

Abstract
The patterns of malignancies in Down syndrome (DS) are unique and highlight the relationship between chromosome 21 and cancer. DS children have a ∼10- to 20-fold higher risk for developing acute lymphoblastic leukemia and acute myeloid leukemia (AML), as compared with non-DS children, although they do not have a uniformly increased risk of developing solid tumors. DS children with acute lymphoblastic leukemia frequently experience higher levels of treatment-related toxicity and inferior event-free survival rates, as compared with non-DS children. DS children also develop AML with unique features and have a 500-fold increased risk of developing the AML subtype, acute megakaryocytic leukemia (AMkL; M7). Nearly 10% of DS newborns are diagnosed with a variant of AMkL, the transient myeloproliferative disorder, which can resolve spontaneously without treatment; event-free survival rates for DS patients with AMkL ranges from 80% to 100%, in comparison with <30% for non-DS children with AMkL. In addition, somatic mutations of the GATA1 gene have been detected in nearly all DS TMD and AMkL cases and not in leukemia cases in non-DS children. GATA1 mutations are key factors linked to both leukemogenesis and the high cure rates of DS AMkL patients. Identifying the mechanisms that account for the high event-free survival rates of DS AMkL patients may ultimately improve AML treatment as well. Examining leukemogenesis in DS children may identify factors linked to the general development of childhood leukemia and lead to potential new therapeutic strategies to fight this disease.
Approximately 350,000 individuals in the United States have Down syndrome (DS) and the Center for Disease Control estimates that the incidence of DS occurs once in 733 live births in the United States (5429 new cases per year).1 Based on the prevalence and recognition of DS individuals in the general population, considerable resources have been directed toward studying the various phenotypic features of DS individuals including learning disabilities, congenital heart disease, early onset Alzheimer's disease, and high risk of developing leukemia.
Although DS children have an increased risk of developing acute leukemias, there is no increased risk of solid tumors in DS individuals. These discrepancies highlight the relationship between chromosome 21 and cancer, and they will be the subject of this review.
DS and Risk of Developing Solid Tumors
Several types of solid tumors, including lymphomas, retinoblastomas, and testicular germ cell tumors, have been reported to occur in DS individuals.2,3,4 However, large population-based and/or tumor registries have shown that solid tumors occur significantly less frequently in DS children and adults in comparison with individuals without trisomy 21. Among 2814 individuals from different age groups with DS registered in a Danish study, only 24 cases of solid tumors were identified, whereas 47.7 cases would have been expected.5
An analysis of 6724 patients with neuroblastoma comprising 54.1 million children reported from 11 European countries identified no cases of neuroblastoma among DS children, although at least five cases would have been expected (P = 0.0045).6 The National Wilms Tumor Study registry reviewed 5854 Wilms tumor cases and did not identify any kidney tumors in DS children, though it was estimated that 7.6 to 9.9 cases would have been expected.7 A British registry of 11,000 childhood solid tumor cases, identified only seven tumors in DS children [lymphoma (3), teratoma (1), glioma (2), and fibrosarcoma (1)].8
Among DS adults, carcinomas occur less frequently than in the general population. Several European studies have reported lower than expected incidences of breast cancer in DS females. Among 1278 women with DS registered in the Danish Cytogenetic and Danish Cancer Registries, no cases of breast cancer were identified, while at least seven cases would have been expected (P = 0.0007).5 Five DS women with breast cancer were identified in a French Registry spanning a 24 year period, though 69 cases would have been expected.
These clinical observations suggest that gene dosage imbalances caused by the presence of an extra copy of chromosome 21 may be protective effect against the development of certain malignancies in DS individuals. The S-100b protein (encoded by a chromosome 21-localized gene) is normally secreted by glial cells and non-neural cells of the peripheral system. It has been shown that S-100b protein can inhibit the growth and induce death of human and murine neuroblastoma cell lines.6,10 DS patients have higher blood levels of serum S-100b protein than controls, which may be one factor accounting for the low incidence of neuroblastoma in DS children.6
The COL18A1 gene (localized to chromosome 21) encodes for the anti-angiogenic agent, endostatin, which inhibits angiogenesis and tumor development in human and animal models. Higher levels of serum endostatin in DS individuals may potentially contribute to the lower occurrence of solid tumors in this group.11 Other candidate chromosome 21-localized genes include: i) The DSCR-1 gene (Down syndrome critical region 1), which blocks blood vessel formation and tumor progression in mice by inhibition of intracellular calcineurin signaling.12 ii)The ETS 2 oncogene, which has been shown to protect against the development of intestinal tumors in a mouse model of DS.13
Epidemiology of Leukemia in DS
In contrast to the potential protective effect of an extra copy of chromosome 21 and the incidence of solid tumors in DS individuals, the presence of trisomy 21 is considered one of the most significant and identifiable risk factors for the development of acute leukemias. It is estimated that DS children have a 10- to 20-fold increased risk of developing acute lymphoblastic leukemia (ALL) or acute myeloid leukemia (AML) compared with non-DS children, such that approximately 1:100 to 1:150 DS children will develop leukemia.14
A number of epidemiology studies have been conducted by the Children's Oncology Group (COG) to identify the basis for the increased incidence of leukemia in DS children. The studies have failed to identify any association between infection,15 maternal or paternal preconception irradiation exposure,16 or presence of congenital anomalies17 and the risk of developing acute leukemias in DS children. Studies have suggested that vitamin use in the periconceptional period may be protective against the development of ALL in DS,18 while maternal exposure to professional pest exterminations, pesticides or chemicals,18 and maternal infertility19 may potentially increase the risk of acute leukemias in DS children. In the future, epidemiological studies should document environmental exposures with the analysis of biological samples,20 as well as correlate these findings with leukemia-specific gene mutations.
Case Study 1
A 4-year-old Caucasian boy with DS presented to the emergency room with worsening right leg pain and problems walking. On examination, the patient appeared to be in pain. There was no lymphadenopathy, though hepatosplenomegaly was present.
The white blood cell (WBC) count was 7200/mm3, with 5% myelocytes, 4% metamyelocytes, 14% bands, 36% neutrophils, 38% lymphocytes, 2% monocytes, 1% basophils, and 3% nucleated red blood cells; the hemoglobin was 10.6g/dL with normal mean corpuscular volume, and the platelet count 330, 000/mm3. The LDH was 653 U/L (normal range from 100 to 225 units per L). Liver-function tests were within normal limits. The uric acid was 6.8 mg/dl (normal range from 3.5 to 7.2 mg/dl). Review of the peripheral smear showed the presence of lymphoblasts. A bone marrow aspirate was performed and revealed 80% L1 lymphoblasts. Immunophenotyping was consistent with CD-45 negative, CD-34 positive, B precursor ALL expressing CD-19, CD-22, CD-10, and HLA-DR. Cytogenetic demonstrated the presence of trisomy 21 without any additional cytogenetic abnormalities. The patient received chemotherapy treatment according to the Pediatric Oncology Group (POG) 9905 ALinC 17: Protocol for Patients with Newly Diagnosed Standard Risk Acute Lymphoblastic Leukemia, consisting of 130 weeks of therapy. Despite receiving reduced dosing intermediate methotrexate (500 mg/m2 instead of 1000 mg/m2) during consolidation therapy, the patient experienced episodes of transient mucositis. The patient currently remains in remission, 18 months from the completion of his therapy.
DS and ALL
DS children are estimated to represent ∼2% of total ALL cases in children21 (Table 1). Interestingly, the two most common chromosomal/cytogenetic subgroups of childhood B-precursor ALL involve qualitative or quantitative abnormalities involving chromosome 21, suggesting a direct or indirect relationship between chromosome 21 and leukemogenesis. Hyperdiploid ALL, in which blast cells contain >50 chromosomes, uniformly contain three to four copies of chromosome 21, while the TEL-AML1 t(12;21)(p13;q22) translocation involves the AML1 oncogene localized to chromosome 21. These ALL subgroups represent ∼25% to 30% and ∼20% to 25% of all B-precursor ALL cases, respectively, and are considered the most favorable ALL subgroups with event-free survival (EFS) rates of >80%.22 Despite this relationship between chromosome 21 and ALL in non-DS children, several studies have reported that hyperdiploid ALL and TEL-AML1 ALL occur at an equal23 or lower frequencies in DS ALL patients.24,25,26,27,28 Interestingly, the supernumerary chromosomes found in DS children with hyperdiploid ALL are similar to the ones found in non-DS patients,27 suggesting similar pathogenesis between the two hyperdiploid subgroups of patients. The t(8;14) is the second most commonly found translocation in DS ALL, after t(12,21),27,29,30 and certain translocations related to an adverse outcome, such as t(4,11), t(1.19), and t(9,22), are rarely found in DS ALL patients as well as the T-cell ALL phenotype.27,30
Table 1.
Clinical Features of Acute Leukemia in Down Syndrome (DS) and Non-DS Children
| DS-ALL | Non-DS-ALL | DS-AML | Non-DS AML | |
|---|---|---|---|---|
| Total % of cases | 2% | 98% | 15% | 85% |
| Age at diagnosis | Peak 2–5 years | Peak 2–7 years | <4 years | Peak: <1 year, and during adolescence |
| Major predisposing risk factors | None | None | Prior history of the transient myeloproliferative disorder (TMD) | None |
| Cytogenetic subgroups | Lower frequency of hyperdiploidy and TEL-AML1 | Hyperdiploidy (25%–30%) | >90% are AMkL (M7) | ∼10% AMkL |
| Occurrence of T-cell | t(12,21) (20%–25%) | |||
| ALL rare | t(1,19) (5%–6%) | |||
| t(4,11) (2%) | ||||
| Associated somatic gene mutation | JAK2 mutations (∼20%) | TEL-AML1 fusion protein | GATA1 mutations (uniform) | Wild-type GATA1 |
| E2A-PBX1 fusion protein | ||||
| AF4-ALL1/MLL/HRX fusion proteins | ||||
| Treatment toxicity | Excessive toxicity to methotrexate Inreased frequency of infections | Chemotherapy associated myelosuppression | Increased risk of anthracycline-related cardiotoxicity | Chemotherapy associated myelosuppression |
| Cure rates | ∼65% | 70%–85% | 80%–100% | 50% (<30% for AMkL cases) |
A recently identified genetic mutation involving the JAK2 gene (Janus kinase 2; localized to chromosome 9p24) has been found in approximately 20% of DS ALL cases, resulting in the substitution of the conserved arginine at position 683 (JAK2R683).31,32 JAK2 protein belongs to the JAK family of non-receptor tyrosine kinase proteins, which are essential for normal intracellular signaling of hematopoietic and growth factor receptors. Gain-of-function mutations of the JAK2 gene have been identified in a high proportion of adults with myeloproliferative disorder, including polycythemia vera, essential thrombocythemia, and idiopathic myelofibrosis.33 However, JAK2R683 mutations have been found exclusively in patients with DS and ALL, and are probably related to leukemogenesis in this group of patients. Different JAK2 mutations have been rarely found in non-DS ALL patients, but they have not been described in DS AML patients.34
Studies have shown that DS ALL patients typically have inferior EFS rates and a greater incidence of treatment-related mortalities.24,25,26,28 In a recent Children's Cancer Group (CCG) trial, DS ALL patients treated on a high risk protocol had equivalent treatment outcomes compared with non-DS ALL patients.35 The lower frequency of DS ALL patients with favorable cytogenetics (eg, hyperdiploid and TEL-AML1), may partly account for the inferior outcomes in some studies in addition to treatment related mortalities.24,25,26,28
Host factors may also account for the differences in toxicities. DS individuals without leukemia have functional deficiencies in B-cell, T-cell, and phagocytic cell systems.36 They also have a higher risk of dying from bacterial sepsis, as well as developing severe viral infections.37 Additionally, the metabolism of drugs in DS patients may also be distinct. Indeed, earlier studies revealed evidence of excessive toxicity including mucositis with methotrexate therapy.38,39 The reduced folate carrier gene (localized to 21q22) encodes the transmembrane protein that transports intracellularly reduced folates including 5-methylhydrofolate (the major dietary folate) and methotrexate.40 The increased expression of the reduced folate carrier gene in various body tissues of DS patients (eg, gastrointestinal tract) may result in increased intracellular methotrexate transport contributing to methotrexate toxicity of DS patients.
Case Study 2A
A newborn baby girl with DS was admitted to the neonatal intensive care unit because of cyanosis and she was found to have Tetralogy of Fallot. Further laboratory investigation revealed a WBC count of 59,000/mm3, hemoglobin of 17 g/dl and platelets of 80,000/mm3. Review of the peripheral smear showed the presence of 80% of large blasts with very basophilic staining cytoplasm, cytoplasmic blebbing, and prominent nucleoli. Total and direct bilirubin levels and transaminase levels were within the normal range. Flow cytometry of the peripheral smear revealed an abnormal CD-45 dim population of blasts, which expressed CD-11B, CD-33, CD-34, CD-117, weakly expressed CD-4, CD-7, showed partial expression of CD-56, CD-40, CD-13, and platelet-associated antigens CD-41, CD-61, CD-36, CD-9, and glycophorin A. Peripheral blood cytogenetics demonstrated constitutional trisomy 21. A diagnosis of the transient myeloproliferative disorder (TMD) was made.
DS and TMD
TMD is an acute megakaryocytic leukemia (AMkL)-related disorder diagnosed in newborn DS babies based on the detection of megakaryoblasts in the peripheral blood and/or bone marrow. TMD megakaryoblasts have the same morphology and surface antigen expression as AMkL cells.41 TMD is a disorder restricted to patients harboring trisomy 21, and it has been found in phenotypically normal infants and children who were trisomy 21 mosaics.42
It is estimated that TMD occur in approximately 10% of newborn DS infants, if careful and systematic review of peripheral blood smears for the presence of megakaryoblasts is performed. However, TMD cases may go undetected, since blood counts are not necessarily done as part of the routine care of DS babies at birth.41 GATA1 mutation analysis (discussed below) can also be used to detect the abnormal TMD clone. A study from the State of New York analyzed 585 Guthrie newborn screening cards from DS infants and found that 3.8% of DS newborns had detectable GATA1 mutations.43 In this cohort, the incidence of TMD was unknown, but two patients harboring the mutation at birth subsequently developed AMkL.
The defining feature of TMD is the spontaneous clinical regression in a high proportion of cases with supportive care alone without the use of chemotherapy.41 However, approximately 20% of the DS children with a previous history of TMD will later develop AMkL usually within the first 4 years of life, and it is often preceded by a myelodysplastic phase.41 The mechanism(s) accounting for the spontaneous regression of TMD, unique among leukemias, as well as factors that account for the progression of TMD cases to AMkL are unknown. Hence, this subgroup of DS patients has one of the highest predicted risks to develop leukemia.
TMD patients can present with various degrees of leukocytosis, thrombocytopenia, anemia, hepatomegaly, splenomegaly, and liver and heart dysfunction. An analysis of 48 DS infants with TMD from the POG revealed important clinical features of the disorder including: i) median WBC: 28,800 per cubic millimeter (range, 4600 to 218,000); peripheral blasts: 25% (range, 0 to 87%), and ALT: 45 U/L (8 to 1319); ii) spontaneously cleared peripheral blasts occurred in 89%, blood counts normalized in 74%, while 64% of patients maintained a complete remission; and, iii) early death occurred in 17% of patients, which were significantly correlated with higher WBC count, elevated bilirubin and liver enzymes, and failure to normalize blood counts. Nine (19%) patients subsequently developed leukemia; eight with AML and one with ALL. The presence of karyotype abnormalities along with trisomy 21 was associated with an increased risk to develop leukemia.44
Additional studies have analyzed factors linked to prognosis in TMD cases. A German study of 146 babies with TMD revealed a correlation between high WBC, ascites, prematurity, bleeding complications, and failure to achieve spontaneous remission being associated with early death.45 The overall survival rate was 85% with 23% of patients subsequently developing AML. A Japanese study of 70 patients had similar clinical findings including 22.9% of patients experiencing an early death due primarily to hepatic and cardiopulmonary failure.46
Based on their clinical presentation and outcome, TMD patients were stratified in risk groups on the COG protocol A2971.47 Patients who presented with early evidence of acute life threatening disease and were classified as a high risk group had a 3-year EFS of 30%; TMD patients who had any evidence of hepatomegaly and hepatic dysfunction, but had no evidence of acute life threatening disease were classified as an intermediate risk group and had a 3-year EFS of 55.2%. The remaining patients were classified as being low risk and a 3-year EFS of 83%. Therapy with low dose cytarabine (ara-C) administered subcutaneously or intravenously every 12 hours for 5 to 7 days maybe beneficial for patients with either high or intermediate risk disease.
Case Study 2B
Three months after the diagnosis of TMD, the patient's blood counts normalized and she did not require any blood transfusions or treatment with chemotherapy agents. However, at the age of eighteen months, she presented with cold-like symptoms for one week, had a decreased appetite, was febrile for several days and she was found to have hepatosplenomegaly on physical examination.
Further laboratory investigation revealed a WBC count of 18,000/mm3, hemoglobin of 5.2 g/dl with normal mean corpuscular volume, and platelets of 22,000/mm3. Liver function tests were normal. The LDH was 865 U/L and uric acid was 5.8 mg/dl. Peripheral smear demonstrated presence of 45% of large blasts with very basophilic staining cytoplasm, cytoplasmic blebbing, and prominent nucleoli. Flow cytometry of the peripheral smear revealed abnormal CD-45 dim population of blasts, which expressed CD-11B, CD-33, CD-34, CD-117, weakly expressed CD-4, CD-7, and showed partial expression of CD-56, CD-40, CD-13, and platelet-associated antigen CD-41, CD-61, CD-36, CD-9, and glycophorin A. Diagnosis of AMkL was made. She received chemotherapy according to the COG clinical trial AAML0431 “Treatment of Down Syndrome Children with AML and MDS Under the Age of 4 Years” and achieved a bone marrow remission after one course of Induction therapy consisting of ara-C, daunorubicin, and 6-thioguanine. She received three additional courses of Induction therapy and two courses of Intensification therapy and remained in remission at the end of scheduled therapy. Her therapy was complicated by myelosuppression requiring transfusions and delays between chemotherapy courses. She was hospitalized for several episodes of fever and neutropenia though did not have any episodes of documented bacterial nor fungal sepsis. She currently is in remission and has completed her therapy.
DS and AML
Overall, approximately 15% of pediatric AML cases occur in DS children. AMkL is the most common French-American-British subtype of DS AML patients, as reported by multiple pediatric cooperative groups with several reporting that the AMkL phenotype represents more than 90% of DS AML cases and the majority of cases are diagnosed before the age of 4 years (Table 1).48,49,50,51,52,53,54,55 Zipursky et al56 have estimated that DS children have a 500-fold increased risk of developing AMkL compared with non-DS children, highlighting the unique relationship between trisomy 21, leukemogenesis, and a specific leukemia phenotype. Other AML French-American-British subtypes have also been described in DS AML including M0, M1/M2, and M6.48,49,50,51,52,53,54,55
The cytogenetics of DS AML cases differs significantly from non-DS patients with AML. DS children with AML more frequently have trisomies 8, 11, and 21, dup(1p), del(6q), del(7p), dup(7q), and del(16q).27 Translocations commonly seen in non-DS AML [eg, t(8;21); t(15;17); inv(16), 11q23 rearrangements] are rare in patients with DS.27 For DS children older than 4 years of age who develop AML, the cytogenetic features, molecular biology findings and response to therapy significantly diverge from younger patients, and are similar to the ones found in non-DS patients with AML.51
Before the diagnosis of AML, DS patients may develop signs of myelodysplasia, characterized by progressive anemia and thrombocytopenia, dysplastic erythroid cells and megakaryocytes in the bone marrow that may be difficult to aspirate due to the presence of myelofibrosis. This myelodysplastic phase almost always precede the development of AML and those disorders are often times refer as “myeloid leukemia of DS.”56 Of 161 DS patients enrolled in the CCG study 2891, 34% of patients had less than 30% bone marrow blasts, meeting the French-American-British criteria for myelodysplastic syndrome.51,57
It is now recognized that DS children with AML, and in particular children with the AMkL phenotype, have exceptionally high cure rates, which typically have been >80%.48,49,50,51,52,53,54,55 These studies have demonstrated a high complete remission rate, low induction failure rate, and a relatively low relapse rate.48,49,50,51,52,53,54,55 DS patients diagnosed with myelodysplastic syndrome have an excellent response to therapy, as well, and patients are not considered candidates for stem cell transplants. The superior outcome of DS patients with AML is in part explained by the increased sensitivity to ara-C and daunorubicin (see below). Interestingly, in a German study, DS patients with a prior history of TMD had superior outcomes (91%) compared with patients with no history of TMD (70%).45 In contrast to DS AMkL patients, the treatment outcome of non-DS patients with AMkL is relatively poor.58
Treatment-associated mortality has varied among studies, indicating that a balance must be struck between curative leukemia therapy for DS AML patients and potential toxicity.48,49,50,51,52,53,54,55 The CCG-2891 study found similar toxicity between DS and non-DS patients when a decreased dose-intensity regimen was administered.51 Importantly, the remission rate and EFS were not affected by the dose-intensity reduction among DS patients younger than 4 years. The EFS for DS patients older than 4 years was similar to the non-DS group. The current COG DS AML protocol AAML0431 (DS patients younger <4 years of age) consists of receiving three cycles of ara-C continuous infusion (200 mg/m2/24 hours for 4 days) combined with daunorubicin and 6-thioguanine, one ara-C dose intensified cycle during induction (3 g/m2/dose every 12 hours for a total of 4 days; Capizzi II arm) and two cycles of intensification therapy consisting of ara-C and etoposide.
In view of the high incidence of congenital cardiac defects in DS children, potential concerns exist, particularly with the use of anthracyclines. A recent report from the Children's Oncology Group (COG) Study POG 9421 found an increased frequency of cardiac-related late effects, which used a total cumulative anthracycline (daunorubicin and mitoxantrone) dose of 375 mg/m2, with 24% of DS patients developing cardiomyopathies.59 The current COG protocol AAML0431 uses a reduced cumulative dose of daunorubicin by 25% compared with the COG A2971 protocol.
GATA1 Mutations in DS AML
A remarkable observation has been the detection of acquired somatic mutations involving the transcription factor gene GATA1 in nearly all cases of DS TMD and AMkL.60,61,62 The GATA1 protein is a zinc finger transcription factor essential for normal erythroid and megakaryocytic differentiation. It acts as an activator or repressor of different target genes by forming distinct activating or repressive complexes with partner proteins, such as “Friend of GATA1,” the essential hematopoietic factor Gfi-1b, and the repressive MeCPI complex.63,64 Mice with selective loss of megakaryocytic GATA1 expression have substantial reduction of platelet numbers, and deregulated megakaryocyte proliferation.65 The GATA1 gene is localized to chromosome Xp11.23 and mutations involving this gene have been found in human X-linked disorders that cause variable degrees of anemia and thrombocytopenia.66,67,68 However, those hereditary conditions have not been associated with the development of leukemias.66,67,68
In DS patients, the somatic GATA1 mutations include deletions, missense, nonsense, and splice site mutations at the exon 2/intron boundary with the net effect of introducing early stop codons and the synthesis of a shorter GATA1 protein (designated GATA1s, 40-kDa), translated from a downstream initiation site and distinguishable from the full-length 50-kDa GATA1 protein60 (Figure 1). The GATA1s protein has altered transactivation activity, potentially contributing to the uncontrolled proliferation of megakaryocytes. G1SKI mice that express GATA1s have a transient, but significant, hyperproliferation of yolk sac and fetal liver progenitors.69 Similar mutations have not been identified in non-DS AML patients and non-AMkL DS leukemia cases.60,61,62 These findings provide a compelling argument for the screening of GATA1 mutations in TMD and AML cases in DS. In the case of DS AML, GATA1 mutations may predict which cases will be responsive to therapy, while in TMD cases, they would confirm the diagnosis. TMD patients with GATA1 mutations will need close monitoring since they have a 20% to 30% risk of developing AMkL. Monitoring the clinical regression of TMD blasts by minimal residual disease detection by either flow-cytometry based assays or detection of GATA1 mutations by PCR, may be used in the future.70 Minimal residual disease monitoring of TMD cases, may also identify cases which ultimately progress to AML.
Figure 1.
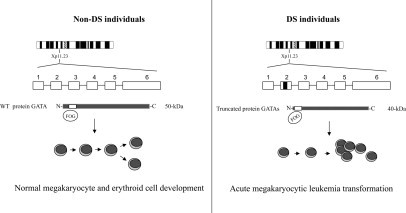
Schematics illustrating the role of GATA1 mutations in leukemogenesis in DS. The transcription factor gene GATA1 is localized to chromosome Xp11.23. GATA1 encodes a protein (50 kDa) that contains two C4-type zinc fingers, which act in DNA binding and protein-protein interactions, and an N-terminal transcription activation domain. The C-terminal zinc finger binds DNA at the consensus sequence (A/T) GATA (A/G). The N-terminal zinc finger can bind the consensus site GATC and also interacts with the partner protein “friend of GATA1.” This process is essential for normal erythroid and megakaryocytic differentiation. Mutations occurring in exon 2 of the transcription factor gene GATA1 result in the synthesis of a shorter GATA1 protein (40 kDa). The short form of the GATA1 protein (GATA1s) shows similar DNA-binding abilities and interactions with “friend of GATA1”; however, GATA1s exhibits altered transactivation capacity due to the loss of the N-terminal activation domain. The GATA1 mutation is likely an essential factor linked to leukemogenesis in DS; however, it is still not clear if this role is due to the loss of function of the wild-type GATA1 protein or if GATA1s has a unique function in the leukemia progression.
A prenatal origin for GATA1 mutations has been supported by several studies: i) A pair of identical twin DS infants with TMD had the identical GATA1 exon 2 mutations, indicating that the GATA1 mutation arose in utero with passage of blast cells from one fetus to the other71; ii) GATA1 mutations have been detected retrospectively in Guthrie newborn screening cards from DS infants who later developed AMkL72; and, iii) GATA1 mutations have also been detected in DS fetal liver samples in the absence of pathological evidence of leukemia, providing additional evidence that the acquisition of GATA1 mutations can occur prenatally.73 The prenatal origin of GATA1 mutations and likely prenatal origin of DS AMkL cases is similar to the prenatal origin of leukemia cases in non-DS children.74 Tunstall-Pedoe et al recently demonstrated the presence of an increased number of megakaryocyte-erythroid progenitors and reduced number of granulocyte-monocyte progenitors in DS fetal livers that did not harbor GATA1 mutations in comparison with gestation-matched normal controls.75 The authors hypothesized that the presence of constitutional trisomy 21 disturbs fetal hematopoiesis, promoting the acquisition of mutations. Similar findings were described by Chou et al.76 The uniform detection of GATA1 mutations in DS TMD and AMkL cases, as well as the presence of JAK2 mutations in DS ALL cases, suggest that the presence of one extra copy of chromosome 21 may be associated with an increased mutation rate, which is also linked to the higher predisposition to develop leukemia.
Why Do DS Children with AML Respond Better to Therapy?
In vitro studies have demonstrated that DS leukemia blasts are more sensitive to a number of different chemotherapy agents than non-DS leukemia blasts, suggesting increased susceptibility to undergo apoptosis.77,78 AMkL blasts are significantly more sensitive to ara-C and daunorubicin (the two most active agents used in AML therapy), as compared with blast cells from non-DS children, suggesting that the high EFS rates of DS AML patients are related to increased chemotherapy drug sensitivities.78,79,80,81,82 DS AMkL blasts also generated significantly higher levels of the active intracellular ara-C metabolite, ara-CTP, than non-DS AML blasts following in vitro incubations with 3H-ara-C, indicating that the metabolism of ara-C in DS AMkL cells is altered.81
The differences in sensitivity of chemotherapy in DS ALL blast cells are not as pronounced as in DS AMkL blast cells. Indeed, several studies have demonstrated that DS lymphoblasts do not have increased in vitro chemotherapy sensitivity, as compared with blasts from non-DS children,79,80 suggesting that the enhanced chemotherapy sensitivity of DS AMkL blasts is linked to the DS AML (ie, AMkL) phenotype.
It has been hypothesized that one or more chromosome 21-localized genes were linked to the increased chemotherapy sensitivities (eg, ara-C) of DS AMkL blasts.78 The trans-sulfuration pathway enzyme, cystathionine-β-synthase (CBS; localized to chromosome 21q22.3), catalyzes the condensation of serine and homocysteine to form cystathionine, an intermediate step in the synthesis of cysteine. Increased CBS activity is associated with significantly lower homocysteine, methionine, and S-adenosylmethionine levels in DS individuals, which may have downstream effects on reduced folate metabolism. Increased CBS activity may also indirectly impact metabolism of nucleosides (including ara-C) and anti-leukemic activity, based on the established synergism of sequential methotrexate and ara-C therapy.82 CBS transcript levels were significantly higher (median 12-fold) in DS AMkL blasts compared with non-DS AML blasts and CBS transcripts correlated with both in vitro ara-C sensitivities and generation of ara-CTP.78
The high frequency of somatic mutations in the GATA1 gene in DS AMkL cases suggests that GATA1 mutations may also be associated with the high EFS rates of DS AML patients. Interestingly, the biological features of AML cases in DS children greater than 4 years of age include a lower frequency of GATA1 mutations, which may account for the low EFS rates in this DS subgroup.83 It is conceivable that GATA1 may regulate the expression of several differentially expressed genes based on the localization of potential GATA1 binding sites in their promoters, leading to altered activity of chemotherapy drugs.84 One likely target is cytidine deaminase (CDA), which deaminates ara-C to the inactive metabolite, uridine arabinoside. It has been demonstrated that both the long and short GATA1 isoforms interact with the CDA promoter.85 CDA transcripts, measured by real-time PCR, were a median 5.1-fold lower in DS megakaryoblasts than in non-DS AML blast cells.84 The CDA transcript is transcribed from a CDA “long form” promoter, while a “short form” promoter acts as an enhancer for the CDA long form promoter. Thus, mutations of the GATA1 gene in DS TMD/AMkL patients that generate a transcriptionally altered GATA1 protein can be envisaged to result in reduced CDA enhancer activity and decreased overall CDA expression, and decreased net conversion of ara-C to uridine arabinoside. This has been demonstrated in a DS AMkL cell line model in which transfection of the full-length GATA1 cDNA resulted in significantly increased CDA expression and reduced ara-C sensitivity.84
Studies have also used microarray analysis to identify differentially expressed genes between DS and non-DS AMkL cases potentially linked to the high EFS rates of DS patients and/or low EFS rates of non-DS patients.86,87 In our recent microarray study, 551 differentially expressed genes (by at least twofold) between DS and non-DS AMkL cases were identified. Several of the genes overexpressed in the non-DS group (eg, Bcl-2 and Hsp-70) would be expected to control apoptosis and to make cells more resistant to chemotherapy, potentially accounting for the low cure rates of AMkL in non-DS children.
Conclusion
Cancer in DS patients offers unique models to improve our understanding of the genetic basis of oncogenesis and leukemogenesis, as well as mechanisms of chemotherapy sensitivity. In the case of DS ALL, further studies are required to optimize therapy and reduce treatment-related morbidity and mortality. Additionally, the occurrence of mutational activation of JAK2 in a proportion of DS patients may have potential therapeutic implications with JAK2 inhibitors. The uniform detection of GATA1 mutations in DS TMD and AML cases has provided important insights in both of these linked conditions. Several unanswered questions remain: i) Why is there a lower frequency of hyperdiploid ALL and TEL-AML1 ALL among DS children? ii) What is the basis for the increased infection-related toxicity for DS ALL patients? iii) Do all DS AMkL cases arise from a preceding case of TMD, which may include subclinical cases? iv) Do GATA1 mutations arise prenatally in all DS AMkL cases? v) Why does TMD resolve spontaneously, though in a proportion of patients, AMkL subsequently develops? vi) What is the association between hematopoiesis in the DS liver and the generation of GATA1 mutations? vii) What are the additional genetic events which must occur for TMD blasts to evolve to AMkL?, and viii) Can new targeted therapies be developed to improve the cure rate of AML cases based on the DS model? Studies designed to answer these questions, may improve our understanding of the biology of leukemia in children, overall.
Footnotes
Supported by grants (RO1 CA120772), from the National Cancer Institute, Leukemia Research Life, BPCT Golf Charity, The Elana Fund, Justin's Gift Charity, Children's Research Center of Michigan, St. Baldrick's Foundation, and The Ring Screw Textron Chair in Pediatric Cancer Research.
This article is partly based on material presented by the authors at the William Beaumont Hospital 17th Annual Symposium on Molecular Pathology: Clinical Applications of Genomic Medicine, which took place on September 10 to 12, 2008, in Troy, MI.
References
- 1.Improved national prevalence estimates for 18 selected major birth defects-United States, 1999–2001. MMWR Morb Mortal Wkly Rep. 2006;54:1301–1305. [PubMed] [Google Scholar]
- 2.Satge D, Schorderet DF, Balmer A, Beck-Popovic M, Addor MC, Beckmann JS, Munier FL. Association Down syndrome-retinoblastoma: a new observation. Ophthalmic Genet. 2005;26:151–152. doi: 10.1080/13816810500228894. [DOI] [PubMed] [Google Scholar]
- 3.Hsiung Stripp DC, Vaughn D, Van Arsdalen K, Whittington R. Three cases of advanced seminoma and Down's syndrome: a possible association. Am J Clin Oncol. 2003;26:197–199. doi: 10.1097/00000421-200304000-00020. [DOI] [PubMed] [Google Scholar]
- 4.Satge D, Le Tourneau A, Verger JP, Lefort S, Geneix A, Malet P, Diebold J, Vekemans M. A case report of Down syndrome and centroblastic lymphoma. Pathol Res Pract. 1996;192:1266–1269. doi: 10.1016/S0344-0338(96)80165-8. [DOI] [PubMed] [Google Scholar]
- 5.Hasle H, Clemmensen IH, Mikkelsen M. Risks of leukaemia and solid tumours in individuals with Down's syndrome. Lancet. 2000;355:165–169. doi: 10.1016/S0140-6736(99)05264-2. [DOI] [PubMed] [Google Scholar]
- 6.Satge D, Sasco AJ, Carlsen NL, Stiller CA, Rubie H, Hero B, de Bernardi B, de Kraker J, Coze C, Kogner P, Langmark F, Hakvoort-Cammel FG, Beck D, von der Weid N, Parkes S, Hartmann O, Lippens RJ, Kamps WA, Sommelet D. A lack of neuroblastoma in Down syndrome: a study from 11 European countries. Cancer Res. 1998;58:448–452. [PubMed] [Google Scholar]
- 7.Olson JM, Hamilton A, Breslow NE. Non-11p constitutional chromosome abnormalities in Wilms' tumor patients. Med Pediatr Oncol. 1995;24:305–309. doi: 10.1002/mpo.2950240507. [DOI] [PubMed] [Google Scholar]
- 8.Narod SA, Stiller C, Lenoir GM. An estimate of the heritable fraction of childhood cancer. Br J Cancer. 1991;63:993–999. doi: 10.1038/bjc.1991.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Satge D. A decreased incidence of neuroblastomas in Down syndrome and overproduction of S-100 b protein: an hypothesis. Med Hypothesis. 1996;46:393–399. doi: 10.1016/s0306-9877(96)90193-0. [DOI] [PubMed] [Google Scholar]
- 11.Zorick TS, Mustacchi Z, Bando SY, Zatz M, Moreira-Filho CA, Olsen B, Passos-Bueno MR. High serum endostatin levels in Down syndrome: implications for improved treatment and prevention of solid tumours. Eur J Hum Genet. 2001;9:811–814. doi: 10.1038/sj.ejhg.5200721. [DOI] [PubMed] [Google Scholar]
- 12.Minami T, Horiuchi K, Miura M, Abid MR, Takabe W, Noguchi N, Kohro T, Ge X, Aburatani H, Hamakubo T, Kodama T, Aird WC. Vascular endothelial growth factor- and thrombin-induced termination factor. Down syndrome critical region-1, attenuates endothelial cell proliferation and angiogenesis. J Biol Chem. 2004;279:50537–50554. doi: 10.1074/jbc.M406454200. [DOI] [PubMed] [Google Scholar]
- 13.Sussan TE, Yang A, Li F, Ostrowski MC, Reeves RH. Trisomy represses Apc(Min)-mediated tumours in mouse models of Down's syndrome. Nature. 2008;451:73–75. doi: 10.1038/nature06446. [DOI] [PubMed] [Google Scholar]
- 14.Taub JW. Relationship of chromosome 21 and acute leukemia in children with Down syndrome. J Pediatr Hematol Oncol. 2001;23:175–178. doi: 10.1097/00043426-200103000-00012. [DOI] [PubMed] [Google Scholar]
- 15.Canfield KN, Spector LG, Robison LL, Lazovich D, Roesler M, Olshan AF, Smith FO, Heerema NA, Barnard DR, Blair CK, Ross JA. Childhood and maternal infections and risk of acute leukaemia in children with Down syndrome: a report from the Children's Oncology Group. Br J Cancer. 2004;91:1866–1872. doi: 10.1038/sj.bjc.6602223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Linabery AM, Olshan AF, Gamis AS, Smith FO, Heerema NA, Blair CK, Ross JA. Exposure to medical test irradiation and acute leukemia among children with Down syndrome: a report from the Children's Oncology Group. Pediatrics. 2006;118:e1499–e1508. doi: 10.1542/peds.2006-0644. [DOI] [PubMed] [Google Scholar]
- 17.Linabery AM, Blair CK, Gamis AS, Olshan AF, Heerema NA, Ross JA. Congenital abnormalities and acute leukemia among children with Down syndrome: a Children's Oncology Group study. Cancer Epidemiol Biomarkers Prev. 2008;17:2572–2577. doi: 10.1158/1055-9965.EPI-08-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ross JA, Blair CK, Olshan AF, Robison LL, Smith FO, Heerema NA, Roesler M. Periconceptional vitamin use and leukemia risk in children with Down syndrome: a Children's Oncology Group study. Cancer. 2005;104:405–410. doi: 10.1002/cncr.21171. [DOI] [PubMed] [Google Scholar]
- 19.Puumala SE, Ross JA, Olshan AF, Robison LL, Smith FO, Spector LG. Reproductive history, infertility treatment, and the risk of acute leukemia in children with Down syndrome: a report from the Children's Oncology Group. Cancer. 2007;110:2067–2074. doi: 10.1002/cncr.23025. [DOI] [PubMed] [Google Scholar]
- 20.Lafiura KM, Bielawski DM, Posecion NC, Jr, Ostrea EM, Jr, Matherly LH, Taub JW, Ge Y. Association between prenatal pesticide exposures and the generation of leukemia-associated T(8;21) Pediatr Blood Cancer. 2007;49:624–628. doi: 10.1002/pbc.21283. [DOI] [PubMed] [Google Scholar]
- 21.Zeller B, Gustafsson G, Forestier E, Abrahamsson J, Clausen N, Heldrup J, Hovi L, Jonmundsson G, Lie SO, Glomstein A, Hasle H. Acute leukaemia in children with Down syndrome: a population-based Nordic study. Br J Haematol. 2005;128:797–804. doi: 10.1111/j.1365-2141.2005.05398.x. [DOI] [PubMed] [Google Scholar]
- 22.Pui CH, Robison LL, Look AT. Acute lymphoblastic leukemia. Lancet. 2008;371:1030–1043. doi: 10.1016/S0140-6736(08)60457-2. [DOI] [PubMed] [Google Scholar]
- 23.Steiner M, Attarbaschi A, Konig M, Nebral K, Gadner H, Haas OA, Mann G. Equal frequency of TEL/AML1 rearrangements in children with acute lymphoblastic leukemia with and without Down syndrome. Pediatr Hematol Oncol. 2005;22:229–234. doi: 10.1080/08880010590921603. [DOI] [PubMed] [Google Scholar]
- 24.Shah N, Al-Ahmari A, Al-Yamani A, Dupuis L, Stephens D, Hitzler J. Outcome and toxicity of chemotherapy for acute lymphoblastic leukemia in children with down syndrome. Pediatr Blood Cancer. 2008;52:14–19. doi: 10.1002/pbc.21737. [DOI] [PubMed] [Google Scholar]
- 25.Arico M, Ziino O, Valsecchi MG, Cazzaniga G, Baronci C, Messina C, Pession A, Santoro N, Basso G, Conter V. Acute lymphoblastic leukemia and Down syndrome: presenting features and treatment outcome in the experience of the Italian Association of Pediatric Hematology and Oncology (AIEOP) Cancer. 2008;113:515–521. doi: 10.1002/cncr.23587. [DOI] [PubMed] [Google Scholar]
- 26.James R, Lightfoot T, Simpson J, Moorman AV, Roman E, Kinsey S. Acute leukemia in children with Down's syndrome: the importance of population based study. Haematologica. 2008;93:1262–1263. doi: 10.3324/haematol.12831. [DOI] [PubMed] [Google Scholar]
- 27.Forestier E, Izraeli S, Beverloo B, Haas O, Pession A, Michalova K, Stark B, Harrison CJ, Teigler-Schlegel A, Johansson B. Cytogenetic features of acute lymphoblastic and myeloid leukemias in pediatric patients with Down syndrome: an iBFM-SG study. Blood. 2008;111:1575–1583. doi: 10.1182/blood-2007-09-114231. [DOI] [PubMed] [Google Scholar]
- 28.Bassal M, La MK, Whitlock JA, Sather HN, Heerema NA, Gaynon PS, Stork LC. Lymphoblast biology and outcome among children with Down syndrome and ALL treated on CCG-1952. Pediatr Blood Cancer. 2005;44:21–28. doi: 10.1002/pbc.20193. [DOI] [PubMed] [Google Scholar]
- 29.Lundin C, Heldrup J, Ahlgren T, Olofsson T, Johansson B. B-cell precursor t(8;14)(q11;q32)-positive acute lymphoblastic leukemia in children is strongly associated with Down syndrome or with a concomitant Philadelphia chromosome. Eur J Haematol. 2009;82:46–53. doi: 10.1111/j.1600-0609.2008.01166.x. [DOI] [PubMed] [Google Scholar]
- 30.Whitlock JA, Sather HN, Gaynon P, Robison LL, Wells RJ, Trigg M, Heerema NA, Bhatia S. Clinical characteristics and outcome of children with Down syndrome and acute lymphoblastic leukemia: a Children's Cancer Group study. Blood. 2005;106:4043–4049. doi: 10.1182/blood-2003-10-3446. [DOI] [PubMed] [Google Scholar]
- 31.Bercovich D, Ganmore I, Scott LM, Wainreb G, Birger Y, Elimelech A, Shochat C, Cazzaniga G, Biondi A, Basso G, Cario G, Schrappe M, Stanulla M, Strehl S, Haas OA, Mann G, Binder V, Borkhardt A, Kempski H, Trka J, Bielorei B, Avigad S, Stark B, Smith O, Dastugue N, Bourquin JP, Tal NB, Green AR, Izraeli S. Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down's syndrome. Lancet. 2008;372:1484–1492. doi: 10.1016/S0140-6736(08)61341-0. [DOI] [PubMed] [Google Scholar]
- 32.Kearney L, Gonzalez De Castro D, Yeung J, Procter J, Horsley SW, Eguchi-Ishimae M, Bateman CM, Anderson K, Chaplin T, Young BD, Harrison CJ, Kempski H, Wai E, So C, Ford AM, Greaves M. Specific JAK2 mutation (JAK2R683) and multiple gene deletions in Down syndrome acute lymphoblastic leukemia. Blood. 2009;113:646–648. doi: 10.1182/blood-2008-08-170928. [DOI] [PubMed] [Google Scholar]
- 33.Kilpivaara O, Levine RL. JAK2 and MPL mutations in myeloproliferative neoplasms: discovery and science. Leukemia. 2008;22:1813–1817. doi: 10.1038/leu.2008.229. [DOI] [PubMed] [Google Scholar]
- 34.Kratz CP, Böll S, Kontny U, Schrappe M, Niemeyer CM, Stanulla M. Mutational screen reveals a novel JAK2 mutation. L611S, in a child with acute lymphoblastic leukemia. Leukemia. 2006;20:381–383. doi: 10.1038/sj.leu.2404060. [DOI] [PubMed] [Google Scholar]
- 35.Hastings C, Whitlock JA, La M, Seibel N. Improved outcome of children with Down syndrome (DS) and high risk acute lymphocytic leukemia (HR-ALL): A report of CCG-1961. Blood (Abstracts) 2007:586. [Google Scholar]
- 36.Ugazio AG, Maccario R, Notarangelo LD, Burgio GR. Immunology of Down syndrome: a review. Am J Med Genet Suppl. 1990;7:204–212. doi: 10.1002/ajmg.1320370742. [DOI] [PubMed] [Google Scholar]
- 37.Garrison MM, Jeffries H, Christakis DA. Risk of death for children with Down syndrome and sepsis. J Pediatr. 2005;147:748–752. doi: 10.1016/j.jpeds.2005.06.032. [DOI] [PubMed] [Google Scholar]
- 38.Peeters M, Poon A. Down syndrome and leukemia: unusual clinical aspects and unexpected methotrexate sensitivity. Eur J Pediatr. 1987;146:416–422. doi: 10.1007/BF00444952. [DOI] [PubMed] [Google Scholar]
- 39.Blatt J, Albo V, Prin W, Orlando S, Wollman M. Excessive chemotherapy-related myelotoxicity in children with Down syndrome and acute lymphoblastic leukaemia. Lancet. 1986;2:914. doi: 10.1016/s0140-6736(86)90429-0. [DOI] [PubMed] [Google Scholar]
- 40.Matherly LH. Molecular and cellular biology of the human reduced folate carrier. Prog Nucleic Acid Res Mol Biol. 2001;67:131–162. doi: 10.1016/s0079-6603(01)67027-2. [DOI] [PubMed] [Google Scholar]
- 41.Zipursky A. Transient leukaemia–a benign form of leukaemia in newborn infants with trisomy 21. Br J Haematol. 2003;120:930–938. doi: 10.1046/j.1365-2141.2003.04229.x. [DOI] [PubMed] [Google Scholar]
- 42.Cushing T, Clericuzio CL, Wilson CS, Taub JW, Ge Y, Reichard KK, Winter SS. Risk for leukemia in infants without Down syndrome who have transient myeloproliferative disorder. J Pediatr. 2006;148:687–689. doi: 10.1016/j.jpeds.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 43.Pine SR, Guo Q, Yin C, Jayabose S, Druschel CM, Sandoval C. Incidence and clinical implications of GATA1 mutations in newborns with Down syndrome. Blood. 2007;110:2128–2131. doi: 10.1182/blood-2007-01-069542. [DOI] [PubMed] [Google Scholar]
- 44.Massey GV, Zipursky A, Chang MN, Doyle JJ, Nasim S, Taub JW, Ravindranath Y, Dahl G, Weinstein HJ. A prospective study of the natural history of transient leukemia (TL) in neonates with Down syndrome (DS): Children's Oncology Group (COG) study POG-9481. Blood. 2006;107:4606–4613. doi: 10.1182/blood-2005-06-2448. [DOI] [PubMed] [Google Scholar]
- 45.Klusmann JH, Creutzig U, Zimmermann M, Dworzak M, Jorch N, Langebrake C, Pekrun A, Macakova-Reinhardt K, Reinhardt D. Treatment and prognostic impact of transient leukemia in neonates with Down syndrome. Blood. 2008;111:2991–2998. doi: 10.1182/blood-2007-10-118810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Muramatsu H, Kato K, Watanabe N, Matsumoto K, Nakamura T, Horikoshi Y, Mimaya J, Suzuki C, Hayakawa M, Kojima S. Risk factors for early death in neonates with Down syndrome and transient leukaemia. Br J Haematol. 2008;142:610–615. doi: 10.1111/j.1365-2141.2008.07231.x. [DOI] [PubMed] [Google Scholar]
- 47.Sharma M, Alonzo TA, Sorrell A, Gerbing RB, Hilden JM, Arceci RJ, Massey G, Doyle JJ, Bayer L, Perentesis JP, Ross J, Smith FO, Brynes R, Hussong J, Hathaway L, Gamis A. Uniform approach better defines natural history of transient myeloproliferative disorder (TMD) in Down syndrome (DS) neonates: outcomes from Children' Oncology Group (COG) study A2971. Blood (ASH Annual Meeting Abstracts) 2006;108 Abstract 376. [Google Scholar]
- 48.Ravindranath Y, Abella E, Krischer JP, Wiley J, Inoue S, Harris M, Chauvenet A, Alvarado CS, Dubowy R, Ritchey AK, Land V, Steuber CP, Weinstein H. Acute myeloid leukemia (AML) in Down's syndrome is highly responsive to chemotherapy: experience on Pediatric Oncology Group AML Study 8498. Blood. 1992;80:2210–2214. [PubMed] [Google Scholar]
- 49.Ravindranath Y, Yeager AM, Chang MN, Steuber CP, Krischer J, Graham-Pole J, Carroll A, Inoue S, Camitta B, Weinstein HJ. Autologous bone marrow transplantation versus intensive consolidation chemotherapy for acute myeloid leukemia in childhood. Pediatric Oncology Group. N Engl J Med. 1996;334:1428–1434. doi: 10.1056/NEJM199605303342203. [DOI] [PubMed] [Google Scholar]
- 50.Lange BJ, Kobrinsky N, Barnard DR, Arthur DC, Buckley JD, Howells WB, Gold S, Sanders J, Neudorf S, Smith FO, Woods WG. Distinctive demography, biology, and outcome of acute myeloid leukemia and myelodysplastic syndrome in children with Down syndrome: children's Cancer Group Studies 2861 and 2891. Blood. 1998;91:608–615. [PubMed] [Google Scholar]
- 51.Gamis AS, Woods WG, Alonzo TA, Buxton A, Lange B, Barnard DR, Gold S, Smith FO. Increased age at diagnosis has a significantly negative effect on outcome in children with Down syndrome and acute myeloid leukemia: a report from the Children's Cancer Group Study 2891. J Clin Oncol. 2003;21:3415–3422. doi: 10.1200/JCO.2003.08.060. [DOI] [PubMed] [Google Scholar]
- 52.Creutzig U, Reinhardt D, Diekamp S, Dworzak M, Stary J, Zimmermann M. AML patients with Down syndrome have a high cure rate with AML-BFM therapy with reduced dose intensity. Leukemia. 2005;19:1355–1360. doi: 10.1038/sj.leu.2403814. [DOI] [PubMed] [Google Scholar]
- 53.Kudo K, Kojima S, Tabuchi K, Yabe H, Tawa A, Imaizumi M, Hanada R, Hamamoto K, Kobayashi R, Morimoto A, Nakayama H, Tsuchida M, Horibe K, Kigasawa H, Tsukimoto I. Prospective study of a pirarubicin, intermediate-dose cytarabine, and etoposide regimen in children with Down syndrome and acute myeloid leukemia: the Japanese Childhood AML Cooperative Study Group. J Clin Oncol. 2007;25:5442–7544. doi: 10.1200/JCO.2007.12.3687. [DOI] [PubMed] [Google Scholar]
- 54.Al-Ahmari A, Shah N, Sung L, Zipursky A, Hitzler J. Long-term results of an ultra low-dose cytarabine-based regimen for the treatment of acute megakaryoblastic leukaemia in children with Down syndrome. Br J Haematol. 2006;133:646–648. doi: 10.1111/j.1365-2141.2006.06097.x. [DOI] [PubMed] [Google Scholar]
- 55.Rao A, Hills RK, Stiller C, Gibson BE, de Graaf SS, Hann IM, O'Marcaigh A, Wheatley K, Webb DK. Treatment for myeloid leukaemia of Down syndrome: population-based experience in the UK and results from the Medical Research Council AML 10 and AML 12 trials. Br J Haematol. 2006;132:576–583. doi: 10.1111/j.1365-2141.2005.05906.x. [DOI] [PubMed] [Google Scholar]
- 56.Zipursky A, Thorner P, De Harven E, Christensen H, Doyle J. Myelodysplasia and acute megakaryoblastic leukemia in Down's syndrome. Leuk Res. 1994;18:163–171. doi: 10.1016/0145-2126(94)90111-2. [DOI] [PubMed] [Google Scholar]
- 57.Hasle H, Niemeyer CM, Chessells JM, Baumann I, Bennett JM, Kerndrup G, Head DR. A pediatric approach to the WHO classification of myelodysplastic and myeloproliferative diseases. Leukemia. 2003;17:277–282. doi: 10.1038/sj.leu.2402765. [DOI] [PubMed] [Google Scholar]
- 58.Athale UH, Razzouk BI, Raimondi SC, Tong X, Behm FG, Head DR, Srivastava DK, Rubnitz JE, Bowman L, Pui CH, Ribeiro RC. Biology and outcome of childhood acute megakaryoblastic leukemia: a single institution's experience. Blood. 2001;97:3727–3732. doi: 10.1182/blood.v97.12.3727. [DOI] [PubMed] [Google Scholar]
- 59.O'Brien MM, Taub JW, Chang MN, Massey GV, Stine KC, Raimondi SC, Becton D, Ravindranath Y, Dahl GV. Cardiomyopathy in children with Down syndrome treated for acute myeloid leukemia: a report from the Children's Oncology Group Study POG 9421. J Clin Oncol. 2008;26:414–420. doi: 10.1200/JCO.2007.13.2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wechsler J, Greene M, McDevitt MA, Anastasi J, Karp JE, Le Beau MM, Crispino JD. Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat Genet. 2002;32:148–152. doi: 10.1038/ng955. [DOI] [PubMed] [Google Scholar]
- 61.Hitzler JK, Cheung J, Li Y, Scherer SW, Zipursky A. GATA1 mutations in transient leukemia and acute megakaryoblastic leukemia of Down syndrome. Blood. 2003;101(11):4301–4304. doi: 10.1182/blood-2003-01-0013. [DOI] [PubMed] [Google Scholar]
- 62.Rainis L, Bercovich D, Strehl S, Teigler-Schlegel A, Stark B, Trka J, Amariglio N, Biondi A, Muler I, Rechavi G, Kempski H, Haas OA, Izraeli S. Mutations in exon 2 of GATA1 are early events in megakaryocytic malignancies associated with trisomy 21. Blood. 2003;102:981–986. doi: 10.1182/blood-2002-11-3599. [DOI] [PubMed] [Google Scholar]
- 63.Crispino JD. GATA1 in normal and malignant hematopoiesis. Semin Cell Dev Biol. 2005;16:137–147. doi: 10.1016/j.semcdb.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 64.Tsang AP, Visvader JE, Turner CA, Fujiwara Y, Yu C, Weiss MJ, Crossley M, Orkin SH. FOG, a multitype zinc finger protein, acts as a cofactor for transcription factor. GATA-1 in erythroid and megakaryocytic differentiation. Cell. 1997;90:109–119. doi: 10.1016/s0092-8674(00)80318-9. [DOI] [PubMed] [Google Scholar]
- 65.Shivdasani RA, Fujiwara Y, McDevitt MA, Orkin SH. A lineage-selective knockout establishes the critical role of transcription factor GATA-1 in megakaryocyte growth and platelet development. EMBO J. 1997;16:3965–3973. doi: 10.1093/emboj/16.13.3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Freson K, Devriendt K, Matthijs G, Van Hoof A, De Vos R, Thys C, Minner K, Hoylaerts MF, Vermylen J, Van Geet C. Platelet characteristics in patients with X-linked macrothrombocytopenia because of a novel GATA1 mutation. Blood. 2001;98:85–92. doi: 10.1182/blood.v98.1.85. [DOI] [PubMed] [Google Scholar]
- 67.Freson K, Matthijs G, Thys C, Marien P, Hoylaerts MF, Vermylen J, Van Geet C. Different substitutions at residue D218 of the X-linked transcription factor GATA1 lead to altered clinical severity of macrothrombocytopenia and anemia and are associated with variable skewed X inactivation. Hum Mol Genet. 2002;11:147–152. doi: 10.1093/hmg/11.2.147. [DOI] [PubMed] [Google Scholar]
- 68.Mehaffey MG, Newton AL, Gandhi MJ, Crossley M, Drachman JG. X-linked thrombocytopenia caused by a novel mutation of GATA-1. Blood. 2001;98:2681–2688. doi: 10.1182/blood.v98.9.2681. [DOI] [PubMed] [Google Scholar]
- 69.Li Z, Godinho FJ, Klusmann JH, Garriga-Canut M, Yu C, Orkin SH. Developmental stage-selective effect of somatically mutated leukemogenic transcription factor GATA1. Nat Genet. 2005;37:613–619. doi: 10.1038/ng1566. [DOI] [PubMed] [Google Scholar]
- 70.Pine SR, Guo Q, Yin C, Jayabose S, Levendoglu-Tugal O, Ozkaynak MF, Sandoval C. GATA1 as a new target to detect minimal residual disease in both transient leukemia and megakaryoblastic leukemia of Down syndrome. Leuk Res. 2005;29:1353–1356. doi: 10.1016/j.leukres.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 71.Shimada A, Xu G, Toki T, Kimura H, Hayashi Y, Ito E. Fetal origin of the GATA1 mutation in identical twins with transient myeloproliferative disorder and acute megakaryoblastic leukemia accompanying Down syndrome. Blood. 2004;103:366. doi: 10.1182/blood-2003-09-3219. [DOI] [PubMed] [Google Scholar]
- 72.Ahmed M, Sternberg A, Hall G, Thomas A, Smith O, O'Marcaigh A, Wynn R, Stevens R, Addison M, King D, Stewart B, Gibson B, Roberts I, Vyas P. Natural history of GATA1 mutations in Down syndrome. Blood. 2004;103:2480–2489. doi: 10.1182/blood-2003-10-3383. [DOI] [PubMed] [Google Scholar]
- 73.Taub JW, Mundschau G, Ge Y, Poulik JM, Qureshi F, Jensen T, James SJ, Matherly LH, Wechsler J, Crispino JD. Prenatal origin of GATA1 mutations may be an initiating step in the development of megakaryocytic leukemia in Down syndrome. Blood. 2004;104:1588–1589. doi: 10.1182/blood-2004-04-1563. [DOI] [PubMed] [Google Scholar]
- 74.Taub JW, Ge Y. The prenatal origin of childhood acute lymphoblastic leukemia. Leuk Lymphoma. 2004;45:19–25. doi: 10.1080/1042819031000149403. [DOI] [PubMed] [Google Scholar]
- 75.Tunstall-Pedoe O, Roy A, Karadimitris A, de la Fuente J, Fisk NM, Bennett P, Norton A, Vyas P, Roberts I. Abnormalities in the myeloid progenitor compartment in Down syndrome fetal liver precede acquisition of GATA1 mutations. Blood. 2008;112:4507–4511. doi: 10.1182/blood-2008-04-152967. [DOI] [PubMed] [Google Scholar]
- 76.Chou ST, Opalinska JB, Yao Y, Fernandes MA, Kalota A, Brooks JS, Choi JK, Gewirtz AM, Danet-Desnoyers GA, Nemiroff RL, Weiss MJ. Trisomy 21 enhances human fetal erythro-megakaryocytic development. Blood. 2008;112:4503–4506. doi: 10.1182/blood-2008-05-157859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yamada S, Hongo T, Okada S, Watanabe C, Fujii Y, Hori H, Yazaki M, Hanada R, Horikoshi Y. Distinctive multidrug sensitivity and outcome of acute erythroblastic and megakaryoblastic leukemia in children with Down syndrome. Int J Hematol. 2001;74:428–436. doi: 10.1007/BF02982087. [DOI] [PubMed] [Google Scholar]
- 78.Taub JW, Huang X, Matherly LH, Stout ML, Buck SA, Massey GV, Becton DL, Chang MN, Weinstein HJ, Ravindranath Y. Expression of chromosome 21-localized genes in acute myeloid leukemia: differences between Down syndrome and non-Down syndrome blast cells and relationship to in vitro sensitivity to cytosine arabinoside and daunorubicin. Blood. 1999;94:1393–1400. [PubMed] [Google Scholar]
- 79.Frost BM, Gustafsson G, Larsson R, Nygren P, Lonnerholm G. Cellular cytotoxic drug sensitivity in children with acute leukemia and Down's syndrome: an explanation to differences in clinical outcome? Leukemia. 2000;14:943–944. doi: 10.1038/sj.leu.2401753. [DOI] [PubMed] [Google Scholar]
- 80.Zwaan CM, Kaspers GJ, Pieters R, Hahlen K, Janka-Schaub GE, van Zantwijk CH, Huismans DR, de Vries E, Rots MG, Peters GJ, Jansen G, Creutzig U, Veerman AJ. Different drug sensitivity profiles of acute myeloid and lymphoblastic leukemia and normal peripheral blood mononuclear cells in children with and without Down syndrome. Blood. 2002;99:245–251. doi: 10.1182/blood.v99.1.245. [DOI] [PubMed] [Google Scholar]
- 81.Taub JW, Matherly LH, Stout ML, Buck SA, Gurney JG, Ravindranath Y. Enhanced metabolism of 1-β-D-arabinofuranosylcytosine in Down syndrome cells: a contributing factor to the superior event free survival of Down syndrome children with acute myeloid leukemia. Blood. 1996;87:3395–3403. [PubMed] [Google Scholar]
- 82.Taub JW, Ge Y. Down syndrome, drug metabolism and chromosome 21. Pediatr Blood Cancer. 2005;44:33–39. doi: 10.1002/pbc.20092. [DOI] [PubMed] [Google Scholar]
- 83.Hasle H, Abrahamsson J, Arola M, Karow A, O'Marcaigh A, Reinhardt D, Webb DK, van Wering E, Zeller B, Zwaan CM, Vyas P. Myeloid leukemia in children 4 years or older with Down syndrome often lacks GATA1 mutation and cytogenetics and risk of relapse are more akin to sporadic AML. Leukemia. 2008;22:1428–1430. doi: 10.1038/sj.leu.2405060. [DOI] [PubMed] [Google Scholar]
- 84.Ge Y, Stout ML, Tatman DA, Jensen TL, Buck S, Thomas RL, Ravindranath Y, Matherly LH, Taub JW. GATA1, cytidine deaminase, and the high cure rate of Down syndrome children with acute megakaryocytic leukemia. J Natl Cancer Inst. 2005;97:226–231. doi: 10.1093/jnci/dji026. [DOI] [PubMed] [Google Scholar]
- 85.Ge Y, Jensen TL, Stout ML, Flatley RM, Grohar PJ, Ravindranath Y, Matherly LH, Taub JW. The role of cytidine deaminase and GATA1 mutations in the increased cytosine arabinoside sensitivity of Down syndrome myeloblasts and leukemia cell lines. Cancer Res. 2004;64:728–735. doi: 10.1158/0008-5472.can-03-2456. [DOI] [PubMed] [Google Scholar]
- 86.Ge Y, Dombkowski AA, LaFiura KM, Tatman D, Yedidi RS, Stout ML, Buck SA, Massey G, Becton DL, Weinstein HJ, Ravindranath Y, Matherly LH, Taub JW. Differential gene expression. GATA1 target genes, and the chemotherapy sensitivity of Down syndrome megakaryocytic leukemia. Blood. 2006;107:1570–1581. doi: 10.1182/blood-2005-06-2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bourquin JP, Subramanian A, Langebrake C, Reinhardt D, Bernard O, Ballerini P, Baruchel A, Cave H, Dastugue N, Hasle H, Kaspers GL, Lessard M, Michaux L, Vyas P, van Wering E, Zwaan CM, Golub TR, Orkin SH. Identification of distinct molecular phenotypes in acute megakaryoblastic leukemia by gene expression profiling. Proc Natl Acad Sci USA. 2006;103:3339–3344. doi: 10.1073/pnas.0511150103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Uncited reference
- 9.Satge D, Sasco AJ, Pujol H, Rethore MO. Breast cancer in women with trisomy 21. Bull Acad Natl Med. 2001;185:1239–1252. [PubMed] [Google Scholar]