

Abstract
Immunotherapy targeting tumor cell surface carbohydrates is a promising approach for cancer treatment. However, the low immunogenecity of carbohydrates presents a formidable challenge. We describe here the enhancement of carbohydrate immunogenicity by an ordered display on the surface of the cowpea mosaic virus (CPMV) capsid. The Tn glycan, which is overexpressed on numerous cancer cell surfaces, was selected as the model antigen for our study. Previously it has been shown that it is difficult to induce a strong T cell-dependent immune response against the monomeric form of Tn presented in several ways on different carriers. In this study, we first synthesized Tn antigens derivatized with either a maleimide or a bromoacetamide moiety that was conjugated selectively to a cysteine mutant of CPMV. The glyco-conjugate was then injected into mice and pre-and post-immune antibody levels in the mice sera were measured by enzyme linked immunosorbant assays. High total antibody titers and, more importantly, high IgG titers specific for Tn were obtained in the post-immune day 35 serum, suggesting the induction of T cell-dependent antibody isotype switching by the glyco-conjugate. The antibodies generated were able to recognize Tn antigens presented in their native conformations on the surfaces of both MCF-7 breast cancer cells and the multi-drug resistant breast cancer cell line NCI-ADR RES. These results suggest that the CPMV capsid can greatly enhance the immunogenicity of weak antigens such as Tn and this can provide a promising tool for the development of carbohydrate based anti-cancer vaccines.
Keywords: antibodies, carbohydrates, Tn antigen, vaccines, viruses
Introduction
Tumor associated carbohydrate antigens (TACAs) are often strongly linked with tumor progression and malignancy.[1, 2] Cancer immunotherapy targeting TACAs is an attractive approach for elimination of circulating tumor cells and eradication of micrometastases which remain after surgery or radiotherapy. However, this has been a highly challenging task as TACAs are often weak T cell-independent antigens. To generate long lasting effective immune surveillance and protection, T cells must be activated to induce antibody isotype switching and immune memory.[3–14]
In order to render a T cell-dependent immune response, TACAs can be conjugated with helper T (Th) cell epitopes such as serum albumin from different sources, keyhole limpet hemocyanin (KLH), or tetanus toxoid (TT).[3–14] In this manner, the Th epitope can be presented by the B cell specific to the TACA, which is required for a Th cell stimulated humoral response.[15, 16] Recently, several other novel innovative approaches have been developed towards carbohydrate based cancer vaccines. Boons and coworkers reported that a three-component covalent construct including TACA, Th epitope and an adjuvant can significantly improve the magnitude and quality of antibodies generated against TACAs.[17, 18] Danishefsky and coworkers synthesized a unimolecular hexavalent construct linking six different TACAs into one glycopeptide, which were then coupled to KLH.[19, 20] This was evaluated as a promising means to target microheterogeneity of TACAs expressed on tumor cell surfaces to prevent escape of tumor cells from immunosurveillence. The Guo group has taken advantage of the higher metabolic rate of tumor cells to selectively modify the TACAs on tumor cell surfaces.[21, 22] Antibodies generated against modified TACAs were found to be effective in killing glyco-engineered melanoma cells. Besides the native O-glycosides, several groups have reported that the usage of S-linked and C-linked glycosides confer higher stabilities to TACAs, which can subsequently result in higher immune responses.[23–26] Another active area of research is to examine novel carriers for TACA, which include dendrimers,[27–29] monoclonal antibodies,[30] influenza virosome,[31] gold nanoparticles[32] as well as direct conjugation with an adjuvant.[33]
It is known that highly patterned displays of antigens can lead to earlier B cell amplification for potent IgM responses as well as efficient switching to IgG.[34–37] Antigen organization has a great influence on B cell tolerance, with B cells unresponsive to poorly organized antigens while responding promptly to the same antigen presented in a highly organized manner.[38] With traditional protein carriers such as KLH and TT, it is impossible to precisely control the display patterns of antigens on the surface. Recently, viral capsids have emerged as a promising platform for antigen presentation.[34, 39–44] Viral capsids are composed of structural proteins that self-assemble in a highly ordered manner. Peptide epitopes presented on the surface of viral capsids can stimulate an adaptive immune response by effective activation of antigen presenting cells as well as stimulation of B-cell mediated response by direct cross-linking of B cell receptors.[34, 45] We became interested in examining whether patterned display of TACAs can also enhance their immune response. Thus we selected the cowpea mosaic virus (CPMV) capsid, which is highly immunogenic yet non-infectious to humans.[43, 44, 46] CPMV capsid is readily available, and can be isolated in gram quantity from cowpea plants.[47] The X-ray structure of CPMV capsid is known to near atomic resolution with sixty copies of protein subunits arranged in a 30 nm icosahedron.[48] Extensive studies have been carried out by Finn, Johnson, and coworkers demonstrating that CPMV capsids can be chemically modified, which allows the attachment of sixty copies of a compound in an icosahedral symmetry to the external surface of the CPMV capsid.[49–53] In addition, the CPMV capsid has been used as a carrier of small peptides through genetic engineering, which was shown to activate Th cells.[46, 54–57] We earlier showed that glycan display on CPMV was effective in chickens to give highly specific anti-carbohydrate IgY antibodies in large quantities.[58] Here we report the results of an initial investigation in mice for IgG generation and applications of CPMV capsids for potential cancer vaccine development.
The Tn antigen (GalNAc-α-O-Ser/Thr) was chosen as the model antigen to be conjugated to CPMV capsid. Tn antigen is a TACA overexpressed on the surface of a variety of cancer cell surfaces including breast, colon and prostate cancer, rendering it an excellent immunotherapy target.[59, 60] Danishefsky and coworkers have reported that the conjugation of multiple copies of monomeric Tn to KLH did not elicit a Tn-specific antibody response.[61] Instead, Tn trimer clusters on KLH were needed to achieve immunological recognition, primarily giving rise to IgM.[62] Similar phenomena were also observed by Lo-Man and coworkers, who reported very low antibody titers when Tn monomer was used as the antigen while Tn trimer was much more effective in active specific immunotherapy.[27, 63] We wished to determine if the low immunogenicity of Tn monomer could be overcome by the highly patterned display of Tn on the CPMV capsid, presenting a new avenue to boost immune responses to weak immunogens such as carbohydrates.
Results and Discussion
Synthesis of Tn Antigens for Bio-conjugation
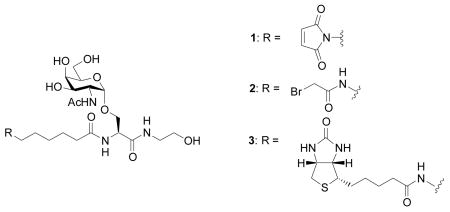
In order to conjugate the Tn antigen to CPMV capsids and for ELISA assays, we designed three Tn analogs, compound 1 with a maleimide linker, compound 2 containing bromoacetamide and 3 terminated with biotin at the N-terminus. The carboxyl ends of all Tn derivatives were capped by ethanolamine. The Tn derivatives 1–3 were synthesized from the common intermediate amine 4 (Scheme 1).
Scheme 1.

Reagents and conditions: a) NIS (2.8 eq), AgOTf (0.2 eq), toluene:dioxane (1:3), MS-AW300; b) Zn dust, AcOH, Ac2O, THF, 0 °C; c) H2, Pd/C, MeOH; d) BOP, DIPEA, THF then ethanolamine; e) NH3, MeOH.
The preparation of amine 4 started from the reaction of thioglycosyl donor 5 with the serine acceptor 7. The amino group in 7 was protected by Fmoc as it is known that a carbamate protective group enhances the nucleophilicity of the serine hydroxyl group. The main challenge in reaction of 5 with 7 is the formation of the α anomer. Boons and coworkers reported the exclusive formation of an α linkage between the thioglycoside donor 6 and a threonine acceptor[64] using the Ph2SO and triflic anhydride promoter system.[65] However, this reaction condition caused extensive decomposition in our case. We found instead that N-iodosuccinimide and silver triflate[66] in diethyl ether, a solvent well known to favor the formation of the axial anomer,[67] provided an α:β ratio of 3:1. Changing the solvent to a mixture of toluene and 1,4-dioxane (1:3)[68] led to enhancement of the ratio to 6:1 with 50–65% yield of the desired anomer 8.
The conversion of the azido group to acetamide can be performed under a variety of conditions including catalytic hydrogenolysis, thioacetic acid reduction or Zn/acetic acid reduction followed by acetylation. Previously we encountered difficulties in simultaneously reducing azide and benzyl groups by catalytic hydrogenolysis.[67, 69] Thus, we tested the direct conversion of azide to acetamide via treatment of thioacetic acid,[62] resulting in amide 9 in 85% yield. However, the cleavage of the benzyl ester from 9 by catalytic hydrogenolysis turned out to be very inconsistent, presumably due to the poisoning of the Pd catalyst by trace amounts of sulfur-containing impurities in 9, despite repeated purifications. Ultimately, the one pot reduction and acetylation of the azide 8 by zinc dust in acetic anhydride and acetic acid was found to give 90–95% of amide 9, which was hydrogenated smoothly with Pd/C under atmospheric pressure of hydrogen within half an hour to yield carboxylic acid 10. The carboxylic acid was capped first with n-butyl amine, but the corresponding amide was very poorly soluble in a variety of solvents. Replacing n-butyl amine with ethanolamine greatly enhanced the product solubility. The coupling of 10 with ethanolamine proceeded smoothly when carboxylic acid 10 was first activated with benzotriazole-1-yl-oxy-tris-(dimethylamino)-phosphonium hexa-fluorophosphate (BOP) prior to the addition of the nucleophile, giving amide 11 in 60–80% yields in a couple of hours. When BOP was added to a mixture of 10 and ethanolamine, the reaction was very slow, with prolonged incubation leading to Fmoc removal under the basic reaction condition.
Compound 11 was deprotected first with catalytic sodium methoxide at pH 9,[70] but slow addition of the base was necessary to carefully control the pH of the reaction mixture in order to prevent base-promoted elimination of the O-glycoside. Instead, we found it more convenient to treat 11 with ammonia in methanol, which cleanly removed all the acetates as well as the Fmoc group in 90–95% yield. The highly polar amino alcohol 4 was purified by a short silica gel column. It is imperative to remove all impurities in 4 in order to facilitate purification of the final products.
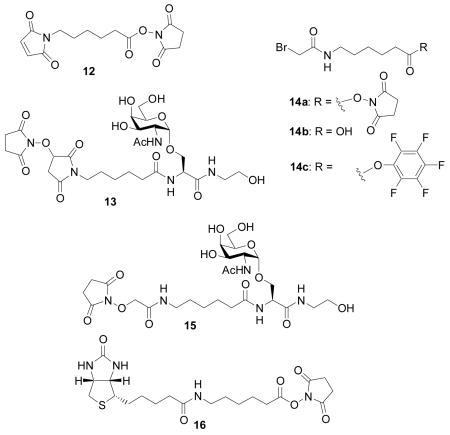
Coupling of maleimide containing N-hydroxysuccinimide ester 12 with 4 was first carried out in N-methylpyrrolidinone (NMP) at room temperature. Besides the desired product 1, a significant amount of N-hydroxysuccinimide-maleimide adduct (compound 13) was obtained, which was very difficult to separate from 1. Extended silica gel chromatography led to decomposition of 1 due to the instability of the maleimide moiety. However, performing the reaction at −20 °C greatly suppressed the formation of 13 in the reaction mixture, from which pure 1 was obtained in 50% yield by selective precipitation upon completion of the reaction and addition of diethyl ether to the mixture.[71, 72]
Amidation of 4 by bromoacetamido containing N-hydroxysuccinimide ester 14a[73] generated desired product 2 mixed with N-hydroxysuccinimide substitution product 15 even at −20°C. Silica gel chromatography, reversed phase HPLC, and size exclusion chromatography all failed to give good separation of the desired product. Dicyclohexylcarbodiimide (DCC) or BOP promoted coupling of acid 14b[73] with 4 again produced large amounts of α-carbon substitution products by the nucleophilic dicyclohexyl urea or hydroxyl benzotriazole side products generated in the reaction. To reduce the nucleophilicity of the side product, we examined pentafluorophenol activated ester 14c. Coupling of amine 4 with 14c in NMP at 0°C produced pure 2 in 59% yield without any pentafluorophenol substitution side product following diethyl ether precipitation. The biotinated Tn analog 3 was prepared in 72% yield by amidation of amine 4 with biotinated amino caproic acid N-hydroxysuccinimide ester 16.
Bioconjugation
Previously it has been reported that native CPMV has no exposed cysteine on the exterior surface. Therefore, the wild type CPMV is fairly inert to thiol-reactive reagents.[52, 53, 74] A number of mutants of CPMV bearing reactive cysteines on the exterior surface have been produced by cassette mutagenesis or site-specific mutagenesis, and a wide range of compounds, including biotin, fluorescent dyes, nanogold, and carbohydrates, can be attached to the thiol groups of such mutant CPMV particles.[52, 53, 74] In this study, the T2102C/T228C chimera (residues 102 and 28 of the large subunits were replaced by cysteines, termed as S-CPMV), was employed as a stable and thiol-reactive particle (Figures S1–S3).[75, 76] Because two cysteine moieties are located close to each other in space (approximately 21A apart), it is difficult to load more than 60 molecules on the surface of the capsid (Figure S4).[75] Both Tn derivatives 1 and 2 were immobilized on S-CPMV through the reported procedures (Scheme 2). Briefly, purified S-CPMV was incubated with an excess of 1 or 2 overnight at 4°C in potassium phosphate buffer (PBS, pH 7.0) with the addition of DMSO (20%) to aid in the solubility of the Tn antigens. The unconjugated Tn antigens were removed by ultracentrifugation over a sucrose gradient and the products were analyzed by transmission electron microscopy (TEM) and fast protein liquid chromatography (FPLC) verifying the presence of intact CPMV capsids (Figure 1). Reactions performed under the same conditions with analogous fluorescein reagents confirmed the assumption that 60±10 attachments are made to the CPMV capsid in each case (Supporting Information).
Scheme 2.

Synthesis of Tn-S-CPMV conjugates.
Figure 1.

Size-exclusion FPLC of S-CPMV (blue line) and purified Tn-1-S-CPMV (red line) showing intact capsids eluting at 10 mL and a small amount of disassembled protein eluting at 19 mL. TEM of Tn-1-S-CPMV verifying the presence of intact capsids (inset, scale bar = 100 nm).
The presence of the Tn antigen on the surface of S-CPMV was confirmed by protein binding studies with Soybean Agglutinin (SBA), a tetrameric plant lectin specific for GalNAc. An increase in absorbance monitored by UV-vis spectroscopy at 600 nm was observed when SBA was mixed with Tn-S-CPMV, indicating the formation of aggregates due to crosslinking of several Tn-S-CPMV units by SBA leading to an increase in light scattering. In contrast, a slight decrease in absorbance resulted from mixing S-CPMV with SBA due to the dilution effect (Figure 2). The UV-visible spectroscopy results were confirmed by TEM, with aggregates clearly observed when SBA was mixed with Tn-1-S-CPMV, while underivatized S-CPMV remained monomeric with SBA (Figure 3) highlighting the specificity of this interaction.
Figure 2.

Binding of SBA (50 mM) and CPMV conjugates (0.2 mg/mL). Tn-1-S-CPMV (square), S-CPMV (diamond).
Figure 3.

TEM images of SBA incubated with S-CPMV (left) and Tn-1-S-CPMV (right). Conditions identical to UV-visible light scattering assay. Scale bars = 100 nm.
Immunological studies
With the Tn-S-CPMV conjugates in hand, immunological studies were carried out on a group of five C57BL/6 female mice. Tn-1-S-CPMV (0.5 mg corresponding to approximately 40 μg of Tn) as an emulsion in complete Freund’s adjuvant was injected subcutaneously into each mouse (day 0). Booster immunizations were performed subcutaneously on days 14 and 28 with the same amount of Tn-1-S-CPMV in incomplete Freund’s adjuvant emulsion. As a control, another group of five C57BL/6 mice received identical vaccine formulations except that S-CPMV (0.5 mg) was used to replace Tn-1-S-CPMV. Blood was collected from both groups of mice on days 0, 7 and 35 and serum was isolated, in which the Tn specific antibody levels were analyzed by Enzyme Linked Immunosorbent Assay (ELISA). To exclude the immune response against the maleimide linker,[77] the biotinated Tn 3 devoid of maleimide was immobilized on a microtiter plate coated with Neutravidin. The sera was added to the microtiter plate at increasing dilutions and the IgG, IgM as well as total antibody titers were determined photometrically using secondary anti-mouse IgG+IgM, IgG, and IgM antibodies conjugated to horseradish peroxidase.
While day 0 and day 7 sera did not show significant antibody titers, the sera collected on day 35 from mice immunized with Tn-1-S-CPMV showed a total titer (IgG+IgM) of 15,000, an IgG titer of 10,500, and IgM titer of 5,000 (Figure 4). Importantly, high IgG titers were obtained indicating that a T cell mediated immune response was produced (Figure 4b). Mice immunized with S-CPMV only minimum anti-Tn antibody titer on day 35 (Figure 4a) as expected. To further ascertain that the linker does not contribute to the immune response much, the total IgG+IgM titer of day 35 serum was measured by ELISA in the presence of 0.1 M free GalNAc. The total antibody titer detected under these conditions dropped by more than two orders of magnitude, indicating that a significant amount of the polyclonal antibodies generated indeed recognized the GalNAc moiety, which is present in all Tn antigens (Figure 4a).
Figure 4.

a) ELISA titers of anti-Tn IgG+IgM antibody in mouse sera prior to immunization (day 0), after immunization with Tn-1-S-CPMV (day 35) in the absence and presence of 0.1 M GalNAc. b) ELISA titers of anti-Tn IgM and IgG antibody in mouse sera after immunization with Tn-1-S-CPMV (day 35). Day 35 serum from mice immunized with the carrier S-CPMV was used as the control. Antibody titers were defined as the maximum folds of dilution resulting in 0.1 OD unit higher than the average absorbance after greater than one hundred thousand folds of dilution.
Finally, the immune response against the natural target, Tn positive tumor cells, was tested. It is known that Tn antigen is overexpressed on MCF-7 breast cancer cell surfaces.[61] Preimmune and postimmune (day 35) sera were analyzed for binding to MCF-7 cells by Fluorescent Assisted Cell Sorting (FACS) using a fluorescein labeled anti-IgG secondary antibody (Figure 5a). A statistically significant increase in binding to MCF-7 cells by IgG was found, which was similar to those obtained from a trivalent vaccine construct.[19] This suggests the IgG antibodies generated in our study were able to recognize Tn in its native configurations on the tumor cell surface. Multi-drug resistant cancer cells present a major obstacle to cancer therapy. We next evaluated the binding of our antibodies with a multidrug resistant breast cancer cell line NCI-ADR RES. Excitingly, the day 35 postimmune serum generated in our studies showed good affinity with NCI-ADR RES cells, suggesting that carbohydrate based tumor associated vaccines can be a potentially novel therapeutic towards multidrug resistant cancer (Figure 5b). The binding of antibodies with cancer cells was greatly reduced in the presence of free GalNAc (Figure S5). The observed differential FACS responses of MCF-7 and NCI-ADR RES cells could be due to different patterns of Tn displayed on these cell surfaces.
Figure 5.

FACS analysis of cell surface reactivity to a) MCF-7 cell and b) NCI-ADR RES cell by IgG antibodies in day 35 mice sera generated against Tn-1-S-CPMV. The control samples are the mice serum from day 0 just prior to immunization. Day 35 serum from mice immunized with S-CPMV showed identical cell surface reactivity to that of the day 0 serum on both cells.
The monomeric form of Tn represents one of the weakest forms of immunogen. Yet, through conjugation with CPMV capsid, a strong humoral immune response with the desired antibody subtype was obtained, indicating that patterned display of Tn can enhance the immunogenicity of this weak antigen. It is possible that the covalent linkage between Tn and CPMV renders Tn epitopes on CPMV to be processed and presented by the Tn-specific B cell, thus allowing B cell stimulation by matched helper T cells. It may also be the case that the spacing between neighboring Tn molecules on the capsid surface (3.2–5 nm, depending on the sites of attachment) can crosslink B cell receptors[78] leading to potent activation of B cells.
CPMV capsids have been successfully adapted to express foreign peptide epitopes on the surface and the resulting chimera induced peptide specific antibodies, which have been shown to provide protection against infections.[46, 54–57] However, the display of each peptide epitope requires cloning and expression of chimera capsids individually. Our approach of post-expression modification gives us greater flexibility in varying antigen structures.
Conclusion
We have demonstrated that by covalently conjugating a weak carbohydrate antigen Tn to CPMV, strong humoral immune responses were induced, which produced high titers of IgG antibodies capable of recognizing Tn on breast cancer cell surfaces. Although our current proof of principle studies are focused on the Tn antigen, this technology can be easily adapted towards the study of other carbohydrate based anti-cancer vaccine constructs, thus presenting a general approach towards carbohydrate based vaccine development. Structurally well-characterized platforms such as CPMV offer different reactive groups, such as the exterior lysine side chains[50, 53, 58, 79] in addition to the inserted cysteines used here, giving us orthogonal ways to introduce multiple antigens onto the carrier surface. Work is in progress to construct these novel conjugates as well as to evaluate in vivo the protective effects of these constructs on tumor models.
Experimental Section
General Procedures
All chemical reactions were carried out under nitrogen with anhydrous solvents in flame-dried glassware, unless otherwise noted. All glycosylation reactions were performed in the presence of molecular sieves, which were flame-dried right before the reaction under high vacuum. Glycosylation solvents were dried using an MBraun solvent purification system and used directly without further drying. Chemicals used were reagent grade as supplied except where noted. Analytical thin-layer chromatography was performed using silica gel 60 F254 glass plates (EM Science); compound spots were visualized by UV light (254 nm) and by staining with a yellow solution containing Ce(NH4)2(NO3)6 (0.5 g) and (NH4)6Mo7O24 4H2O (24.0 g) in 6% H2SO4 (500 mL). Flash column chromatography was performed on silica gel 60 (230 – 400 Mesh, EM Science). 1H-NMR, 13C-NMR, 1H–1H gCOSY, 1H–13C gHMQC and 1H–13C gHMBC spectra were recorded on a Varian VXRS-400 or Inova-600 instrument and were referenced using Me4Si (0 ppm), residual CHCl3 (δ1H-NMR 7.26 ppm, 13C-NMR 77.0 ppm). Optical rotations were measured at 25 C. ESI mass spectra were recorded on ESQUIRE LC–MS operated in positive ion mode. High-resolution mass spectra were recorded on a Micromass electrospray Tof™ II (Micromass, Wythenshawe, UK) mass spectrometer equipped with an orthogonal electrospray source (Z-spray) operated in positive ion mode, which is located at the Mass Spectrometry and Proteomics Facility, the Ohio State University.
N-(9-Fluorenylmethyloxycarbonyl)-O-(3,4,6-tri-O-acetyl-2-azido-2-deoxy-α-D-galactopyranosyl)-L-serine benzylester (8)
Donor 5 (1 g, 2.29 mmol) and acceptor 7 (1.05 g, 2.51 mmol, 1.1 eq) were first coevaporated with anhydrous toluene and pumped under vacuum for one hour. The mixture was then dissolved in a toluene/1,4-dioxane mixture (1:3, 40 mL total) and stirred at 0 °C in the presence of activated MS AW-300 (5g) for 45 minutes. Silver triflate (295 mg, 1.15 mmol, 0.5 eq) was added followed by the addition of N-iodosuccinimide (1.03 g, 4.58 mmol, 2 eq). The reaction mixture was allowed to warm up to room temperature and was stirred for up to 32 hours when complete consumption of donor 5 was confirmed by TLC analysis. The reaction was then quenched by filtration through Celite. Dichloromethane was added to the reaction mixture and extracted with a saturated solution of sodium bicarbonate. The organic layer was dried over anhydrous sodium sulfate, concentrated to dryness and the residue was purified by flash chromatography (Hexanes/EtOAc, 3:1 → 2:1). The desired α isomer was obtained in its pure form as an oil with a 50–65% yield and a 6:1αβ selectivity. Comparison with literature data[62] confirms its identity. 1H-NMR (400 MHz, CDCl3):α anomer δ 7.79–7.73 (2H, m), 7.65–7.59 (2H, m), 7.41–7.30 (9H, m), 5.96 (1H, d, 3J = 8.0 Hz), 5.39 (1H, d, 3J = 2.4 Hz), 5.27–5.23 (3H, m), 4.86 (1H, d, 3J = 3.6 Hz), 4.62–4.59 (1H, m), 4.41 (1H, d, 3J = 7.2 Hz), 4.24 (1H, t, 3J = 7.2 Hz), 4.17–3.99 (9H, m), 3.58 (1H, dd, 3J = 3.6, 11.2 Hz), 2.14 (3H, s), 2.07 (3H, s), 1.96 (3H, s).
N-(9-Fluorenylmethyloxycarbonyl)-O-(2-acetamido-3,4,6-tri-O-acetyl-2-deoxy-α-D-galactopyranosyl)-L-serine benzylester (9)
A mixture of compound 8 (1 g, 1.37 mmol), zinc dust (2.6 g, 27.4 mmol, 20 eq), acetic acid (0.8 mL,13.7 mmol, 10 eq) and acetic anhydride (1.3 mL, 13.7 mmol, 10 eq) in THF (30 mL) was stirred overnight at room temperature. Complete reaction of compound 8 was confirmed by TLC analysis. The reaction was then quenched by filtration through Celite followed by extraction with ethyl acetate and a saturated solution of sodium bicarbonate. The organic layer was dried over anhydrous sodium sulfate, concentrated to dryness and the residue was purified by flash chromatography (Hexanes/EtOAc, 2:1). The desired product was obtained in its pure form as an oil with a 90–95% yield. Comparison with literature data[62] confirms its identity. 1H-NMR (600 MHz, CDCl3): δ 7.79-7.73 (2H, m), 7.64-7.57 (2H, m), 7.41-7.30 (9H, m), 5.86 (1H, d, 3J = 7.8 Hz), 5.59 (1H, d, 3J = 9.6 Hz), 5.29 (1H, d, 3J = 3.0 Hz), 5.22-5.15 (2H, m), 5.02 (1H, dd, 3J = 2.4, 10.8 Hz), 4.75 (1H, d, 3J = 3.0 Hz), 4.59-4.57 (1H, m), 4.54-4.50 (1H, m), 4.42 (1H, d, 3J = 7.2 Hz), 4.22 (1H, t, 3J = 7.2 Hz), 4.06-3.93 (5H, m), 2.15 (3H, s), 1.99 (3H, s), 1.96 (3H, s), 1.89 (3H, s).
N-(9-Fluorenylmethyloxycarbonyl)-O-(2-acetamido-3,4,6-tri-O-acetyl-2-deoxy-α-D-galactopyranosyl)-L-serine (10)
A mixture of compound 9 (1 g, 1.3 mmol) and palladium on activated carbon (1 g) in methanol was stirred under a hydrogen atmosphere for 30 minutes. Completion of the reaction was confirmed by TLC analysis (dichloromethane/methanol, 95:5). The reaction mixture was then filtered through Celite and concentrated under vacuum. Compound 10 was obtained in its pure form with a 90–95% yield and was used without further purification. 1H-NMR (600 MHz, CDCl3): δ 7.79-7.72 (2H, m), 7.54-7.49 (2H, m), 7.37-7.30 (4H, m), 5.40 (1H, br s), 5.23 (1H, d, 3J = 11.4 Hz), 4.93 (1H, d, 3J = 3.0 Hz), 4.56 (1H, dd, 3J = 3.0, 11.4 Hz), 4.28 (1H, t, 3J = 6.0 Hz), 4.18-4.09 (3H, m), 3.95-3.87 (2H, m), 3.75 (1H, t, 3J = 9.0 Hz), 2.18 (3H, s), 2.07 (3H, s), 2.01 (3H, s), 1.99 (3H, s).
N-(9-Fluorenylmethyloxycarbonyl)-O-(2-acetamido-3,4,6-tri-O-acetyl-2-deoxy-α-D-galactopyranosyl)-L-serine ethanolamide (11)
Compound 10 (0.75 g, 1.1 mmol), BOP (973 mg, 2.2 mmol, 2 eq) and diisopropylethylamine (0.38 ml, 2.2 mmol, 2 eq) were dissolved in THF/dichloromethane (1:1, 25 mL: 25 mL). The mixture was stirred at room temperature for one hour and then ethanolamine (0.33 mL, 5.5 mmol, 5 eq) was added. The reaction mixture was stirred at room temperature for two hours or until completion of the reaction was confirmed by TLC analysis (dichloromethane/methanol, 90:10). The reaction mixture was diluted with dichloromethane and extracted with a saturated solution of ammonium chloride. The organic layer was dried over anhydrous sodium sulfate, concentrated to dryness and the residue was purified via flash column chromatography (dichloromethane/methanol, 90:10). The desired product was obtained in its pure form as a white solid with a 60–80% yield. 1H-NMR (600 MHz, CDCl3): δ 7.79-7.78 (2H, m), 7.67-7.65 (2H, m), 7.42-7.40 (2H, m), 7.35-7.32 (2H, m), 6.37 (1H, d, 3J= 3.0 Hz), 5.93 (1H, d, 3J= 7.2 Hz), 5.10-5.02 (1H, m), 4.60-4.41 (3H, m), 4.25-4.17 (2H, m), 4.13-4.06 (2H, m), 3.94-3.78 (2H, m), 3.67-3.65 (2H, m), 3.47-3.43 (m, 2H), 2.19 (3H, s), 2.07 (3H, s), 2.00 (3H, s); 1.96 (3H, s) 13C-NMR (150 MHz, CDCl3) δ 171.8, 170.9, 170.6, 143.8, 141.5, 128.1, 127.3, 125.1, 120.3, 98.9, 69.4, 68.5, 67.3, 67.2, 61.9, 61.3, 47.7, 47.3, 41.9; ESI [M + Na]+ m/z calcd for C34H39N2NaO14 722.2, found 722.3.
O-(2-Acetamido-2-deoxy-α-D-galactopyranosyl)-L-serine ethanolamide (4)
Compound 11 (0.5 g, 0.71 mmol) was dissolved in a solution of ammonia in methanol (20 mL) and the reaction was stirred at 0oC for six hours. Completion of the reaction was confirmed by TLC analysis (dichloromethane/methanol, 80:20). The reaction mixture was concentrated to dryness and the residue was purified via short flash column chromatography (dichloromethane/methanol, 80:20→20:80). The desired product was obtained in its pure form as a white solid with a 90–95% yield. 1H-NMR (600 MHz, CDCl3): δ 4.76 (1H, d, 3J= 3.0 Hz), 4.26 (1H, dd, 3J= 3.0, 10.8 Hz), 3.86 (1H, br s), 3.81 (2H, m), 3.76-3.67 (3H, m), 3.59 (1H, t, 3J= 5.4 Hz), 3.53-3.49 (2H, m), 3.33-3.28 (4H, m), 1.98 (3H, s), ESI [M + Na]+ m/z calcd for C13H23N2NaO9 374.2, found 374.2.
N-6-Maleimido hexanoyl-O-(2-acetamido-2-deoxy-α-D-galactopyranosyl)-L-serine ethanolamide (1)
Compound 4 (30 mg, 0.085 mmol) and compound 12 (29 mg, 0.094 mmol, 1.1 eq) were dissolved in N-methylpyrrolidinone (5 mL). The reaction mixture was stirred at −20°C for 40 minutes. Completion of the reaction was confirmed by mass spectroscopy. The reaction was quenched by addition of diethyl ether to the mixture in order to precipitate the product. The solid residue was obtained by filtration. It was then redissolved in water and lyophilized. The desired product was obtained as a white solid with a 50% yield. [α]25D + 238 (c 2.5, H2O) 1H-NMR (600 MHz, D2O): δ 6.65 (2H, s), 4.72 (1H, d, 3J = 3.6 Hz), 4.39 (1H, t, 3J = 5.4 Hz), 3.98 (1H, dd, 3J = 3.6, 10.8 Hz), 3.79 (1H, d, 3J = 2.4 Hz), 3.74-3.69 (3H, m), 3.47-4.45 (2H, m), 3.31 (2H, t, 3J = 7.2 Hz), 3.19-3.17 (2H, m), 3.16 (1H, s), 2.53 (1H, s), 2.14 (2H, t, 3J = 7.2 Hz), 1.86 (3H, s), 1.44-1.38 (4H, m), 1.12-1.09 (2H, m); 13C-NMR (150 MHz, D2O) δ 177.2, 174.6, 173.5, 171.7, 134.4, 97.9, 71.4, 68.5, 67.8, 67.4, 61.3, 59.9, 53.9, 49.9, 41.7, 37.5, 35.3, 27.4, 25.6, 24.8, 22.1; HRMS [M + Na]+ m/z calcd for C23H35N4NaO11 567.2278, found 567.2278.
Pentafluorophenyl 6-N-bromoacetamido hexanoate (14c)
Compound 14b (0.5 g, 1.98 mmol)[73] was dissolved in CH2Cl2 (20 mL) along with N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (0.43 g, 2.78 mmol, 1.4 eq). Pentafluorophenol (0.51 g, 2.78 mmol, 1.4 eq) was then added and the reaction mixture was stirred at room temperature for five hours. Completion of the reaction was confirmed by TLC analysis (dichloromethane/methanol, 90%:10%). The reaction was quenched by extraction with water and dichloromethane. The organic layer was then dried over anhydrous sodium sulfate, concentrated to dryness and the residue was purified by flash column chromatography (Hexanes/EtOAc, 3:1→1:1). The desired product was obtained in its pure form as a white solid with a 71 % yield. 1H-NMR (600 MHz, CDCl3): δ 6.56 (1H, br s), 3.85 (2H, s), 3.31-3.28 (2H, m), 2.66 (2H, t, 3J= 7.2 Hz), 1.81-1.76 (2H, m), 1.62-1.57 (2H, m), 1.47-1.42 (2H, m); 13C-NMR (150 MHz, CDCl3) δ 169.5, 165.6, 142.1, 140.4, 138.8, 137.2, 40.1, 33.3, 29.5, 29.1, 26.2, 24.4. ESI-MS [M + Na]+ m/z calcd for C14H13BrF5NNaO3 440.0, found 440.1.
6-N-Bromoacetamido-hexanoyl-O-(2-acetamido-2-deoxy-α-D-galactopyranosyl)-L-serine ethanolamide (2)
Compound 4 (30 mg, 0.085 mmol) and compound 14c (39 mg, 0.094 mmol, 1.1 eq) were dissolved in N-methylpyrrolidinone (5 mL). The reaction mixture was stirred at −20 °C for 45 minutes. Completion of the reaction was confirmed by mass spectroscopy. The reaction was quenched by addition of diethyl ether to the mixture in order to precipitate the product. The solid residue was obtained by filtration. It was then redissolved in water and lyophilized. The desired product was obtained as a white solid with a 59 % yield. [α]25D + 254.8 (c 2.1, H2O) 1H-NMR (600 MHz, D2O): δ 4.70 (1H, d, 3J = 3.6 Hz), 4.38 (1H, d, 3J = 3.6 Hz), 3.95 (1H, dd, 3J = 3.6, 10.8 Hz), 3.77 (1H, d, 3J = 2.4 Hz), 3.73-3.69 (1H, m), 3.61-3.58 (1H, m), 3.55 (1H, t, 3J = 7.2 Hz), 3.46-3.43 (2H, m), 3.05-3.00 (2H, m), 2.14 (2H, t, 3J = 7.2 Hz), 1.84 (3H, s), 1.45-1.33 (4H, m), 1.16-1.13 (2H, m) 13C-NMR (150 MHz, D2O) δ 177.2, 174.5, 171.7, 169.9, 97.9, 71.4, 68.5, 67.7, 67.3, 61.2, 59.9, 53.8, 49.8, 41.7, 39.7, 35.3, 28.2, 27.8, 25.5, 24.9, 22.1; HRMS [M + Na]+ m/z calcd for C21H35BrN3NaO11 607.1585, found 607.1585.
6-N-Biotinamido-hexanoyl-O-(2-acetamido-2-deoxy-α-D-galactopyranosyl)-L-serine ethanolamide (3)
Compound 4 (30 mg, 0.085 mmol) and compound 16 (42 mg, 0.094 mmol, 1.1 eq) were dissolve in N-methylpyrrolidinone (5 mL). The reaction mixture was stirred at room temperature for 30 minutes. Completion of the reaction was confirmed by mass spectroscopy. The reaction was quenched by addition of diethyl ether to the mixture in order to precipitate the product. The solid residue was obtained by filtration. It was then redissolved in water and lyophilized. The desired product was obtained as a white solid with a 72% yield. [α]25D + 26.2 (c 1.3, H2O) 1H-NMR (600 MHz, D2O): δ 4.68 (1H, d, 3J = 3.6 Hz), 4.40-4.36 (2H, m), 4.21 (1H, dd, 3J = 4.2, 7.8 Hz), 3.93 (1H, dd, 3J = 4.2, 11.4 Hz), 3.76 (1H, d, 3J = 3 Hz), 3.71-3.65 (2H, m), 3.59-3.51 (3H, m), 3.44-3.41 (2H, m), 3.16-3.10 (4H, m), 2.98-2.95 (2H, m), 2.7 (1H, dd, 3J = 4.8, 13.2 Hz), 2.56 (1H, d, 3J = 12.6 Hz), 2.13 (2H, t, 3J = 7.8 Hz), 2.01 (2H, t, 3J = 7.2 Hz), 1.83 (3H, s), 1.51-1.28 (8H, m), 1.21-1.11 (4H, m); 13C-NMR (150 MHz, D2O) δ 177.2, 176.7, 174.5, 171.7, 97.9, 71.4, 68.5, 67.7, 67.4, 62.2, 61.3, 60.4, 59.9, 55.5, 53.8, 49.9, 41.7, 39.8, 39.2, 35.6, 35.4, 28.2, 27.9, 27.8, 25.7, 25.3, 24.9, 22.1 HRMS [M + Na]+ m/z calcd for C28H47N6NaO11S 713.3156, found 713.3158.
Synthesis of Tn-S-CPMV
The cysteine mutant of CPMV (S-CPMV) was generated by modifying the viral RNA to mutate T2102 and T228 into cysteines as reported.[75] S-CPMV was incubated at 1 mg/mL with 50 molar equivalents of maleimide-Tn or 200 molar equivalents of bromoacetamide-Tn overnight at 4°C in buffer with 20% DMSO. The reaction was purified by ultracentrifugation over a sucrose gradient (0–40%) at 27,000 rpm for 2 hours at 4°C using a Beckman SW28 rotor. The band corresponding to intact CPMV was collected and pelleted by ultracentrifuge at 42,000 rpm using Beckman 50.2Ti rotor for 2.5 hours. The pellet was resuspended in buffer. The conjugate was analyzed by UV-visible spectroscopy, TEM and FPLC.
General procedure for handling and analysis of CPMV
The virus was stored in buffer at a concentration of about 10 mg/mL. Unless otherwise indicated, “buffer” refers to 0.1 M PBS (pH 7.0). Virus concentrations were determined by measuring the UV-visible absorbance at 260 nm; virus at 0.1 mg/mL gives a standard absorbance of 0.8. Ultracentrifugation was performed at the indicated rpm values using a Beckman OptimaTM L-90K Ultracentrifuge equipped with either SW28 or 50.2 Ti rotors. TEM analyses were carried out by depositing 20 μL aliquots of each sample onto 100-mesh carbon-coated copper grids for 2 minutes. The grids were then stained with 20 μL of 2% uranyl acetate and viewed with Hitachi H-8000 electron microscope.
UV-visible binding studies with Soybean Agglutinin (SBA)
SBA was purchased from Sigma-Aldrich and used without further purification. It was stored as a lyophilized powder at −20°C until use. Tn-S-CPMV was incubated at a concentration of 0.2 mg/mL with 50 mM SBA in buffer. Binding was monitored over time by the absorbance caused by light scattering at 600 nm. UV-visible spectroscopy measurements were made using an Agilent 8400 UV-visible spectrometer. TEM analysis was performed after incubation under these conditions for 30 minutes as described above under the general procedure for handling and analysis of S-CPMV.
Immunization of mice
Pathogen-free C57BL/6 female mice age 6–10 weeks were obtained from Jackson Laboratory (Bar Harbor, ME, USA) and maintained in the Animal Care Facility of the University of Toledo. All animal care procedures and experimental protocols have been approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Toledo. Groups of five C57BL/6 mice were injected subcutaneously under the scruff on day 0 with 0.5 mg of Tn-1-S-CPMV (containing 40 μg of the glycoconjugate 1) or 0.5 mg of S-CPMV as emulsions in complete Freund’s adjuvant (0.1 mL, Fisher). Boosters were given subcutaneously under the scruff on days 14 and 28 with 0.5 mg of Tn-1-S-CPMV or S-CPMV as emulsions in incomplete Freund’s adjuvant (0.1 mL). Blood (~ 0.2 mL/mouse) was collected from both groups of mice on days 0, 7 and 35. Sera from each group of mice were isolated and pooled.
Enzyme-Linked Immunosorbent Assay (ELISA)
A 96-well microtiter plate was first coated with a solution of Neutravidin in PBS buffer (8 μg/mL) and then incubated overnight at 4 °C. The plate was then washed four times with PBS/0.5% Tween-20 (PBST), followed by the addition of 1% (w/v) BSA in PBS to each well and incubation at room temperature for one hour. The plate was washed again with PBST and a solution of biotinylated Tn 3 (5 μg/mL) in 0.1% BSA in PBS was added. The plate was incubated for one hour at 37 °C. The plate was then washed and mice sera were added in a 1:3 serial dilution in 0.1% BSA/PBS. The plate was incubated for two hours at 37 °C and washed. A 1:2000 diluted horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG+IgM, IgG or IgM antibody (Jackson ImmunoResearch Laboratory IgG+IgM catalog #115-035-068, IgM #115-035-075, IgG #115-035-071) in 0.1% BSA/PBS was added to each well respectively. The plate was incubated for one hour at 37 °C, washed and a solution of 3,3′,5,5′-tetramethylbenzidine (TMB) was added. Color was allowed to develop for 20 minutes and then a solution of 0.5 M H2SO4 was added to quench the reaction. The opticaldensity was then measured at 450 nm. Each experiment was repeated at least four times and the average of the quadruplicate was used to calculate the titer. Errors of each measurement were typically within 10%. Antibody titers were defined as the maximum folds of dilution resulting in 0.1 OD unit higher than the average absorbance after greater than one hundred thousand folds of dilution.
FACS
MCF-7 and NCI-ADR RES human breast cancer cells were obtained from the National Cancer Institute and maintained in RPMI-1640 containing 2.0 mM L-glutamine and 20 mM HEPES supplemented with 5% FBS. Both cells were trypsinized. Single-cell suspensions of 4*106 cells/mL (25 μL) were washed with FACS buffer (PBS-1% BSA-0.1% NaN3) and incubated with 1:10 diluted test sera (75 μL) for 30 minutes at 4 °C. The cells were washed twice with FACS buffer (1 mL) and then 1:100 diluted goat antimouse IgG labeled with FITC (100 μL, Jackson ImmunoResearch Laboratory, catalog #115-095-164) was added. The samples were incubated for 30 minutes at 4 °C. The cells were washed again twice with FACS buffer (1 mL) and re-suspended in FACS buffer (0.2 mL). Samples were kept in the dark at 4 °C till time of analysis. Analyses of the percentage positive cells and mean fluorescence intensity of stained cells were done using FACScalibur (BD Biosciences).
Supplementary Material
Acknowledgments
The work was supported by National Institute of General Medical Sciences, NIH (R01-GM 72667 to XH and R01-EB000432 to MGF), an Interdisciplinary Research Initiation Award from the University of Toledo (XH and KAW), the National Science Foundation (CHEM-0748690 and BES-0508419 to QW), DoD BCRP Synergy Award, DURIP and the W. M. Keck Foundation (QW and MGF). We would like to thank Profs. John E. Johnson and Tianwei Lin (The Scripps Research Institute) for providing the initial stocks of S-CPMV, Prof. Anthony Quinn (Univ. of Toledo) for help on FACS experiments, Brandon Slotterbeck (Univ. of Toledo) for ELISA, Prof. Viranga Tillekeratne, Prof. Max Funk and Ms. Nicole Ellis (Univ. of Toledo) for generously providing the MCF-7 and NCI-ADR RES cells and other experimental help.
Footnotes
Supporting information for this article is available on the WWW under http://www.chemeurj.org/ or from the author.
References
- 1.Dwek MV, Brooks SA. Curr Cancer Drug Targets. 2004;4:425–442. doi: 10.2174/1568009043332899. [DOI] [PubMed] [Google Scholar]
- 2.Hakomori S, Zhang Y. Chem Biol. 1997;4:97–104. doi: 10.1016/s1074-5521(97)90253-2. [DOI] [PubMed] [Google Scholar]
- 3.Livingston PO, Ragupathi G. Human Vaccines. 2006;2:137–143. doi: 10.4161/hv.2941. [DOI] [PubMed] [Google Scholar]
- 4.Vliegenthart JFG. FEBS Lett. 2006;580:2945–2950. doi: 10.1016/j.febslet.2006.03.053. [DOI] [PubMed] [Google Scholar]
- 5.Wang LX. Curr Opin Drug Discovery Dev. 2006;9:194–206. [PubMed] [Google Scholar]
- 6.Ouerfelli O, Warren JD, Wilson RM, Danishefsky SJ. Exp Rev Vaccines. 2005;4:677–685. doi: 10.1586/14760584.4.5.677. [DOI] [PubMed] [Google Scholar]
- 7.Roy R. Drug Disc Today: Technologies. 2004;1:327–336. doi: 10.1016/j.ddtec.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 8.Werz DB, Seeberger PH. Chem Eur J. 2005;11:3194–3206. doi: 10.1002/chem.200500025. [DOI] [PubMed] [Google Scholar]
- 9.Danishefsky SJ, Allen JR. Angew Chem. 2000;112:882–912. doi: 10.1002/(sici)1521-3773(20000303)39:5<836::aid-anie836>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2000;39:836–863. doi: 10.1002/(sici)1521-3773(20000303)39:5<836::aid-anie836>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 10.Galonic DP, Gin DY. Nature. 2007;446:1000–1007. doi: 10.1038/nature05813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liakatos A, Kunz H. Curr Opin Mol Therap. 2007;9:35–44. [PubMed] [Google Scholar]
- 12.Freire T, Bay S, Vichier-Guerre S, Lo-Man R, Leclerc C. Mini-Rev Med Chem. 2006;6:1357–1373. doi: 10.2174/138955706778992996. [DOI] [PubMed] [Google Scholar]
- 13.Slovin SF, Keding SJ, Ragupathi G. Immunol Cell Biol. 2005;83:418–428. doi: 10.1111/j.1440-1711.2005.01350.x. [DOI] [PubMed] [Google Scholar]
- 14.Kuberan B, Linhardt RJ. Curr Org Chem. 2000;4:635–677. [Google Scholar]
- 15.Dziadek S, Hobel A, Schmitt E, Kunz H. Angew Chem. 2005;117:7803–7808. doi: 10.1002/anie.200501594. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2005;44:7630–7635. doi: 10.1002/anie.200501594. [DOI] [PubMed] [Google Scholar]
- 16.Keil S, Claus C, Dippold W, Kunz H. Angew Chem. 2001;113:379–382. doi: 10.1002/1521-3773(20010119)40:2<366::AID-ANIE366>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2001;40:366–369. [Google Scholar]
- 17.Ingale S, Wolfert MA, Gaekwad J, Buskas T, Boons GJ. Nature Chem Biol. 2007;3:663–667. doi: 10.1038/nchembio.2007.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buskas T, Ingale S, Boons G-J. Angew Chem. 2005;117:6139–6142. doi: 10.1002/anie.200501818. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2005;44:5985–5988. doi: 10.1002/anie.200501818. [DOI] [PubMed] [Google Scholar]
- 19.Ragupathi G, Coltart DM, Williams LJ, Koide F, Kagan E, Allen J, Harris C, Glunz PW, Livingston PO, Danishefsky SJ. Proc Nat Acad Sci USA. 2002;99:13699–13704. doi: 10.1073/pnas.202427599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ragupathi G, Koide F, Livingston PO, Cho YS, Endo A, Wan Q, Spassova MK, Keding SJ, Allen J, Ouerfelli O, Wilson RM, Danishefsky SJ. J Am Chem Soc. 2006;128:2715–2725. doi: 10.1021/ja057244+. [DOI] [PubMed] [Google Scholar]
- 21.Wang Q, Zhang J, Guo Z. Bioorg Med Chem. 2007;15:7561–7567. doi: 10.1016/j.bmc.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu J, Guo Z. Bioconjuate Chem. 2006;17:1537–1544. doi: 10.1021/bc060103s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bundle DR, Rich JR, Jacques S, Yu HN, Nitz M, Ling C-C. Angew Chem. 2005;117:7903–7907. doi: 10.1002/anie.200502179. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2005;44:7725–7729. doi: 10.1002/anie.200502179. [DOI] [PubMed] [Google Scholar]
- 24.Kuberan B, Sikkander SA, Tomiyama H, Linhardt RJ. Angew Chem. 2003;115:2119–2121. doi: 10.1002/anie.200351099. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2003;42:2073–2075. doi: 10.1002/anie.200351099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cipolla L, Rescigno M, Leone A, Peri F, La Ferla B, Nicotra F. Bioorg Med Chem. 2002;10:1639–1646. doi: 10.1016/s0968-0896(01)00433-3. [DOI] [PubMed] [Google Scholar]
- 26.Bousquet E, Spadaro A, Pappalardo MS, Bernardini R, Romeo R, Panza L, Ronsisvalle G. J Carbohydr Chem. 2000;19:527–541. [Google Scholar]
- 27.Lo-Man R, Vichier-Guerre S, Perraut R, Deriaud E, Huteau V, BenMohamed L, Diop OM, Livingston PO, Bay S, Leclerc C. Cancer Res. 2004;64:4987–4994. doi: 10.1158/0008-5472.CAN-04-0252. [DOI] [PubMed] [Google Scholar]
- 28.Lo-Man R, Bay S, Vichier-Guerre S, Deriaud E, Cantacuzene D, Leclerc C. Cancer Res. 1999;59:1520–1524. [PubMed] [Google Scholar]
- 29.Roy R, Baek MG. J Biotechnol. 2002;90:291–309. doi: 10.1016/s1389-0352(01)00065-4. [DOI] [PubMed] [Google Scholar]
- 30.Kircheis R, Vondru P, Nechansky A, Ohler R, Loibner H, Himmler G, Mudde GC. Bioconjugate Chem. 2005;16:1519–1528. doi: 10.1021/bc050157m. [DOI] [PubMed] [Google Scholar]
- 31.Liu X, Siegrist S, Amacker M, Zurbriggen R, Pluschke G, Seeberger PH. ACS Chem Biol. 2006;1:161–164. doi: 10.1021/cb600086b. [DOI] [PubMed] [Google Scholar]
- 32.Ojeda R, de Paz JL, Barrientos AG, Martin-Lomas M, Penades S. Carbohydr Res. 2007;342:448–459. doi: 10.1016/j.carres.2006.11.018. [DOI] [PubMed] [Google Scholar]
- 33.Toyokuni T, Dean B, Cai S, Boivin D, Hakomori S, Singhal AK. J Am Chem Soc. 1994;116:395–396. [Google Scholar]
- 34.Denis J, Majeau N, Acosta-Ramirez E, Savard C, Bedard M-C, Simard S, Lecours K, Bolduc M, Pare C, Willems B, Shoukry N, Tessier P, Lacasse P, Lamarre A, Lapointe R, Lopez Macias C, Leclerc D. Virology. 2007;363:59–68. doi: 10.1016/j.virol.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 35.Jegerlehner A, Storni T, Lipowsky G, Schmid M, Pumpens P, Bachmann MF. Eur J Immunol. 2002;32:3305–3314. doi: 10.1002/1521-4141(200211)32:11<3305::AID-IMMU3305>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 36.Bachmann MF, Zinkernagel RM. Annu Rev Immunol. 1997;15:235–270. doi: 10.1146/annurev.immunol.15.1.235. [DOI] [PubMed] [Google Scholar]
- 37.Bachmann MF, Hengartner H, Zinkernagel RM. Eur J Immunol. 1995;25:3445–3451. doi: 10.1002/eji.1830251236. [DOI] [PubMed] [Google Scholar]
- 38.Bachmann MF, Rohrer UH, Kundig TM, Burki K, Hengartner H, Zinkernagel RM. Science. 1993;262:1448–1451. doi: 10.1126/science.8248784. [DOI] [PubMed] [Google Scholar]
- 39.Jennings GT, Bachmann MF. Curr Mol Med. 2007;7:143–155. doi: 10.2174/156652407780059140. [DOI] [PubMed] [Google Scholar]
- 40.Chackerian B. Expert Rev Vaccines. 2007;6:381–390. doi: 10.1586/14760584.6.3.381. [DOI] [PubMed] [Google Scholar]
- 41.Grgacic EVL, Anderson DA. Methods. 2006;40:60–65. doi: 10.1016/j.ymeth.2006.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCormick AA, Corbo TA, Wykoff-Clary S, Palmer KE, Pogue GP. Bioconjugate Chem. 2006;17:1330–1338. doi: 10.1021/bc060124m. [DOI] [PubMed] [Google Scholar]
- 43.Porta C, Spall VE, Lin T, Johnson JE, Lomonossoff GP. Intervirology. 1996;39:79–84. doi: 10.1159/000150478. [DOI] [PubMed] [Google Scholar]
- 44.Usha R, Rohll JB, Spall VE, Shanks M, Maule AJ, Johnson JE, Lomonossoff GP. Virology. 1993;197:366–374. doi: 10.1006/viro.1993.1598. [DOI] [PubMed] [Google Scholar]
- 45.Fehr T, Skrastina D, Pumpens P, Zinkernagel RM. Proc Natl Acad Sci USA. 1998;95:9477–9481. doi: 10.1073/pnas.95.16.9477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brennan FR, Jones TD, Hamilton WDO. Mol Biotechnol. 2001;17:15–26. doi: 10.1385/MB:17:1:15. [DOI] [PubMed] [Google Scholar]
- 47.Lin T, Porta C, Lomonossoff G, Johnson JE. Fold Des. 1996;1:179–187. doi: 10.1016/s1359-0278(96)00030-2. [DOI] [PubMed] [Google Scholar]
- 48.Lin T, Chen Z, Usha R, Stauffacher CV, Dai JB, Schmidt T, Johnson JE. Virology. 1999;265:20–34. doi: 10.1006/viro.1999.0038. [DOI] [PubMed] [Google Scholar]
- 49.Gupta SS, Kuzelka J, Singh P, Lewis WG, Manchester M, Finn MG. Bioconjuate Chem. 2005;16:1572–1579. doi: 10.1021/bc050147l. [DOI] [PubMed] [Google Scholar]
- 50.Chatterji A, Ochoa WF, Paine M, Ratna BR, Johnson JE, Lin T. Chem Biol. 2004;11:855–863. doi: 10.1016/j.chembiol.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 51.Chatterji A, Ochoa W, Shamieh L, Salakian SP, Wong SM, Clinton G, Ghosh P, Lin T, Johnson JE. Bioconjugate Chem. 2004;11:807–813. doi: 10.1021/bc0402888. [DOI] [PubMed] [Google Scholar]
- 52.Wang Q, Lin T, Tang J, Johnson JE, Finn MG. Angew Chem. 2002;114:477–480. doi: 10.1002/1521-3773(20020201)41:3<459::aid-anie459>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2002;41:459–462. [Google Scholar]
- 53.Wang Q, Kaltgrad E, Lin T, Johnson JE, Finn MG. Chem Biol. 2002;9:805–811. doi: 10.1016/s1074-5521(02)00165-5. [DOI] [PubMed] [Google Scholar]
- 54.Liu L, Canizares MC, Monger W, Perrin Y, Tsakiris E, Porta C, Shariat N, Nicholson L, Lomonossoff GP. Vaccine. 2005;23:1788–1792. doi: 10.1016/j.vaccine.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 55.Nicholas BL, Brennan FR, Martinez-Torrecuadrada JL, Casal JI, Hamilton WDO, Wakelin D. Vaccine. 2002;20:2727–2734. doi: 10.1016/s0264-410x(02)00200-1. [DOI] [PubMed] [Google Scholar]
- 56.Rennermalm A, Li YH, Bohaufs L, Jarstrand C, Brauner A, Brennan FR, Flock JI. Vaccine. 2001;19:3376–3383. doi: 10.1016/s0264-410x(01)00080-9. [DOI] [PubMed] [Google Scholar]
- 57.Langeveld JPM, Brennan FR, Martinez-Torrecuadrada JL, Jones TD, Boshuizen RS, Vela C, Casal JI, Kamstrup S, Dalsgaard K, Meloen RH, Bendig MM, Hamilton WDO. Vaccine. 2001;19:3661–3670. doi: 10.1016/s0264-410x(01)00083-4. [DOI] [PubMed] [Google Scholar]
- 58.Kaltgrad E, Sen Gupta S, Punna S, Huang CY, Chang A, Wong CH, Finn MG, Blixt O. Chembiochem. 2007;8:1455–1462. doi: 10.1002/cbic.200700225. [DOI] [PubMed] [Google Scholar]
- 59.Springer GF. J Mol Med. 1997;75:594–602. doi: 10.1007/s001090050144. [DOI] [PubMed] [Google Scholar]
- 60.Springer GF. Science. 1984;224:1198–1206. doi: 10.1126/science.6729450. [DOI] [PubMed] [Google Scholar]
- 61.Kagan E, Ragupathi G, Yi SS, Reis CA, Gildersleeve J, Kahne D, Clausen H, Danishefsky SJ, Livingston PO. Cancer Immunol Immunother. 2005;54:424–430. doi: 10.1007/s00262-004-0584-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kuduk SD, Schwarz JB, Chen XT, Glunz PW, Sames D, Ragupathi G, Livingston PO, Danishefsky SJ. J Am Chem Soc. 1998;120:12474–12485. [Google Scholar]
- 63.Lo-Man R, Vichier-Guerre S, Bay S, Deriaud E, Cantacuzene D, Leclerc C. J Immunol. 2001;166:2849–2854. doi: 10.4049/jimmunol.166.4.2849. [DOI] [PubMed] [Google Scholar]
- 64.Cato D, Buskas T, Boons GJ. J Carbohydr Chem. 2005;24:503–516. [Google Scholar]
- 65.Codee JDC, Litjens REJN, den Heeten R, Overkleeft HS, van Boom JH, van der Marel GA. Org Lett. 2003;5:1519–1522. doi: 10.1021/ol034312t. [DOI] [PubMed] [Google Scholar]
- 66.Konradsson P, Udodong UE, Fraser-Reid B. Tetrahedron Lett. 1990;30:4313–4316. [Google Scholar]
- 67.Miermont A, Zeng Y, Jing Y, Ye XS, Huang X. J Org Chem. 2007;72:8958–8961. doi: 10.1021/jo701694k. and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Demchenko A, Stauch T, Boons GJ. Synlett. 1997:818–820. [Google Scholar]
- 69.Wang Z, Zhou L, El-boubbou K, Ye X-S, Huang X. J Org Chem. 2007;72:6409–6420. doi: 10.1021/jo070585g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liebie B, Kunz H, Liebie B, Kunz H. Angew Chem. 1997;109:629–631. [Google Scholar]; Angew Chem Int Ed. 1997;36:618–621. [Google Scholar]
- 71.Shao N, Guo Z. Org Lett. 2005;7:3589–3592. doi: 10.1021/ol0514193. [DOI] [PubMed] [Google Scholar]
- 72.Wen S, Guo Z. Org Lett. 2001;3:3773–3776. doi: 10.1021/ol0101988. [DOI] [PubMed] [Google Scholar]
- 73.Zaitsu K, Ohnishi M, Hosoya H, Sugimoto H, Ohkura Y. Chem Pharm Bull. 1987;35:1991–1997. [PubMed] [Google Scholar]
- 74.Raja KS, Wang Q, Finn MG. Chembiochem. 2003;4:1348–1351. doi: 10.1002/cbic.200300759. [DOI] [PubMed] [Google Scholar]
- 75.Lin T. J Mater Chem. 2006;16:3673–3681. [Google Scholar]
- 76.Blum AS, Soto CM, Wilson CD, Cole JD, Kim M, Gnade B, Chatterji A, Ochoa WF, Lin T, Johnson JE, Ratna BR. Nano Lett. 2004;4:867–870. [Google Scholar]
- 77.Buskas T, Li Y, Boons GJ. Chem Eur J. 2004;10:3517–3524. doi: 10.1002/chem.200400074. [DOI] [PubMed] [Google Scholar]
- 78.Mond JJ, Lees A, Snapper CM. Annu Rev Immunol. 1995;13:655–692. doi: 10.1146/annurev.iy.13.040195.003255. [DOI] [PubMed] [Google Scholar]
- 79.Kaltgrad E, O’Reilly MK, Liao L, Han S, Paulson J, Finn MG. J Am Chem Soc. 2008 doi: 10.1021/ja077801n. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.