

Abstract
The repair of oxidative DNA lesions (ODLs) in the nucleus of ischemic cortical brain cells was examined following experimentally induced stroke by occluding the right middle cerebral artery and both common carotid arteries for 60–90 min followed by reperfusion in male long-Evans hooded rats. The control group consisted of sham-operated animals undergoing the same surgery without vessel occlusion. Using a gene-specific assay based upon the presence of Escherichia coli Fpg protein-sensitive sites, we noted that animals with stroke exhibited six and four ODLs per gene in the actin and DNA polymerase-β genes, respectively. This was increased from one per four copies of each gene in the sham-operated control (p < 0.01). One half of the initial ODLs was repaired within 30 min, and 83% of them were repaired as early as 45 min of reperfusion. There was no further increase when gene repair was measured again at 2 h of reperfusion. The rates of active repair within 45 min of reperfusion were the same in these two genes (p = 0.103, anova). BrdU (10 mg/kg) was administered via intraperitoneal injection at least one day before surgery. We observed that there was no significant incorporation of BrdU triphosphates into genomic DNA during active repair, but there were significant amounts of BrdU triphosphate in nuclear DNA after active repair. The result indicates that genomic repair of ODLs in the brain did not significantly incorporate BrdU, and the initiation of neurogenesis probably starts after the completion of repair in the brain.
Keywords: brain repair, bromodeoxyuridine, cerebral ischemia-reperfusion, neurogenesis, oxidative stress, stroke
Oxidative stress in the brain induces mitochondrial dysfunction and an elevation in extracellular glutamate, intracellular calcium, reactive oxygen species (ROS), oxidative lesions in proteins, lipids and nucleic acids (Lipton 1999; Lewen et al. 2000). These changes often induce cell death that can be detected as necrosis and programmed cell death (apoptosis). Apoptosis is a natural process during development, but can also be activated in disease or following cellular injury. One way to reduce cell death is to decrease the level of ROS by endogenous scavengers.
In addition to scavenger enzymes that quench ROS and reduce the formation of ODLs, oxidative damage is minimized by gene repair (Liu et al. 1996; Chen et al. 2000; Lin et al. 2000). The brain is known to be deficient in tissue regeneration after brain injury except in specific populations of cells (Liu et al. 1998; Takagi et al. 1999; Gu et al. 2000; Arvidsson et al. 2001; Jiang et al. 2001; Jin et al. 2001). To express correct transcripts from the remainder of the tissue, the brain would have to contain an extremely accurate mechanism that repairs its genes (molecular repair of genes), so that oxidative stress that occurs in an oxygen-utilizing organ will be kept to a minimum. One report suggests that different regions of the normal brain have different rates of repair activities (Cardozo-Pelaez et al. 2000). Our knowledge concerning gene repair in the brain after brain injury is very limited.
Generally speaking, gene repair pathways include the recognition and the excision of lesions from the DNA double helix by endonucleases or glycosylases, generating single-stranded breaks (SSBs), followed by repair synthesis accomplished by DNA polymerases. The repair process is completed by DNA ligase which connects the new and old DNA stands (Liu et al. 2001). The nucleotide excision repair pathway excises the damaged and the adjacent nucleotides, and synthesizes at least 20 nucleotides in one repair patch. Base-excision repair generally excises and replaces one nucleotide, although an incorporation of 6-10 nucleotides in one repair patch has been reported using a cell culture system (Fortini et al. 1998; Lindahl and Wood 1999). The repair synthesis is mediated by DNA-dependent DNA polymerases (α, β, δ, ε and γ) acting on the SSB with a 3′-OH end using the appropriate dNTP as directed by the sequence in the template (Sobol et al. 1996; Wilson and Singhal 1998). In mitochondria, DNA polymerase-γ is responsible for both DNA replication and repair syntheses (Naviaux et al. 1999; Chen et al. 2000).
5-Bromo-2′-deoxyuridine (BrdU) is an analog of thymidine that is metabolized to BrdU-triphosphate (BrdUTP). Bromo-dUTP can be incorporated into DNA in place of thymidine triphosphate. This property of BrdU has been used to determine the replication activity of tissue repair, or neurogenesis in the brain (Gould et al. 1999). Furthermore, BrdU-mediated DNA labeling can also be used to distinguish DNA synthesized as part of mitosis from DNA undergoing global repair synthesis (Hwang et al. 1999). In this study, the properties of BrdU-incorporation are used to determine the relationship between gene maintenance by molecular repair and tissue repair by neurogenesis using BrdU-immunoreactivity (IR) in the ischemic brain. We also aimed to determine whether nuclear genes were targeted differently by ROS, and whether genes with different transcription activities in the brain are repaired equally.
Materials and methods
Experimental stroke model using focal cerebral ischemia-reperfusion (FCIR)
Male long-Evans hooded rats (250-300 mg) were anesthetized using sodium Nembutal [80 mg/kg, intraperitoneal injection (i.p.), Abbott Laboratories, Chicago, IL, USA] according to a published protocol (Chen et al. 1986; Cui et al. 1999a, 1999b, 2000; Cui and Liu 2001a). The right middle cerebral artery (MCA) was exposed using microsurgical techniques. First, a 2-cm vertical skin incision was made from the midpoint between the right eye and the right ear and the temporalis muscle was split. Next, a 2-mm burr hole was drilled at the junction of the zygoma and the squamosal bone. A 10-0 suture was then placed, but not yet tied, around the MCA trunk. Both common carotid arteries (CCA) were exposed using micro-surgical techniques and were occluded using nontraumatic aneurysm clips, followed by immediate tightening of the 10-0 suture previously placed on the MCA trunk. Complete interruption of blood flow was confirmed using an operating microscope. The control group received the same surgery except the suture was never tightened to occlude the vessels (sham-operation). After 60- or 90-min of ischemia, the MCA ligature and the aneurysm clips were removed. Restoration of blood flow (reperfusion) in all three arteries was observed directly under the microscope. Rectal temperature was monitored and maintained at 37 ± 0.5°C via an electronic temperature controller (Versa-Therm 2156, Cole-Pharmer, Chicago, IL, USA) connected to a heating lamp. Animals were allowed free access to food and water after recovery from anesthesia. Animals were kept in air-ventilated incubators at 24 ± 0.5°C for different reperfusion periods. Animals were housed three to a cage in large rectangular cages. Veterinarian care was provided as needed. Housing and anesthesia concurred with guidelines established by the institutional animal welfare committee in accordance with the NIH Guide for the Care and Use of Laboratory Animals, USDA Regulations, and with the American Veterinary Medical Association Panel on Euthanasia guidelines.
At the designated time points, animals used for the isolation of nucleic acid or for the immunohistochemical detection of BrdU were placed under general anesthesia using sodium Nembutal. For DNA isolation, the animal was killed by decapitation and the brain was dissected. The entire cerebral cortex and hippocampus from the right hemisphere were immediately separated and frozen in liquid nitrogen before being transferred to − 80°C for storage.
Gene-specific assay to detect damage and repair after experimental stroke using FCIR
Cortical DNA of high molecular weight was isolated from the right (ipsilateral) cortex of sham-operated (n = 10) and cerebral ischemia-induced animals (n = 4 per time point) using silica gel method (Liu et al. 1996; Cui et al. 1999a, 2000). Brain DNA (48 μg) was treated with restriction endonuclease EcoRI (5 U/μg DNA, 37°C 2 h) producing fragments of a defined length in the target genes. After treatment with EcoRI, the DNA was separated into two equal parts. One part (24 μg) was mixed with buffer only and served to determine the amount of DNA that was present from each animal (lanes with N, Fig. 1). The other part was further digested using Escherichia coli Fpg protein (Novus Biologicals, Littleton, CO, 8 U/μg DNA repeated twice) at 37°C for 40 min. The E. coli Fpg protein contains two catalytic activities: the glycosylase activity that removes 8-hydroxy-2′-deoxyguanosine [oh8dG], 2,6-diamino-4-hydroxy-5N-methylformamidopyrimidine [FapyGua], 5′-hydroxycy-tosine, and the lyase activity that cuts DNA without bases [AP sites] (Demple and Harrison 1994; Krokan et al. 1997; Liu et al. 2001). The DNA in both parts was placed in adjacent lanes as a pair in an alkaline agarose gel; and was resolved using electrophoresis. The DNA in the gel was transferred to a nylon blot using the vacuum-transfer technique.
Fig. 1.

ODLs (measured as Fpg-sensitive sites) in DNA polymerase-β gene from the ipsilateral cortex before and after experimental stroke. Ten control animals were sham-operated and without focal cerebral ischemia (no FCI control) shown in the top row. Each pair of parallel lanes are DNA from the same animal, where N = not treated with Fpg protein, Y = treated with Fpg protein (N in the right and Y in the left lanes of the adjacent pair). The second and the third rows are DNA from four animals treated with experimental stroke using FCIR.
To detect the amount of DNA after Fpg protein digestion, a riboprobe of the actin or DNA polymerase-β cDNA was transcribed in the presence of [32P]UTP (specific activity of the probe was 109 dpm/pg). The ribo-probe was hybridized to the DNA blot. The intensity of the hybrid signal was developed using autoradiographic film, and analyzed using the Alphalmager (Alpha Innotech, San Leandro, CA, USA) and statistic software program (GraphPad PRISM2, San Diego, CA, USA). If the DNA contains any of the ODLs listed above, they would be removed in samples treated with Fpg protein, and the DNA strand will not migrate with the intact strand of the gene in alkaline gel. DNA without ODLs will not be digested by Fpg protein and will migrate along as the intact gene. An intact or repaired gene will show no change in its migration in the gel between samples. Therefore a decrease in the target DNA intensity with Fpg protein indicated the presence of ODLs in that gene. The number of ODLs per gene was calculated as natural log (In) of 1/R, where R is the ratio of the intensity of Fpg-protein-treated DNA (lane Y) to the intensity from sample not treated with the protein (lane N).
The rate of DNA repair in the nuclear genes of the brain
To calculate the rate of DNA repair in the brain, the initial number of ODLs per gene without reperfusion (time zero) was considered as 100% oxidative lesions. The rate of DNA repair, expressed in percent, in different reperfusion time points was determined as following: The difference between the initial number of ODLs per gene (at time zero) and the number of ODLs per gene at a particular reperfusion time was divided by the initial number of ODLs per gene, multiplied by 100.
Injection and quantitative measurement of BrdU
Unless otherwise indicated, animals were injected with BrdU (10 mg/kg, i.p) one day before surgical operation. The location where BrdU was taken up was determined using immunohisto-chemistry (n = 8). The amount of BrdU that was incorporated into nuclear DNA was measured using the South-western assay, which measure DNA in the blot using antibodies against BrdU (n = 11). Three 20-μg samples of purified high-molecular weight DNA from the right (ipsilateral) cortex of each animal were denatured using NaOH/EDTA (0.4 N/10 mm, 100°C for 10 min). The DNA was neutralized using an equal volume of ammonium acetate (2 m, pH 7) before being blotted onto a nitrocellulose membrane (Hybond ECL, Amersham Pharmacia Biotech, Piscataway, NJ, USA) using a Bio-Dot SF Microfiltration apparatus (Bio-Rad Laboratories, Hercules, CA, USA). The membrane was washed using 2 × saline sodium citrate buffer (SSC; 0.3 m NaCl and 0.03 m trisodium citrate) and dried at 80°C for 2 h. The membrane was incubated with monoclonal antibodies against BrdU (Sigma Chem. Co. St Louis, MO, USA) followed by horseradish peroxidase-conjugated anti-mouse IgG and the ECL Immunodetection system (Amersham Pharmacia Biotech) as described (Lin et al. 2000). The intensity of BrdU signal in the DNA was measured in the blot and analyzed using the Alphalmager and GraphPad Prism software. Each measurement was repeated once by each of three research personnel.
Immunoreactivity of BrdU
Anesthetized rats were retrograde-perfused with 4% paraformaldehyde in 0.1 m phosphate-buffered saline (PBS; pH 7.4). The brains were cryo-protected in 20% sucrose/4% paraformaldehyde overnight at 4°C and then embedded in paraffin. Five micrometer thick sections were sequentially de-paraffin-treated using xylene (10 min), chloroform (15 min), xylene (10 min), twice with 100% ethanol (10 min each), and 95% and 75% ethanol (3 min each) and PBS (5 min). The tissue was treated with proteinase K (20 μg/mL, 15 min at 37°C) and washed with PBS six times. The tissue was treated with DNase-free RNase A (5 μg/mL in H2O for 1 h, 37°C). The tissue was then treated with acid (4 N HC1; Liu et al. 1998) for 30 min at 37°C followed by neutralization in 2 m ammonium acetate before incubation with the antibody against BrdU [Sigma, St Louis, MO, USA; 1/600 in 1% normal goat serum, 1% normal mouse serum, 1% bovine serum albumin (BSA) in PBS, at 4°C overnight]. The tissue was washed in 0.1% triton X 100 in PBS three times. The negative control (no primary antibody) was washed in a separate jar from those with primary antibody. The brain tissue was then incubated with a bodipy-conjugated goat anti-mouse IgG (1/800), or alkaline phosphatase-conjugated goat anti-mouse IgG (1/500 in 1% BSA in PBS), followed by three washes in 0.1% triton X 100 in PBS. The tissue with alkaline phosphatase-IgG was detected using 4-nitro blue tetrazolium (NBT) and BCIP staining (Roche Molecular Biochem, Indianapolis, IN, USA).
Results
DNA damage occurs in all nuclear genes that were measured
We have previously reported that oxidative damage to nuclear genes occurs equally in both strands of the c-fos gene after experimental stroke (Cui et al. 1999a). We do not know whether the c-fos gene is specifically targeted. To determine whether DNA damage occurs in more than one gene, we measured ODLs in the nontranscribed strand of the DNA polymerase-β gene and the actin gene using a gene specific assay to detect damage/repair as reflected by the presence/absence of sensitive sites to E. coli Fpg protein (Liu et al. 1996). Both of the actin and DNA polymerase-β genes are constitutively expressed, except that mRNA of DNA polymerase-β gene is induced five-fold without an increase in protein synthesis in cells exposed to genotoxic agents under cell culture conditions (Kedar et al. 1991). DNA damage/repair in the nontranscribed strand has been measured because the strand has no transcription complex, and no interference of transcription to repair is expected immediately after brain injury. We observed that cerebral ischemia also induced ODLs in the DNA polymerase-β and actin genes. DNA polymerase-β gene (Fig. 1) or the actin gene (Fig. 2) from 10 sham-operated animals (no FCI, control) was resistant to Fpg protein treatment (the Y lanes). The intensity in each lane without Fpg protein (N) various due to the presence of alkaline-sensitive lesions in DNA that has been applied. The alkaline sensitive lesions include SSBs of various sizes due to randomly distributed ODLs. The presence of SSBs should not affect the calculation of ODLs based on the presence of Fpg protein sensitive sites because the intensities in adjacent lanes of the same animal have been measured.
Fig. 2.

ODLs (measured as Fpg-sensitive sites) in the actin gene. DNA from 10 control animals and 20 animals treated with experimental stroke using the same DNA samples as in Fig. 1. The radioactive riboprobe was stripped by boiling and was hybridized with a radioactive riboprobe transcribed from actin cDNA.
We calculated the average number of ODLs per gene (frequency of background ODL) in the non-transcribed strand of DNA polymerase-β and the actin genes to be 0.17 (DNA polymerase-β) or 0.24 (the actin gene) in the control animals (Fig. 3). At the end of cerebral ischemia with no reperfusion (zero time), these two genes showed an increase in the sensitivity to the treatment with Fpg protein. Sensitive sites were demonstrated by the loss of the ability to hybridization to a riboprobe of DNA polymerase-β and actin gene after treatment with the Fpg protein and subsequent alkaline electrophoresis (Figs 1 and 2). The frequency at this time point was significantly (p < 0.01, t-test) elevated to 6 and 4 in the DNA polymerase-β and the actin gene, respectively (Fig. 3). This represented at least a 16-fold increase in the burden of repair induced by experimental stroke. The ODLs remained elevated for the next 15 min, and started declining at 30 min, though they remained significantly (p < 0.01, t-test) elevated in both genes. The ODLs disappeared and the intact gene re-appeared at 45 and 120 min of reperfusion. However, the pattern of changes in the number of ODLs between these two genes was not different over time (p = 0.103, one way anova). The data suggest that the nuclear genes that had been examined were not differentially targeted.
Fig. 3.
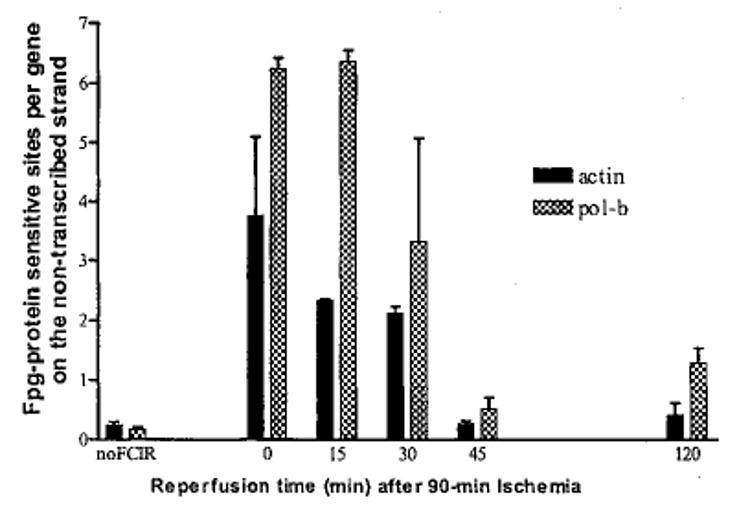
The frequency of ODLs in the actin and DNA polymerase-β genes. We observed no significant difference in the frequency of ODLs at all time points between these two genes (p = 0.103, one way anova), although there is a significant elevation in the frequency of ODLs in animals during 0–30 min of reperfusion compared to the frequency of ODLs in the control animals (p < 0.01, t-test).
The rate of DNA repair in the nuclear genes of the brain
To determine the rate at which ODLs appear and disappear (repair) in these nuclear genes, we calculated the rate of DNA repair using data presented in Fig. 3 (the actin and DNA polymerase-β genes) and from the data previously reported in the c-fos gene (Cui et al. 1999a). The c-fos gene is a silent gene under normal conditions, but is induced immediately after cerebral ischemia (An et al. 1993; Cui and Liu 2001). There was no significant difference in the rate of repair among these three genes, despite a possible difference in the rate of transcription in them. The rates of repair (mean ± standard deviation, %) in these three genes after 90 min of focal cerebral ischemia are 0, 25 ± 17, 46 ± 3, 83 ± 14 and 80 ± 7 at 0, 15, 30, 45 and 120 min of reperfusion, respectively (Fig. 4). We noted that approximately 50% of the initial ODLs were repaired by 30 min of reperfusion. The percent of repaired lesions in each gene reached a steady state at 45 and 120 min of reperfusion. We concluded that experimental stroke induces ODLs in nuclear genes, and that they are repaired at a same rate in all three genes examined.
Fig. 4.
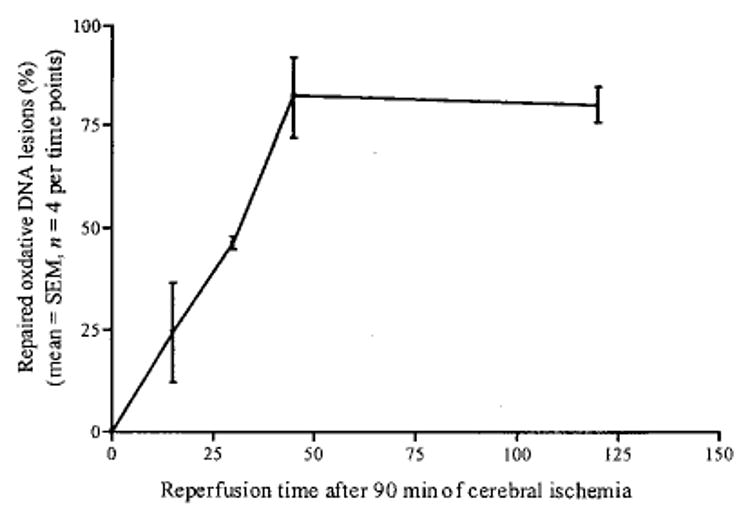
The kinetics of in vivo DNA repair in three nuclear genes from rodent brain after experimental stroke.
The uptake of BrdU after brain injury
We tested whether BrdU could be used to distinguish mitotic activity from repair activity after FCIR. We tested the route of BrdU administration (10 mg/kg, i.p.) and its uptake into the brain. BrdU was injected at 1, 3, 5 and 7 days before sham-operation. Ninety minutes after suture was placed around the MCA, the animals were killed to determine BrdU uptake using BrdU-IR. The contralateral hemisphere served as the control for BrdU uptake. BrdU-IR was observed in the cytosolic region around the suture wound in the ipsilateral site due to the placement of the 10–0 suture during sham-operation regardless the time of BrdU injection (Fig. 5). There was no BrdU-IR in the contralateral site (not shown). We noted a gradient of BrdU-IR from the suture wound into the cerebral parenchyma. When the brain tissue sections were treated with DNase-free RNase A, we observed a disappearance in the cytosolic BrdU-IR (data not shown). The results suggest that seven days after the administration of BrdU at a dose of 10 mg/kg, free BrdU remained present in the animals.
Fig. 5.
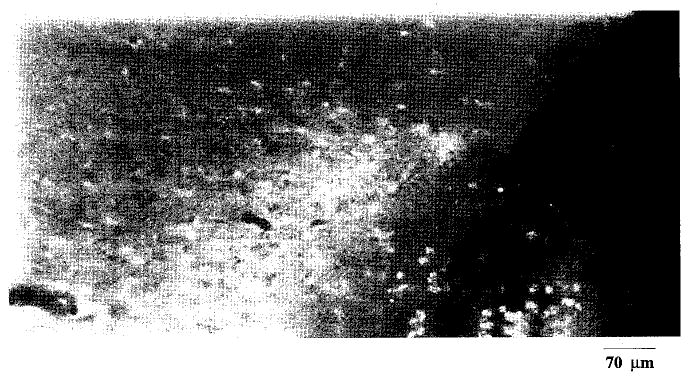
The uptake of BrdU in the brain. BrdU-IR is shown around the site of injury in the right cerebral cortex from one of the eight animals that received treatment of BrdU (10 mg/kg, i.p.). In this animal, BrdU was injected 7 days before sham-operation. Animals were killed 90 min after sham-operation for determination of BrdU-IR. BrdU-IR was noted in the cytosolic fraction of the sham-operated cortex in all eight animals. Two mitotic cells (green) are noted on the surface of the injured site. Bar = 70 μm.
The incorporation of BrdU into nuclear DNA
To determine mitotic activity immediately after experimental stroke, BrdU was injected one day before the surgery. Experimental stroke was induced using cerebral ischemia for 60 min, followed by reperfusion for up to 4 h before sacrificing the animal. This assay requires high molecular weight DNA for Southern–western blots. To isolate high molecular weight DNA from the ischemic cortex, 60 min of vessel occlusion was used to minimize cells with DNA strand breaks (Chen et al. 1997; Clark et al. 2001). Furthermore, we used silica gel filtration to isolate high molecular DNA of the nuclear origin that is free of mitochondrial DNA (data not shown). This procedure reduces a loss of signal due to low molecular weight DNA. Figure 6 shows the results from BrdU-IR assay on purified DNA from ipsilateral cortices of two control groups [sham-operated and normal animals both injected with BrdU (n = 4)] and there was no significant difference in BrdU-IR between these two control groups. Figure 6 also shows BrdU-IR in DNA from the ischemic cortices (n = 7). There was a slight, but significant increase in BrdU-IR in DNA from four animals that sustained longer than one hour of reperfusion. The temporal incorporation of BrdU into nuclear DNA shown in Fig. 6 coincided with the reappearance of intact DNA in Figs 1 and 2, suggesting neurogenesis occurs after the completion of ODL repair.
Fig. 6.
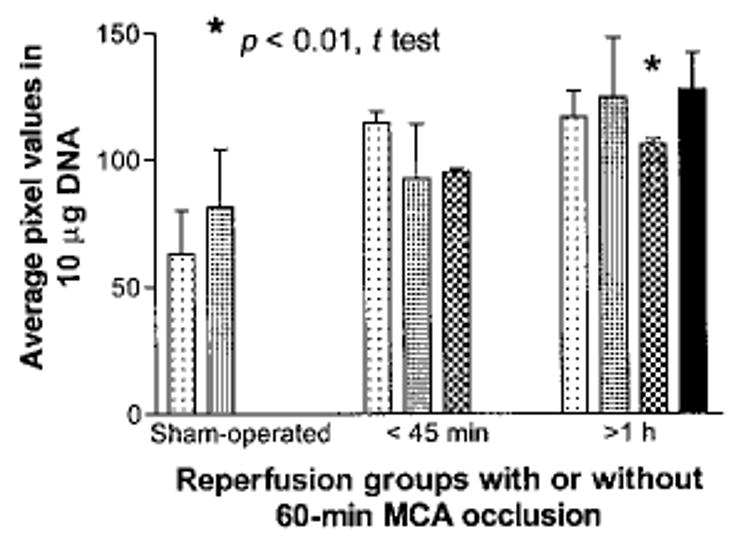
The presence of BrdU-IR in high molecular weight DNA purified from ipsilateral cortex in control animals (normal without surgery [n = 2] and sham-operation without ischemia [n = 2]) and animals with vessel occlusion for 60 min with reperfusion for 45 min (n = 3) and > 60 min (n = 4). Bars represent the average of at least four determinations from each animal for each of the treatments.
To determine whether the elevated BrdU-IR was from cells undergoing replication, we attempted to detect mitotic cells using an in situ BrdU-IR assay. We injected BrdU into six animals one day before surgery. Three animals were sham-operated and the other three underwent vessel occlusion for the induction of experimental stroke. After one day of reperfusion, all animals were killed for BrdU-IR examination. We examined 30 15-μm sections (separated by 100 μm between sections) in each of six animals. We observed no cells with BrdU-IR in the penumbral region of the ischemic cortex or the contralateral cortex. Nor did we observe BrdU-IR positive cells in the brains of the sham-operated animals. However, we did observe an increase in the number of cells that were BrdU-IR positive in the ischemic core of animals one-day after stroke. The core that contained positive BrdU-IR cells also contained cells with DNA fragmentation at this time (data not shown). The BrdU-IR was present in the nucleus and the perinuclear regions, but none of these BrdU-IR-positive cells expressed neuro-peptide Y or glial fibrillary acidic protein (not shown). BrdU-IR was resistant to treatment with acid, but was sensitive to treatments with DNase I. Because most of these mitotic cells are negative in GFAP or neuro-peptide Y antigen, we could not definitively conclude the origin of these mitotic cells. They could have been macrophages that are associated with reaction to inflammatory reaction in the site of necrosis (the ischemic core). Our results indirectly support the conclusion that in situ BrdU-IR does not detect ischemia-induced DNA repair activity in the brain (Liu et al. 1998; Gould et al. 1999).
Discussion
Brain repair is one of many potential therapies that are being explored to assist diseased brain to regain functions. We have considered both gene repair and tissue repair in this communication. We have compared the repair of ODLs in three nuclear genes after experimental stroke using an FCIR model. Although the initial frequency of ODLs is different among these genes, the brain repairs ODLs with the same rate in all nuclear genes: 50% of ODLs are repaired by 30 min and more than 80% of ODLs are repaired by 45 min of reperfusion. No significant increase in the incorporation of BrdU in nuclear DNA was noted during active repair. A significant increase of BrdU in nuclear DNA from injured-cortex after one hour of reperfusion, however, suggests that the incorporation of BrdUTP into nuclear DNA did not coincide with the repair activity (Figs 3 and 4). Therefore, the data suggest an initiation of DNA replication or neurogenesis after the completion of gene repair. The significance of this finding suggests that neurogenesis or tissue repair occurs after the majority of nuclear gene damage is no longer present. This is a safekeeping process that reduces expression of mutant message in future generation of cells. Therefore, gene repair plays an important role in the biology of neurogenesis.
In order to study the repair mechanism in the brain, we investigated the appearance and disappearance of ODLs in the actin and DNA polymerase- β genes from the ischemic cortex in rats. Experimental stroke in the rat induces ODLs in the same nuclear genes that have been reported using another model of brain injury in the mouse (Liu et al. 1996; Lin et al. 2000). It appears that all genes that have been examined thus far contain ODLs after cerebral ischemia-reperfusion regardless of experimental models.
Oxidative damage to nucleic acids is often determined by detecting base modifications in DNA or RNA. We did not measure single stranded breaks with 3′-OH termini as ODLs, because they also occur as intermediates during gene repair and during normal cellular process of DNA replication, and they can not be easily distinguished from DNA damage or fragmentation elicited by oxidative stress after brain injury. There are at least nine oxidative lesions in nucleic acids that have been shown to be increased significantly after cerebral oxidative stress in animal brains using experimental models that simulate seizure (Schulz et al. 1995; Lan et al. 2000), cardiac arrest (Floyd and Carney 1992; Liu et al. 1996), and stroke (Cui et al. 1999a, 2000; Nagayama et al. 2000; Cui and Liu 2001). Mitochondrial gene damage has recently been demonstrated after brain injury of ischemia-reperfusion type (Englander et al. 1999; Cui et al. 1999a, 2000; Huang et al. 2000). Cardozo-Pelaez and colleagues reported higher basal levels of oh8dG in total DNA from the midbrain, caudate putamen, and hippocampus than in the DNA from the cerebellum, cortex, pons and medulla (Cardozo-Pelaez et al. 2000). They observed that these regional differences in basal levels of DNA damage inversely correlate with the regional capacity to remove oh8dG from DNA. We have not detected a significant difference in the basal oh8dG content from different regions of the rat brain (Cui et al. 1999a, 2000), but we have observed repair of more than 80% of ODLs in nuclear genes within 1 h of reperfusion after cerebral ischemia in both the mouse and the rat using different injury models (Cui et al. 1999a; Liu et al. 2000). The fate of the remaining 20% or less of ODLs in the brain is not known. The ODLs, most likely, will become DNA fragmentation if not repaired.
We observed that the rate of repair in three nuclear genes of the brain was the same and appeared linear during the first 45 min. Anson et al. (1998) reported a homogenous repair of the same ODLs in different mitochondrial genes from human cells. Our observation of a homogenous rate of repair on ODLs of the brain is consistent with their observation. DNA repair in mitochondrial genome of the brain has been demonstrated (Chen et al. 2000), but the rate of DNA repair in the mitochondria in the brain after injury is not known, except hypoxia-induced DNA damage in mitochondrial and nuclear genes is no longer present within 3 h after animals are exposed to normoxia (Englander et al. 1999). The rate of DNA repair in nuclear and mitochondrial genes of the brain needs be elucidated.
We have noted that the half-life of ODLs in at least three genes of an injured brain is 30 min of reperfusion. The result agrees with the data previously reported in the α-actin gene of the brain after γ-irradiation (Ploskonosova et al. 1999). In addition, we have shown that both neurons and astrocytes contain ODLs, which are removed one hour after brain injury (Cui et al. 1999a, 2000; Huang et al. 2000). The appearance of intact genes that have been repaired after 45 min of reperfusion suggested that repair in the brain is rapid. On the other hand, it takes 20 h to repair 80% of the ODLs in human mitochondrial DNA under culture conditions (Anson et al. 1998). The same could be true for experimental stroke (Chen et al. 2000). Global repair of ODLs in nuclear genes of human cells grown in culture is also completed by approximately 24 h (Hwang et al. 1999). The basis of this difference in the rate of ODL repair in the brain and in the tissue culture conditions is not understood. Nevertheless, our data presented here suggest that the brain has an efficient molecular repair of its genes.
What was not known is the mechanism by which the brain repairs these genes. We have shown that DNA repair of both strands of an active gene (the c-fos gene) is completed in one hour using mouse and rat cerebral ischemia models (Cui et al. 1999a; Lin et al. 2000). Two repair pathways (a global nucleotide-excision repair pathway and or base-excision repair pathway) may mediate ODL repair in the brain (Reardon et al. 1997; Brooks 1998). Both pathways do not exhibit strand-specific mechanism, except a global repair generally takes longer than a few hours to complete (Hanawalt 1994; Hwang et al. 1999). A fast rate of repair within one hour in the injured brain does not suggest global nucleotide-excision repair activities.
Our data suggest that the re-appearance of intact DNA after brain injury does not result from replication activity, because there was no significant elevation in BrdU incorporation during active repair within 45 min of reperfusion. The repair activity in the brain during reperfusion excises one base per patch in the mouse brain (Lin et al. 2000). This observation is consistent with the first step of base-excision repair (Liu et al. 2001). There are two alternative routes after the first step: one that incorporates one nucleotide, and the alternative one that incorporates 6–10 nucleotides per patch. Growing evidence suggests that DNA polymerase-β incorporates one nucleotide, and DNA polymerase-ε or -δ along with proliferating cell-nuclear antigen (PCNA) and flip-endonuclease-1 (FEN-1) incorporate 6–10 nucleotides (Liu et al. 2001). The ODL in the brain identified thus far are abnormal bases of guanine, adenine, and cytosine and E. coli Fpg-protein does not excise modified thymidine (Liu et al. 1996). The lack of BrdU-incorporation during active repair of ODLs is consistent with the classical form of base-excision repair pathway in the brain. In conclusion, we show here a process of molecular repair of ODLs in nuclear genes of the brain after experimental stroke. This process is rapid and is homogeneous in all nuclear genes regardless of transcription activities. The molecular repair of genes appears to precede neurogenesis after stroke.
Acknowledgments
We thank Mr T. Arora for his assistance. The research are supported by grants from NINDS (NS34810). PKL is an Established Investigator of American Heart Association.
Abbreviations used
- BSA
bovine serum albumin
- FCIR
focal cerebral ischemia-reperfusion
- ODL
oxidative DNA lesion
- PBS
phosphate-buffered saline
- ROS
reactive oxygen species
- SSB
single-stranded break
- SSC
saline sodium citrate buffer
References
- An G, Lin TN, Liu JS, Xue JJ, He YY, Hsu CY. Expression of c-fos and c-jun family genes after focal cerebral ischemia. Ann Neurol. 1993;33:457–464. doi: 10.1002/ana.410330508. [DOI] [PubMed] [Google Scholar]
- Anson RM, Croteau DL, Stierum RH, Filburm C, Parsell R, Bohr VA. Homogenous repair of singlet oxygen-induced DNA damage in differentially transcribed regions and strands of human mitochondrial DNA. Nucleic Acids Res. 1998;26:662–668. doi: 10.1093/nar/26.2.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvidsson A, Kokaia Z, Lindvall O. N-Methyl-D-Aspartate receptor-mediated increase of neurogenesis in adult rat dentate gyrus following stroke. Eur J Neurosci. 2001;14:10–18. doi: 10.1046/j.0953-816x.2001.01611.x. [DOI] [PubMed] [Google Scholar]
- Brooks PJ. Detection of excision nuclease in cell-free extracts from the adult mammalian brain. Mutat Res. 1998;408:37–46. doi: 10.1016/s0921-8777(98)00018-4. [DOI] [PubMed] [Google Scholar]
- Cardozo-Pelaez F, Brooks PJ, Stedeford T, Song S, Sanchez-Ramos J. DNA damage, repair, and antioxidant systems in brain regions: a correlative study. Free Radic Biol Med. 2000;28:779–785. doi: 10.1016/s0891-5849(00)00172-6. [DOI] [PubMed] [Google Scholar]
- Chen ST, Hsu CY, Hogan EL, Maricq H, Balentine JD. A model of focal ischemic stroke in the rat: reproducible extensive cortical infarction. Stroke. 1986;17:738–743. doi: 10.1161/01.str.17.4.738. [DOI] [PubMed] [Google Scholar]
- Chen J, Jin K, Chen M, Pei W, Kawaguchi K, Greenberg DA, Simon RP. Early detection of DNA strand breaks in the brain after transient focal ischemia. Implications for the role of DNA damage in apoptosis and neuronal cell death. J Neurochem. 1997;69:232–245. doi: 10.1046/j.1471-4159.1997.69010232.x. [DOI] [PubMed] [Google Scholar]
- Chen D, Lan J, Pei W, Chen J. Detection of DNA base-excision repair activity for oxidative lesions in adult rat brain mitochondria. J Neurosci Res. 2000;61:225–236. doi: 10.1002/1097-4547(20000715)61:2<225::AID-JNR13>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Clark RS, Chen M, Kochanek PM, Watkins SC, Jin KL, Draviam R, Nathaniel PD, Pinto R, Marion DW, Graham D. Detection of single- and double-strand DNA breaks after traumatic brain injury in rats, comparison of in situ labeling techniques using DNA polymerase I, and terminal deoxynucleotidyl transferase. J Neurotrauma. 2001;18:675–689. doi: 10.1089/089771501750357627. [DOI] [PubMed] [Google Scholar]
- Cui JK, Liu PK. Neuronal NOS inhibitor that reduces oxidative DNA lesions and neuronal sensitivity increases the expression of intact c-fos transcripts after brain injury. J Biomed Sci. 2001;8:336–341. doi: 10.1007/BF02258375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui JK, Holmes EH, Liu PK. Oxidative damage to the c-fos gene and reduction of its transcription after focal cerebral ischemia. J Neurochem. 1999a;73:1164–1174. doi: 10.1046/j.1471-4159.1999.0731164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui JK, Hsu CY, Liu PK. Suppression of postischemic hippocampal nerve growth factor expression by a c-fos antisense oligodeoxynucleotide. J Neurosci. 1999b;19:2784–2893. doi: 10.1523/JNEUROSCI.19-04-01335.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui JK, Holmes EH, Cao ST, Greene TG, Liu PK. Oxidative DNA damage precedes DNA fragmentation after experimental stroke in rat brain. FASEB J. 2000;14:955–967. doi: 10.1096/fasebj.14.7.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demple B, Harrison L. Repair of oxidative damage to DNA. Enzymol Biol Annu Rev Biochem. 1994;63:915–948. doi: 10.1146/annurev.bi.63.070194.004411. [DOI] [PubMed] [Google Scholar]
- Englander EW, Greeley GH, Jr, Wang G, Pere-Polo JR, Lee H. Hypoxia-induced mitochondrial and nuclear DNA damage in the rat brain. J Neurosci Res. 1999;58:262–269. [PubMed] [Google Scholar]
- Floyd RA, Carney JM. Free radical damage to protein and DNA. Mechanisms involved and relevant observations on brain undergoing oxidative stress. Ann Neurol. 1992;32:22–27. doi: 10.1002/ana.410320706. [DOI] [PubMed] [Google Scholar]
- Fortini P, Pascucci B, Parlanti E, Sobol RW, Wilson SH, Doglioli E. Different DNA polymerases are involved in the short- and long-patch base excision repair in mammalian cells. Biochemistry. 1998;37:3575–3580. doi: 10.1021/bi972999h. [DOI] [PubMed] [Google Scholar]
- Gould E, Reeves AJ, Graziano MSA, Gross CG. Neurogenesis in the neocortex of adult primates. Science. 1999;286:548–552. doi: 10.1126/science.286.5439.548. [DOI] [PubMed] [Google Scholar]
- Gu W, Brannstrom T, Wester P. Cortical neurogenesis in adult rats after reversible photothrombotic stroke. J Cereb Blood Flow Metab. 2000;20:1166–1173. doi: 10.1097/00004647-200008000-00002. [DOI] [PubMed] [Google Scholar]
- Hanawalt PC. Transcription-coupled repair and human disease. Science. 1994;266:1957–1958. doi: 10.1126/science.7801121. [DOI] [PubMed] [Google Scholar]
- Huang D, Shenoy A, Huang W, Cao S, Cui J, Liu PK. Nitric oxide mediates oxidative DNA damage in cell nuclei and neuronal death in cerebral cortex after experimental forebrain ischemia. FASEB J. 2000;14:407–417. [Google Scholar]
- Hwang BJ, Ford JM, Hanawalt PC, Chu G. Expression of the p48 xeroderma pigmentosum gene is p53-dependent and is involved in global genomic repair. Proc Natl Acad Sci USA. 1999;96:424–428. doi: 10.1073/pnas.96.2.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Gu W, Brannstrom T, Rosqvist R, Wester P. Cortical neurogenesis in adult rats after transient middle cerebral artery occlusion. Stroke. 2001;32:1201–1207. doi: 10.1161/01.str.32.5.1201. [DOI] [PubMed] [Google Scholar]
- Jin K, Minami M, Lan JQ, Mao XO, Batteur S, Simon RP, Greenberg DA. Neurogenesis in dentate subgranular zone and rostral subventricular zone after focal cerebral ischemia in the rat. Proc Natl Acad Sci USA. 2001;98:4710–4715. doi: 10.1073/pnas.081011098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedar PS, Widen SG, Englander EW, Fornace AJ, Jr, Wilson SH. The ATF/CREB transcription factor-binding site in the polymerase beta promoter mediates the positive effect of N-methyl-N'-nitro-N-nitrosoguanidine on transcription. Proc Natl Acad Sci USA. 1991;88:3729–3733. doi: 10.1073/pnas.88.9.3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krokan HE, Standal R, Slupphaug G. DNA glycosylases in the base excision repair of DNA. Biochem J. 1997;325:1–16. doi: 10.1042/bj3250001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan J, Hanshall DC, Simon RP, Chen J. Formation of the base modification 8-hydroxyl-2′-deoxyguanosine and DNA fragmentation following seizures induced by systemic kainic acid in the rat. J Neurochem. 2000;74:302–309. doi: 10.1046/j.1471-4159.2000.0740302.x. [DOI] [PubMed] [Google Scholar]
- Lewen A, Matz P, Chan PH. Free radical pathways in CNS injury. J Neurotraum. 2000;17:871–890. doi: 10.1089/neu.2000.17.871. [DOI] [PubMed] [Google Scholar]
- Lin L, Cao S, Yu L, Cui J, Hamilton WJ, Liu PK. Up-regulation of base excision repair activity for 8-hydroxy-2′-deoxyguanosine in the mouse brain after forebrain ischemia-reperfusion. J Neurochem. 2000;74:1098–1105. doi: 10.1046/j.1471-4159.2000.741098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl T, Wood RD. Frontiers in cell biology: quality control by DNA repair. Science. 1999;286:1897–1905. doi: 10.1126/science.286.5446.1897. [DOI] [PubMed] [Google Scholar]
- Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- Liu J, Solway K, Messing RO, Sharp FR. Increased neurogenesis in the dentate gyrus after transient global ischemia in gerbils. J Neurosci. 1998;18:7768–7778. doi: 10.1523/JNEUROSCI.18-19-07768.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu PK, Hsu CY, Dizdaroglu M, Floyd RA, Kow YW, Karakaya A, Rabow LE, Cui JK. Damage, repair and mutagenesis in nuclear genes after mouse forebrain ischemia-reperfusion. J Neurosci. 1996;16:6795–6806. doi: 10.1523/JNEUROSCI.16-21-06795.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu PK, Grossman RG, Hsu CY, Robertson CS. Ischemic injury and faulty gene transcripts in the brain. Trends Neurosci. 2001;24:581–588. doi: 10.1016/s0166-2236(00)01918-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagayama T, Lan J, Henshall DC, Chen D, O'Horo C, Simon RP, Chen J. Induction of oxidative DNA damage in the peri-infarct region after permanent focal cerebral ischemia. J Neurochem. 2000;75:1716–1728. doi: 10.1046/j.1471-4159.2000.0751716.x. [DOI] [PubMed] [Google Scholar]
- Naviaux RK, Nyhan WL, Barrshop BA, Poulton J, Markusic D, Karpinski NC, Haas RH. Mitochondrial DNA polymerase γ deficiency and mtDNA depletion in a child with Alpers' Syndrome. Ann Neurol. 1999;45:54–58. doi: 10.1002/1531-8249(199901)45:1<54::aid-art10>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Ploskonosova I, Baranov V, Gaziev A. PCR assay of DNA damage and repair at the gene level in brain and spleen of γ-irradiated young and old rats. Mut Res. 1999;434:109–117. doi: 10.1016/s0921-8777(99)00019-1. [DOI] [PubMed] [Google Scholar]
- Reardon JT, Bessho T, Kung HC, Bolton PH, Sancar A. In vitro repair of oxidative DNA damage by human nucleotide excision repair system: Possible explanation for neurodegeneration in Xeroderma pigmentosum patients. Proc Natl Acad Sci USA. 1997;94:9463–9468. doi: 10.1073/pnas.94.17.9463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz JB, Mathews RT, Jenkins BG, Ferrante RJ, Siwek D, Henshaw DR, Cipolloni PB, Mecocci P, Kowall NW, Rosen BR, Beal MF. Blockade of neuronal nitric oxide synthase protects against excitotoxicity in vivo. J Neurosci. 1995;15:8419–8429. doi: 10.1523/JNEUROSCI.15-12-08419.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobol RW, Horton JK, Kuhn R, Gu H, Singhal RK, Prasad R, Rajewsky K, Wilson SH. Requirement of mammalian DNA polymerase-β in base-excision repair. Nature. 1996;379:183–186. doi: 10.1038/379183a0. [DOI] [PubMed] [Google Scholar]
- Takagi Y, Nozaki K, Takahashi J, Yodoi J, Ishikawa M, Hashimoto N. Proliferation of neuronal precursor cells in the dentate gyrus is accelerated after transient forebrain ischemia in mice. Brain Res. 1999;831:283–287. doi: 10.1016/s0006-8993(99)01411-0. [DOI] [PubMed] [Google Scholar]
- Wilson SH, Singhal RK. Mammalian DNA repair and the cellular DNA polymerases. In: Nickoloff JA, Hoekstra MF, editors. DNA Damage and Repair, Vol. 2, DNA Repair in Higher Eukaryotes. Humana Press Inc; Totowa NJ: 1998. pp. 161–180. [Google Scholar]