy of substituting nonbridging methylphosphonate (MeP) groups for normal phosphodiester linkages (OxP) in DNA and RNA has been recently reviewed (1). This conservative substitution (i) ablates the negative charge at the site regardless of the MeP diastereomer, (ii) makes the nonbridging oxygen atoms slightly more electropositive by increasing the P–O bond order (19), (iii) and increases the van der Waals radius of the substituted nonbridging atom from 1.44 Å (O) to 1.74 Å (CH3) (20). In addition, theoretical studies suggest that MeP substitution may alter the conformational preferences of the linkage and give rise to small changes in the minor groove width (21, 22). Despite the possibility of complex kinetic effects upon MeP substitution, a comprehensive examination of many such substitutions in the literature has concluded that large stereospecific methyl effects (kOxP/kMeP ≫ 1) correlate very well with the presence of direct hydrogen bonds between the protein and the corresponding phosphate oxygen (1). In contrast, methyl effects that are not stereoselective may arise from the ablation of an electrostatic interaction with the phosphate anion. A non-stereoselective effect is anticipated because substitution of either oxygen effectively removes the charge of the linkage. In addition to removing the charge associated with the nonbridging oxygen, a methyl group can also introduce unfavorable steric hindrance in the ground state or transition state. Thus, in general, any observed methyl effect must be interpreted as an upper limit for the electrostatic contributions of the negatively charged phosphate ester to ground state or transition state stabilization (2). We use this framework to conservatively interpret the methyl effects measured here.
The duplex DNA substrate (U) in Table 1 was synthesized with OxP and MeP linkages at the +2 and +1 positions. For the +1 and +2 substitutions, the two MeP diastereoisomers were not stereospecifically assigned but are termed early and late migrating isomers, respectively, corresponding to their elution order from a reverse phase HPLC column (Figure 2). The absence of stereospecific assignments has no impact on the interpretation of the data obtained in this or our previous study with ssDNA substrates.
Figure 2.
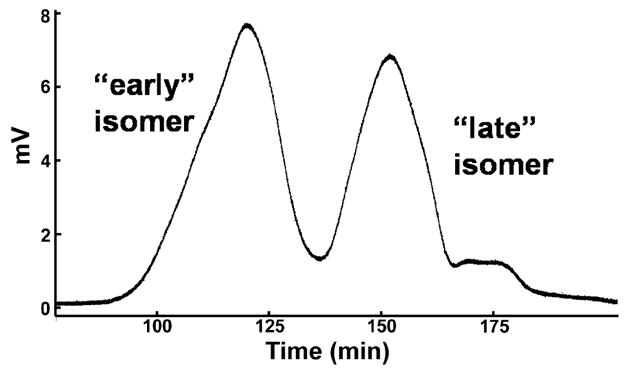
Resolution of the early and late eluting +1 methylphosphonate stereoisomers of the U oligonucleotide using reverse phase HPLC (see Table 1).
Methyl Effects at the +1 Position
We first sought to determine whether the methyl effect at the +1 position was similar to that previously measured using the ssDNA ApA+1mUpApA using single-turnover uracil cleavage rate measurements. In these experiments, the abasic site products were quantified by first thermally cleaving the phosphodiester backbone 3′ to the abasic site via β-elimination. The substrate and labeled product fragment were then separated on a 19% polyacrylamide denaturing gel (Figure 3). The single-turnover cleavage rate (kcl) using U(OxP) dsDNA was 110s−1 (Figure 3A), which is identical to that previously reported for ssDNA (2). This finding has important implications: identical activation barriers for ds- and ssDNA require that significantly different catalytic strategies be employed for these substrates if the +2 phosphate contributes 5 kcal/mol to electrostatic catalysis with the dsDNA substrate only. To the best of our knowledge, this would be novel, as we are not aware of an enzyme system in which an identical catalytic power is achieved with two related substrates without employing the same catalytic machinery.
Figure 3.

Single-turnover kinetics of the eUNG reaction measured by the rapid chemical quench method and processed using the piperidine-induced abasic site cleavage assay. The data points were fit to a first-order exponential decay (see Table 1 for rate constants). (A) Reaction of 10 μM wild-type UNG with +2 MeP-substituted U dsDNA (20 nM) compared with the phosphodiester form (OxP). (B) Reaction of 10 μM wild-type UNG with +1 MeP-substituted U dsDNA (20 nM) compared with the OxP form.
When a MeP substitution was introduced at the +1 position in the U dsDNA, methyl effects (kclOxP/kclMeP) of 180 and 5 × 108 were measured for the late and early isomers, respectively (Figure 3A). These results may be compared with the 52- and 420-fold +1 methyl effects, respectively, on kcat for the corresponding isomers in ssDNA (Table 2). The much larger damaging effect of the early isomer in dsDNA, which to the best of our knowledge is the largest methyl effect ever reported, indicates a direct interaction between the enzyme and the corresponding oxygen in the phosphate ester linkage in dsDNA and, likely, a significant steric clash with the methyl substituent. The methyl effect is largely on the activation barrier ES → ES‡ because the rate increased only 40% when the UNG concentration was doubled to 20 μM, indicating that most of the substrate was enzyme-bound (not shown). It is interesting that the early MeP isomer in ssDNA exhibited a 1 million-fold smaller methyl effect, which may arise from the increased backbone rigidity of duplex DNA as compared to more flexible ssDNA substrates. The nature of the potential clash can be surmised from the crystal structures of UNG bound to DNA containing a deoxypseudouridine substrate analogue and the 1-azadeoxyribose glycosyl cation analogue [ES and ES‡ (Figure 1)] (23, 24). In these structures, the +1 pro-Sp nonbridging oxygen interacts with two water molecules, while the +1 pro-Rp oxygen is in close contact with both the γOH and αNH groups of Ser169. Thus, the early isomer likely corresponds to methyl substitution at the pro-Rp oxygen of the +1 phosphate. Since the contribution of a purely electrostatic effect should be independent of stereo-chemistry (i.e., equal effects for both charge-neutral MeP diastereomers), a conservative estimate of the electrostatic contribution of the +1 phosphate anion to transition state stabilization is provided by the smaller methyl effect of 180 for the late isomer, corresponding to a 3.1 kcal/mol increase in the activation barrier. This stabilization coincides well with the previously reported contribution of the +1 phosphate of −2.4 and −3.7 kcal/mol based on the stereospecific methyl effects on kcat for ssDNA substrates (2).
Table 2.
Effects of Methylphosphonate Substitution on Uracil Glycosidic Bond Cleavage by eUNG
| DNA | kcl(ssDNA) (s−1)a | kcl(dsDNA) (s−1) | kclOxP/kclMeP (ssDNA)a | kclOxP/kclMeP(dsDNA) |
|---|---|---|---|---|
| OxP | 110.0 | 100.3 | – | – |
| + 1 MePlate | 2.1 | 0.56 | 50 | 180 |
| + 1 MePearly | 0.26 | 2 × 10−7 | 420 | 5 × 108 |
| +2 MePlate | 220 | 54 | 0.5 | 1.8 |
| +2 MePearly | 220 | 180 | 0.5 | 0.6 |
Rates of uracil cleavage in ssDNA were previously reported in ref 2.
Methyl Effects at the +2 Position
When MeP substitutions were made at the controversial +2 position in the U dsDNA, only a small methyl effect of 1.8 was measured for the late isomer, and the early isomer was actually more reactive than the OxP form, giving rise to an inverse methyl effect of 0.6 (Figure 3B and Table 2). These measurements recapitulate those previously reported with ssDNA (Table 2) and provide no evidence of a catalytically important interaction with the +2 linkage, electrostatic or otherwise.
The lack of an effect of removal of either nonbridging oxygen at the +2 position is surprising given that the +2 phosphate forms a 2.65 Å hydrogen bond with the side chain NH group of His 148 in the ES complex (Figure 1). However, in the complex with the 1-azadeoxyribose glycosyl cation analogue [ES‡ (Figure 1)], the +2 phosphate moves away from His 148, the O–HN distance increasing to 3.8 Å. In this structure, which was not used in the theoretical studies even though it is the best available structural mimic of the high-energy glycosyl cation, the +2 phosphate does not interact with any enzyme group. The small +2 methyl effect on the activation barrier has been previously attributed to the equivalent stabilization of the ground and transition states by the +2 phosphate oxygens (the stabilization is estimated to be −0.5 to −1.6 kcal/mol based on which oxygen was substituted with methyl) (2). In summary, we find no significant differences in the catalytic contributions of the +1 and +2 DNA phosphates between ss- and dsDNA substrates.
Kinetic Effects of the H67A Mutation2
The theoretical proposal that doubly protonated His 148 destabilizes the glycosyl cation by 6–8 kcal/mol and the previous report that the H148L mutation resulted in an only 2-fold decrease in kcat (10) prompted us to reexamine the catalytic role of this absolutely conserved residue. Structural studies reveal that His 148 has significantly different interactions at different steps along the reaction coordinate (Figure 1). In the free enzyme (E), it shares a water molecule with the catalytic aspartate group (Asp 145). [Asp 145 has previously been shown to stabilize the glycosyl cation and position the nucleophilic water (3, 23, 25).] In the early extrahelical complex (EI) (26), His 148 hydrogen bonds with O2 of the emerging pyrimidine base, and also to its own backbone carbonyl. This arrangement strongly suggests that His 148 is doubly protonated at this step. In the catalytically poised ES complex, His 148 now hydrogen bonds to the +2 phosphate and an ordered water molecule that is very well positioned to serve as the nucleophile. However, as noted above, the hydrogen bond to the +2 phosphate is broken in the complex with the glycosyl cation mimic (ES‡), while the water is still held in place by both His 148 and Asp145, poised nicely for attack at C1′. Finally, in the product complex (EP), His148 returns to its hydrogen bond with the +2 phosphate. These apparently important interactions at multiple steps along the reaction coordinate make the lack of a mutagenic effect enigmatic.
We measured the steady state kinetic parameters of the corresponding H67A mutation in the E. coli enzyme using a continuous fluorescence assay (Figure 4) (15). Contrary to the previous report using the H148L mutant hUNG (10), a 21-fold damaging effect on kcat was measured for H67A [kcat(H67A) = 0.15 ± 0.1 s−1, and kcat(wt) = 3.2 ± 0.2 s−1], corresponding to a increase in the activation barrier of 1.9 kcal/mol. Moreover, substrate binding was also weakened by the removal of His67, as evidenced by a 52-fold increase in Km[Km(H67A) = 1.6 ± 0.2 μM, and Km(wt) = 0.03 ± 0.01 μM], which corresponds to a +2.4 kcal/mol destabilization of the ground state. Thus, like the +2 phosphate with which it interacts, His67 stabilizes both the ground state and transition state. However, unlike the +2 phosphate, His67 stabilizes the transition state to a greater extent than the ground state, leading to a 1.9 kcal/mol lowering of the activation barrier. These measurements provide no evidence of an anticatalytic, destabilizing interaction between His67 and the glycosyl cation transition state, as dissected further below.
Figure 4.
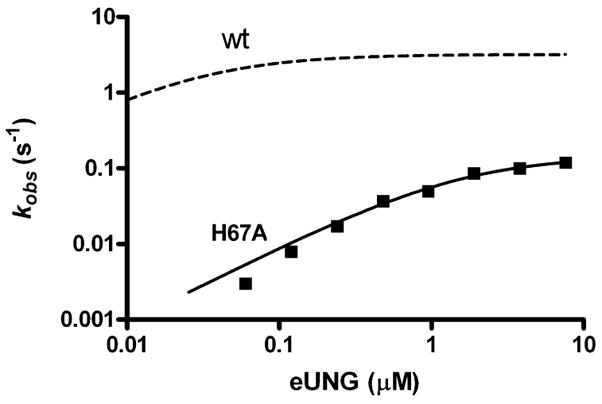
Steady state kinetics of cleavage of uracil from U-19 dsDNA by wild-type (– – –) or H67A eUNG (—) using the 2-AP continuous fluorescence assay. Curves are nonlinear regression best fits to eq 1 (note the log scale on both axes). The data for the wild-type enzyme have previously been reported, and the dashed line shows the theoretical curve using the measured kinetic parameters: kcat = 3.2 s−1, and Km = 0.03 μM(15).
NMR Studies of the Ionization and Tautomeric State of His 148 of hUNG
To determine the ionization state of His 148 in free and DNA-bound hUNG, 2D heteronuclear NMR experiments were performed. The lH–15N LR-HMQC experiment is optimized for histidine ring spin systems such that the Nδ1 and Nε2 resonances may be correlated with the carbon-attached Hδ2 and Hε1 resonances via scalar couplings (13). The peak patterns unambiguously distinguish whether a given histidine is singly or doubly protonated and, in addition, reveal the tautomeric state of the neutral ring form (i.e., whether Nδ or Nε carries the single proton).
In the LR-HMQC spectrum for 15N-labeled wild-type hUNG, all 13 of the expected histidine spin systems were resolved and identified (Figure 6A, black peaks). Inspection of the LR-HMQC spectra of H148A UNG shows only 12 histidine spin systems, each unchanged from those of the wild-type spectrum, and one system (6) that was absent, allowing the assignment of His 148 (Figure 6A, red peaks). From examination of the His 148 spin system, and the canonical chemical shift patterns for neutral and cationic histidines (17, 27), we conclude that the side chain of His 148 is neutral in free UNG at pH 7.0, with Nε carrying the single proton.
Figure 6.

Binding of P, F, and Z duplexes to wild-type and H67A eUNG–uracil complexes at pH 8.0. (A) Binding of the P duplex to the wild type and H67A. The signal is the decrease in 2-aminopurine fluorescence upon binding of P to the EU complex ([E] = 0.3 μM, [P] = 0.2 μM, and [U] = 1 mM). The curves are the best fits to eq 2. (B) Displacement of P from the EU complexes by F. (C) Displacement of P from the EU complexes by Z. Curves in panels B and C are from computer simulations using Dynafit (see the text).
The LR-HMQC experiments were then repeated after the addition of 1.1 equiv of dsDNA that contained a tetrahydrofuran abasic product analogue (F, Table 1), and also in the presence of excess uracil base, so that the enzyme was fully bound as the ternary EUF complex (this is structurally similar to the EP complex in Figure 1). LR-HMQC spectra for the wild-type complex contained 10 histidine spin systems that were superimposable with the free enzyme, but three systems were missing, including that of His 148. This is not surprising because in addition to His 148, two other histidines are located in the active site (His268 and His215). Unfortunately, the disappearance of these signals in the EUF complex prevents any determination of the ionization state of His 148 while DNA is bound. Nevertheless, the binding measurements below strongly suggest that His 148 is neutral in the presence of the 1-azadeoxyribose glycosyl cation mimic.
H67A Destabilizes Binding of a Glycosyl Cation Mimic
To further probe the ionization state of this histidine in the presence of the high-energy glycosyl cation intermediate, we measured the affinity of both wild-type and H67A eUNG for the 1-azadeoxyribose glycosyl cation mimic (Z) and a neutral tetrahydrofuran analogue (F) in the presence of excess uracil (Scheme 1).
Scheme 1.
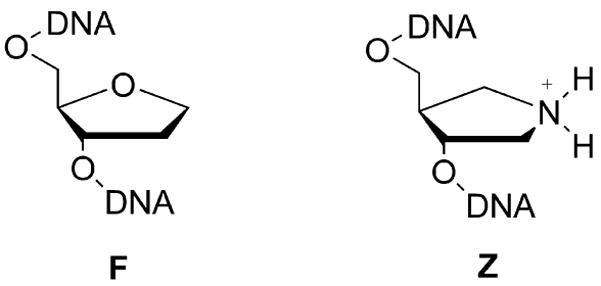
These analogues have similar structures and steric properties and differ most notably by substitution of a methylene group in F for the secondary ammonium group of Z, thereby removing the positive charge of Z. The logic in this experimental approach is as follows. If His67 interacts unfavorably with the positive charge in the glycosyl cation, there should be an unfavorable coupling energy for the binding of Z in the presence of His67 (i.e., the His67 cation will clash with the ammonium cation). This prediction may be tested by evaluating whether the removal of His67 has a larger damaging energetic effect on binding of cationic Z as compared to neutral F, as defined explicitly in the difference free energies in eqs 5 and 6.
| (6) |
| (7) |
In these equations, ΔGM,Z, ΔGW,Z, ΔGM,F, and ΔGW,F are the binding free energies of the mutant (M) and wild-type (W) enzymes for Z and F in the presence of a saturating level of uracil. For an unfavorable electrostatic interaction, ΔΔGM,Z would be expected to be negative, indicating relief from electrostatic strain upon removal of His67, and ΔΔGM,F would be expected to be more positive than ΔΔGM,Z because no electrostatic strain was present in the wild-type complex with neutral F.
To measure the binding affinities, we used a competition assay, where either a Z or an F containing 11-mer DNA duplex displaced a fluorescent abasic dsDNA analogue (P, Table 1) from the EUP ternary complex in the presence of excess uracil (2). The first step in these experiments was to measure the KD value of P for the wild-type and H67A EU complex by following the quenching of the 2-aminopurine fluorescence upon binding (Figure 6A) (15). From these data, similar binding affinities were measured for the wild type (KD = 25 ± 5 nM) and H67A (KD = 78 ± 19 nM). The KD values of neutral F and cationic Z for the wild type and H67A were then determined from the concentration dependence of the fluorescence increase upon displacement of P from the EU complex (Figure 6B, C). The binding data were simulated using Dynafit (28), employing the measured values for the binding affinities of P for the wild-type and mutant EU complexes. From this analysis, similar KD values of 437 ± 23 and 395 ± 20 nM were obtained for binding of the neutral F construct to the wild type and H67A, respectively, providing a value for ΔΔGM,F of −0.06 ± 0.04 kcal/mol. In contrast, the KD values for binding of the cationic Z construct differed by ~200-fold [KD(wt) = 0.4 ± 0.25 nM, and KD(H148A) = 82 ± 6 nM], corresponding to a value for ΔΔGM,Z of 3.2 ± 0.2 kcal/mol. The large positive value for ΔΔGM,Z and the negligible effect of H67A on binding of the neutral F are inconsistent with the proposed anticatalytic interaction between this histidine and the glycosyl cation. Instead, the data strongly suggest that His67 specifically stabilizes the glycosyl cation analogue. These findings are not easily reconciled with the theoretical proposal that this histidine is doubly protonated in the presence of the glycosyl cation.3
CONCLUSIONS
Catalytic Contributions of DNA Phosphates and His 148
These results, in combination with previous findings with single-stranded DNA, now effectively exclude a catalytic role for the +2 phosphate in both types of UNG substrates. Thus, any differences in solvation of this site between ss- and dsDNA are not important for catalysis, and the computed differences in electrostatic stabilization of the glycosyl cation between these substrates remain experimentally elusive. The simple function of the +2 phosphate, as determined from numerous experiments, is to promote ground state binding. In contrast, the +1 phosphate is found to be catalytically important for both substrates. The smaller of the two +1 MeP effects in the context of dsDNA provides an estimate of the electrostatic contribution of the +1 phosphate (3.2 kcal/mol), which parallels the importance of this phosphate in ssDNA (2.4 kcal/mol). The 0.8 kcal/mol greater stabilization in the context of dsDNA may arise from a greater electrostatic contribution as compared to ssDNA, but the extra amount of stabilization is small. The 3 million-fold stereoselectivity for the +1 methyl effect in dsDNA is dramatic compared to the 8-fold stereoselectivity seen with ssDNA (Table 2). The increased stereoselectivity in dsDNA could result from one or more of the following effects: (i) a more stringent requirement for positioning of the +1 phosphate in the transition state through interactions with the pro-Sp nonbridging oxygen and Ser169 (Figure 1), (ii) increased steric demands arising from the increased rigidity of the duplex DNA scaffold, and (iii) greater electrostatic stabilization of the transition state through Coulombic interactions with this site.
In general, we propose that long-range electrostatic interactions from phosphate esters are not ideally suited to providing specific stabilization of transition states of the magnitude suggested by the theoretical studies of Dinner (5), which is consistent with conclusions derived from previous computational analyses in other systems (7). We base our conclusion on the consideration that these anionic groups will almost certainly be close to complementary enzyme side chains, which will always tend to stabilize ground states as well. This contention is supported by comparing the previously measured effects of +1 and −1 methyl groups on binding of the neutral substrate and glycosyl cation analogues, where phosphate charge was found to contribute significantly to stabilization of both species (2). Nevertheless, electrostatic stabilization by the +1 or − 1 phosphates was greater for the glycosyl cation analogue (and the transition state), ultimately lowering the activation barrier by approximately 2 kcal/mol for each position. For comparison, UNG uses the combined action of the uracil and Asp145 anions to specifically stabilize the glycosyl cation by 7 kcal/mol with no apparent anticatalytic stabilization of the ground state (3). In this regard, Asp145 actually destabilizes the ground state by approximately +0.7 kcal/mol compared to the D145N mutation, presumably because of charge repulsion with the DNA phosphates (3).
These findings provide the first insights into the function and protonation state of the absolutely conserved His 148 (His67). The first function of this group is to interact with O2 of thymine or uracil in the early intermediate during base flipping (EI, Figure 1), providing specificity for these two bases. Previous NMR dynamic studies have shown that the H148A mutant has an only 2-fold effect at this early step (26), indicating that its major function lies later along the reaction coordinate. The structure of this trapped intermediate strongly suggests that His 148 is protonated (Figure 1). In contrast to the small H148A mutational effect on the early intermediate, the steady state uracil excision measurements demonstrate a strong effect of the corresponding H67A mutation on the activation barrier for uracil excision (1.9 kcal/mol). A favorable transition state interaction of His67 has been established on thr basis of the 4.3 kcal/mol damaging effect of H67A at the transition state, the 3.2 kcal/mol mutational effect on binding of the glycosyl cation mimic (Figure 6C), and the negligible effect of the H67A mutation on binding of neutral abasic DNA (Figure 6B). These findings suggest a neutral ionization state for this group in the presence of the glycosyl cation and an oscillating protonation state for this group as the reaction proceeds.
The specific stabilization afforded by His 148 (His67) at the chemical step may arise in several ways. First, neutral His 148 with the assistance of Asp145 could help orient the catalytic water for attack at C1′ as suggested in the structure of the glycosyl cation mimic shown in Figure 1. Second, His 148 may serve to exclude general solvent from the active site and thereby enhance the electrostatic stabilization afforded by Asp145 and the uracil anion, which have been described as an electrostatic sandwich that stabilizes the glycosyl cation (3, 14, 23). These findings make His 148, along with Asp145 and His268, one of the more important active site groups involved in transition state stabilization. These highly polar interactions of UNG with the bound transition state provide a preeminent example of active site preorganization and specific solvation (8).
Figure 5.

2D 1H–15N LR-HMQC spectra of wild-type hUNG (black peaks) and H148A hUNG (red peaks) at pH 7.0 in the presence and absence of abasic DNA and uracil. Spin systems are connected by solid black lines and numbered. (A) 2D NMR spectra for free wild-type and H148A hUNG (0.5 mM). Spin system 6 corresponds to His 148. (B) 2D NMR spectra for wt and H148A hUNG (0.5 mM) complexed to F (0.6 mM) and uracil (1 mM).
Footnotes
This research was supported by National Institutes of Health Research Grant GM56834 (to J.T.S.) and a Ruth L. Kirschstein National Research Service Award (F31 GM083623) (to J.B.P.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIGMS or the NIH.
Abbreviations: UNG, uracil DNA glycosylase; MeP, methylphosphonate linkage; OxP, phosphodiester linkage; EDTA, ethylenediaminetetraacetic acid.
The H148A mutation of hUNG corresponds to the H67A mutation in E. coli UNG.
It has been argued by Dinner et al. that the +2 phosphate is required for maintenance of the doubly protonated form of His 148 and that the lack of a +2 methyl effect may arise from fortuitous cancellation of the unfavorable interaction of His 148 and the favorable interaction of +2 anion with the glycosyl cation. However, this scenario cannot reasonably account for the 3 kcal/mol favorable interaction of His 148 in binding to the glycosyl cation mimic (Figure 6C). It was also argued that MeP substitution, although electronically neutral, is not equivalent to computational ablation of charge, since electron density remains on the remaining nonbridging oxygen. However, in this work, both +2 stereoisomers showed no significant effect which makes any explanation based on residual charge untenable.
References
- 1.Stivers JT, Nagarajan R. Probing Enzyme Phosphoester Interactions by Combining Mutagenesis and Chemical Modification of Phosphate Ester Oxygens. Chem Rev. 2006;106:3443–3467. doi: 10.1021/cr050317n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jiang YL, Ichikawa Y, Song F, Stivers JT. Powering DNA Repair through Substrate Electrostatic Interactions. Biochemistry. 2003;42:1922–1929. doi: 10.1021/bi027014x. [DOI] [PubMed] [Google Scholar]
- 3.Jiang YL, Drohat AC, Ichikawa Y, Stivers JT. Probing the Limits of Electrostatic Catalysis by Uracil DNA Glycosylase using Transition-State Mimicry and Mutagenesis. J Biol Chem. 2002;277:15385–15392. doi: 10.1074/jbc.M200634200. [DOI] [PubMed] [Google Scholar]
- 4.Jiang YL, Stivers JT. Reconstructing the Substrate for Uracil DNA Glycosylase: Tracking the Transmission of Binding Energy in Catalysis. Biochemistry. 2001;40:7710–7719. doi: 10.1021/bi010622c. [DOI] [PubMed] [Google Scholar]
- 5.Dinner AR, Blackburn GM, Karplus M. Uracil-DNA Glycosylase Acts by Substrate Autocatalysis. Nature. 2001;413:752–755. doi: 10.1038/35099587. [DOI] [PubMed] [Google Scholar]
- 6.Ma A, Hu J, Karplus M, Dinner AR. Implications of Alternative Substrate Binding Modes for Catalysis by Uracil-DNA Glycosylase: An Apparent Discrepancy Resolved. Biochemistry. 2006;45:13687–13696. doi: 10.1021/bi061061y. [DOI] [PubMed] [Google Scholar]
- 7.Warshel A, Sharma PK, Kato M, Xiang Y, Liu H, Olsson MH. Electrostatic Basis for Enzyme Catalysis. Chem Rev. 2006;106:3210–3235. doi: 10.1021/cr0503106. [DOI] [PubMed] [Google Scholar]
- 8.Braun-Sand S, Olsson MHM, Warshel A. In: Computer Modeling of Enzyme Catalysis and its Relationship to Concepts in Physical Organic Chemistry. Richard JP, editor. 2005. pp. 201–245. [Google Scholar]
- 9.Varshney U, van de Sande JH. Specificities and Kinetics of Uracil Excision from Uracil-Containing DNA Oligomers by Escherichia coli Uracil DNA Glycosylase. Biochemistry. 1991;30:4055–4061. doi: 10.1021/bi00230a033. [DOI] [PubMed] [Google Scholar]
- 10.Mol CD, Arvai AS, Slupphaug G, Kavli B, Alseth I, Krokan HE, Tainer JA. Crystal Structure and Mutational Analysis of Human Uracil-DNA Glycosylase: Structural Basis for Specificity and Catalysis. Cell. 1995;80:869–878. doi: 10.1016/0092-8674(95)90290-2. [DOI] [PubMed] [Google Scholar]
- 11.Drohat AC, Jagadeesh J, Ferguson E, Stivers JT. Role of Electrophilic and General Base Catalysis in the Mechanism of Escherichia coli Uracil DNA Glycosylase. Biochemistry. 1999;38:11866–11875. doi: 10.1021/bi9910878. [DOI] [PubMed] [Google Scholar]
- 12.Neidhardt FC, Bloch PL, Smith DF. Culture Medium for Enterobacteria. J Bacteriol. 1974;119:736–747. doi: 10.1128/jb.119.3.736-747.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Drohat AC, Xiao G, Tordova M, Jagadeesh J, Pankiewicz KW, Watanabe KA, Gilliland GL, Stivers JT. Hetronuclear NMR and Crystallographic Studies of Wild-Type and H187Q Escherichia coli Uracil DNA Glycosylase: Electrophilic Catalysis of Uracil Expulsion by a Neutral Histidine 187. Biochemistry. 1999;38:11876–11886. doi: 10.1021/bi9910880. [DOI] [PubMed] [Google Scholar]
- 14.Jiang YL, Ichikawa Y, Stivers JT. Inhibition of Uracil DNA Glycosylase by an Oxacarbenium Ion Mimic. Biochemistry. 2002;41:7116–7124. doi: 10.1021/bi025694y. [DOI] [PubMed] [Google Scholar]
- 15.Stivers JT. 2-Aminopurine Fluorescence Studies of Base Stacking Interactions at Abasic Sites in DNA: Metal-Ion and Base Sequence Effects. Nucleic Acids Res. 1998;26:3837–3844. doi: 10.1093/nar/26.16.3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drohat AC, Stivers JT. Escherichia coli Uracil DNA Glycosylase: NMR Characterization of the Short Hydrogen Bond from His187 to Uracil O2. Biochemistry. 2000;39:11865–11875. doi: 10.1021/bi000922e. [DOI] [PubMed] [Google Scholar]
- 17.Pelton JG, Torchia DA, Meadow ND, Roseman S. Tautomeric States of the Active-Site Histidines of Phosphorylated and Unphosphorylated IIIGlc, a Signal-Transducing Protein from Escherichia coli, using Two-Dimensional Hetero-nuclear NMR Techniques. Protein Sci. 1993;2:543–558. doi: 10.1002/pro.5560020406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: A Multidimensional Spectral Processing System Based on UNIX Pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 19.Florian J, Strajbl M, Warshel A. Conformational Flexibility of Phosphate, Phosphonate, and Phosphorothioate Methyl Esters in Aqueous Solution. J Am Chem Soc. 1998;120:7959–7966. [Google Scholar]
- 20.Creighton TE. Proteins. W. H. Freeman and Co; New York: 1997. [Google Scholar]
- 21.Thiviyanathan V, Vyazovkina KV, Gozansky EK, Bichenchova E, Abramova TV, Luxon BA, Lebedev AV, Gorenstein DG. Structure of Hybrid Backbone, Methylphosphonate DNA Heteroduplexes: Effect of R and S Stereo-chemistry. Biochemistry. 2002;41:827–838. doi: 10.1021/bi011551k. [DOI] [PubMed] [Google Scholar]
- 22.Mukherjee S, Bhattacharyya D. Effect of Phosphorothioate Chirality on the Grooves of DNA Double Helices: A Molecular Dynamics Study. Biopolymers. 2004;73:269–282. doi: 10.1002/bip.10550. [DOI] [PubMed] [Google Scholar]
- 23.Bianchet MA, Seiple LA, Jiang YL, Ichikawa Y, Amzel LM, Stivers JT. Electrostatic Guidance of Glycosyl Cation Migration Along the Reaction Coordinate of Uracil DNA Glycosylase. Biochemistry. 2003;42:12455–12460. doi: 10.1021/bi035372+. [DOI] [PubMed] [Google Scholar]
- 24.Parikh SS, Walcher G, Jones GD, Slupphaug G, Krokan HE, Blackburn GM, Tainer JA. Uracil-DNA Glycosylase-DNA Substrate and Product Structures: Conformational Strain Promotes Catalytic Efficiency by Coupled Stereo-electronic Effects. Proc Natl Acad Sci USA. 2000;97:5083–5088. doi: 10.1073/pnas.97.10.5083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Drohat AC, Jagadeesh J, Ferguson E, Stivers JT. The Role of Electrophilic and Base Catalysis in the Mechanism of Escherichia coli Uracil DNA Glycosylase. Biochemistry. 1999;38:11866–11875. doi: 10.1021/bi9910878. [DOI] [PubMed] [Google Scholar]
- 26.Parker JB, Bianchet MA, Krosky DJ, Friedman JI, Amzel LM, Stivers JT. Enzymatic Capture of an Extrahelical Thymine in the Search for Uracil in DNA. Nature. 2007;449:433–438. doi: 10.1038/nature06131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pelton JG, Torchia DA, Remington SJ, Murphy KP, Meadow ND, Roseman S. Structures of Active Site Histidine Mutants of IIIGlc, a Major Signal-Transducing Protein in Escherichia coli: Effects on the Mechanism of Regulation and Phosphoryl Transfer. J Biol Chem. 1996;271:33446–33456. doi: 10.1074/jbc.271.52.33446. [DOI] [PubMed] [Google Scholar]
- 28.Kuzmic P. Program DYNAFIT for the Analysis of Enzyme Kinetic Data: Application to HIV Proteinase. Anal Biochem. 1996;237:260–273. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]