

Abstract
We report an oxaziridine-mediated enantioselective aminohydroxylation of olefins catalyzed by a chiral copper(II) bis(oxazoline) complex. A variety of styrenic olefins undergo efficient aminohydroxylation with excellent regioselectivity and synthetically useful levels of enantioselectivty (up to 84% ee). The reaction can be conducted on multi-gram scale with as little as 2 mol% of the copper(II) catalyst. Hydrolysis of the resulting 1,3-oxazolines under acidic conditions produces N-sulfonyl amino alcohols that can be purified by recrystallization to afford very high levels of enantioselectivity.
1. Introduction
Vicinal aminoalcohols are substructures found frequently in a diverse range of natural products and other biologically active compounds. In addition, many of the most frequently utilized chiral reagents for asymmetric synthesis employ 1,2-aminoalcohols as key stereocontrolling structural elements. Due to the importance of this valuable structural motif, a great number of methods have been developed for the synthesis of enantiopure chiral 1,2-aminoalcohols.1 Among the most powerful of these methods is the osmium-catalyzed asymmetric aminohydroxylation reported by Sharpless in 1996,2 which enables the direct oxidative functionalization of a variety of structurally diverse alkenes. The utility of this reaction has become widely recognized,3 and numerous enantioselective total syntheses have been reported that feature this method.4
Nevertheless, several research groups have continued to investigate alternative methods for the addition of nitrogen- and oxygen-containing moieties across carbon-carbon double bonds.5 This interest has been motivated in part by the cost and toxicity associated with the use of osmium catalysts as well as by the relatively modest regioselectivities observed when styrenes and other non-polarized alkenes are subjected to aminohydroxylation using Sharpless’ methodology.3 A key advance has recently reported by Chemler and co-workers, who have developed a method for the asymmetric synthesis of pyrrolidines and indolines using an intramolecular oxyamination of olefins catalyzed by a chiral copper(II) bis(oxazoline) complex.6 However, to the best of our knowledge, enantioselective intermolecular oxyaminations under osmium-free conditions have not yet been reported.
Over the past several years, our group has been investigating the unique reactivity observed when N-sulfonyl oxaziridines (Davis’ oxaziridines)7 are used in oxidative transformations of organic substrates in the presence of various transition metal catalysts.8 A particular focus of our research has been the development of a novel strategy for copper(II)-catalyzed aminohydroxylation of olefins that utilize oxaziridines as the terminal oxidant.8a,c Given the broad range of styrenes, dienes, and electron-rich olefins that successfully undergo aminohydroxylation using our methodology, the excellent regioselectivity we observe in this process, and the ability to perform these reactions on reasonably large scales, we reasoned that an enantioselective variant of this new reaction using a chiral copper(II) catalyst could be a valuable supplement to the well-established Sharpless methodology. Herein, we report that synthetically useful levels of enantioselectivity can be achieved in oxaziridine-mediated aminohydroxylations of styrenes in the presence of a copper(II) bis(oxazoline) catalyst.
2. Results and Discussion
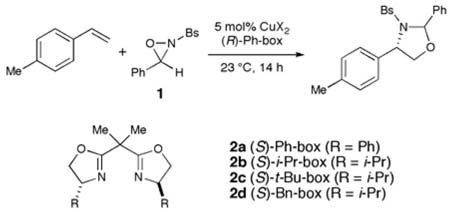
We began our investigations by studying the reaction between N-sulfonyloxaziridine 1 and 4-methylstyrene in the presence of various chiral copper salts (Table 1). A screen of chiral ligands commonly utilized in copper(II)-catalyzed asymmetric reactions (entries 1–4) indicated that the complex formed in situ from Cu(TFA)2 and (R,R)-isopropylidene bis(4-phenyloxazoline) (Ph-box, 2a) afforded modest levels of enantioselectivity for both diastereomers of the 1,3-oxazolidine product (entry 1). Importantly, although the relative diastereoselectivity was poor (2:1 cis:trans), the reaction proceeded with exclusive regioselectivity, and the major enantiomer of each diastereomer formed in this reaction possesses S stereochemistry at the stereogenic benzylic carbon. Hydrolysis of both aminals thus affords the same major enantiomer of the desired 1,2-aminoalcohol. In further optimizations of this reaction, therefore, we found it useful to consider the combined enantioselectivity for formation of the benzylic stereocenter as a weighted average of the enantiomer ratios for each diastereomer.
Table 1.
 | ||||||
|---|---|---|---|---|---|---|
| Entry | X | ligand | Solvent | Yieldc | d.r. syn:anti | %eed,e |
| 1 | TFA | 2a | CH2Cl2 | 64% | 2.0 : 1 | 71/68 (70) |
| 2 | TFA | 2b | CH2Cl2 | 43% | 0.5 : 1 | 30/21 (24) |
| 3 | TFA | 2c | CH2Cl2 | 33% | 0.8 : 1 | 31/13 (21) |
| 4 | TFA | 2d | CH2Cl2 | 54% | 0.3 : 1 | −11/20 (13) |
| 5f | TFA | 2a | CH2Cl2 | 66% | 2.1 : 1 | 31/32 (31) |
| 6g | TFA | 2a | CH2Cl2 | 70% | 2.0 : 1 | 53/47 (51) |
| 7h | TFA | 2a | CH2Cl2 | 62% | 2.3 : 1 | 75/64 (72) |
| 8 | OTf | 2a | CH2Cl2 | 67% | 1.0 : 1 | 53/53 (53) |
| 9 | Cl | 2a | CH2Cl2 | 54% | 2.0 : 1 | 79/18 (59) |
| 10 | F6acac | 2a | CH2Cl2 | 67% | 2.1 : 1 | 75/65 (72) |
| 11 | F6acac | 2a | Toluene | 46% | 1.7 : 1 | 76/59 (70) |
| 12 | F6acac | 2a | EtOAc | 62% | 1.7 : 1 | 84/47 (70) |
| 13 | F6acac | 2a | Acetone | 72% | 2.4 : 1 | 87/71 (82) |
Unless otherwise noted, reactions were performed using 5 mol% of the copper salt, 15 mol% of the ligand, and 2.5 equiv of oxaziridine. Data reported are the average of two reproducible experiments.
Bs = benzenesulfonyl.
Total isolated yield of both diastereomers.
ee values reported as: syn/anti (weighted average).
Product ratios determined by chiral SFC analysis.
6 mol% ligand.
10 mol% ligand.
20 mol% ligand.
The ratio of ligand to copper was an important variable in the optimization of this reaction. We found that a 3:1 ratio was optimal; lower ratios led to a dramatic diminution in enanantioselectivity (entries 5–6), while higher ratios afforded no significant benefit to the rate or selectivity of this reaction (entry 7). These results suggest that the aminohydroxylation is not strongly ligand-accelerated, and that competition between free and ligand-bound copper(II) can adversely impact the enantioselectivity of the reaction. Additional optimization of the copper source (entries 8–10) indicated that reactions performed with copper(II) bis(hexafluoroacetylacetonate) enjoyed a modest increase in yield compared to our initial lead experiments. Finally, variation of the solvent (entries 11–13) indicated that the use of acetone increases the enantioselectivity of this process to synthetically useful levels.
We also examined a variety of other modifications to these reaction conditions. Other ligands commonly utilized in asymmetric copper-catalyzed processes9 failed to give comparable levels of enantioselectivity. Similarly, steric and electronic perturbation of the oxaziridine also failed to improve the selectivity. In each of these extensive studies, we observed that structural changes to the ligand or oxaziridine often had opposing effects on the enantioselectivities of each of the two diastereomers produced in the reaction. Finally, we found that the efficiency of this process is dramatically reduced at lower temperatures without a significant corresponding increase in enantioselectivity. It seems likely, therefore, that additional experiments to control the diastereoselectivity and improve the overall reactivity of this system will be necessary to further improve the enantioselectivity of this process.
Although the enantioselectivities of aminohydroxylations performed using the (Ph-box)copper(II) catalyst are not as high as those observed under Sharpless conditions, they are nevertheless synthetically useful, and the regioselectivites observed are significantly higher than those typically observed in osmium-catalyzed oxyaminations of styrenes. Given these considerations and the attractiveness of an osmium-free asymmetric aminohydroxylation protocol, we next elected to investigate the scope of the oxaziridine-mediated asymmetric aminohydroxylation in greater detail.
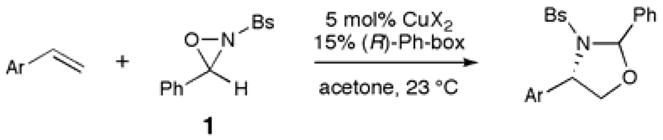
The results of experiments that probe the effect of substrate modification on the yield and selectivity of the aminohydroxylation under our optimized conditions are summarized in Table 2. Substitutions at the para (entry 2) and meta (entry 3 and 4) positions are tolerated; however, substitution at the ortho position significantly reduces both the efficiency and selectivity of the reaction (entry 5). Sterically bulky (entry 5 and 6) and electron-withdrawing groups (entries 7–10) can be incorporated at the para position, although in the latter case the enantioselectivity of the reaction is somewhat diminished. Strongly electron-donating groups at the para position, on the other hand, are not tolerated and result in a dramatic decrease in selectivity (entry 11). The sensitivity of the aminohydroxylation reaction to substituents at the meta position is less pronounced; substrates bearing both electron-donating (entry 12) and electron-withdrawing (entry 13) substituents at the meta position on the aryl ring are aminohydroxylated with good levels of enantioselectivity. Finally, we were delighted to observe that reactions can be performed on gram scale without any significant impact on the yield or selectivity of the process; the aminohydroxylation of 4-methylstyrene conducted on 20 mmol scale, using 2 mol% of Cu(F6acac)2 and 6 mol% of Ph-box, afforded the desired 1,3-oxazoline in essentially the same yield and enantiomeric excess as small-scale reactions with higher catalyst loading (entry 14).
Table 2.
Scope of the aminohydroxylationa
 | |||||
|---|---|---|---|---|---|
| Entry | Ar | Time | Yieldb | d.r. (syn:anti) | %eec,d |
| 1 | Ph | 24 h | 81% | 1.5 : 1 | 85/79 (82) |
| 2 | 4-MePh | 15 h | 72% | 2.2 : 1 | 86/74 (82) |
| 3 | 3-MePh | 18 h | 78% | 1.6 : 1 | 84/72 (79) |
| 4 | 3,5-Me2Ph | 16 h | 65% | 1.6 : 1 | 85/70 (79) |
| 5 | 2-MePh | 26 h | 44% | 0.2 : 1 | 12/47 (20) |
| 6 | 4-t-BuPh | 17 h | 67% | 2.3 : 1 | 87/78 (84) |
| 7 | 4-PhPh | 20 h | 71% | 1.9 : 1 | 87/62 (78) |
| 8 | 4-BrPh | 24 h | 61% | 1.0 : 1 | 83/47 (65) |
| 9 | 4-ClPh | 24 h | 70% | 0.8 : 1 | 89/45 (69) |
| 10 | 4-FPh | 44 h | 57% | 1.4 : 1 | 83/71 (78) |
| 11 | 4-MeOPh | 17 h | 70% | 0.4 : 1 | 0/24 (9) |
| 12 | 3-MeOPh | 18 h | 77% | 1.3 : 1 | 83/65 (75) |
| 13 | 3-ClPh | 29 h | 51% | 0.8 : 1 | 83/39 (60) |
| 14e | 4-MePh | 14 h | 67% | 2.5 : 1 | 86/76 (82) |
Unless otherwise noted, reactions were performed using 1 mmol olefin, 5 mol% Cu(F6acac)2, 15 mol% (S)-Ph-box, and 2.5 equiv oxaziridine. Data reported are the average of two reproducible experiments.
Total isolated yield of both diastereomers.
ee values reported as: syn/anti (weighted average).
Product ratios determined by chiral SFC analysis.
Reaction performed using 20 mmol olefin, 2 mol% Cu(F6acac)2, and 6 mol% (S)-
Ph-box.
The ability to conduct the enantioselective aminohydroxylation on gram scale is important for practical applications of our new methodology. The aminal protecting group can be easily removed from the mixture of diastereomers in high yield under standard acidic hydrolysis conditions. In all cases, the enantiomeric excess of the deprotected N-sulfonyl amino alcohol is in agreement with the calculated weighted average of the two diastereomers, which indicates that the benzylic stereocenter is not racemized under these acidic conditions and that the rate of hydrolysis for the two diastereomers are comparable. In addition, the N-sulfonyl amino alcohols are highly crystalline solids, and the enantiopurity of these compounds can be improved to high levels upon recrystallization.
Examples of this process are summarized in Scheme 1. The 1,3-oxazolidine products arising from reaction of 4-methylstyrene (3) and 3,5-dimethylstyrene (5) can be hydrolyzed on gram scale upon treatment with TFA in aqueous dioxane (86% and 88% yield of 4 and 6, respectively), and a single recrystallization from EtOAc/hexanes affords the N-sulfonyl amino alcohol products in very high enantiomeric purity (>99% ee and 98% ee, respectively). In a similar fashion, the 1,3-oxazoline product derived from 4-phenylstyrene (7) undergoes hydrolysis in 86% yield, and recrystalization affords 3.4 grams of the N-sulfonyl amino alcohol 8 in 86% ee.
Scheme 1.

Large-scale hydrolysis of the diastereomeric 1,3-oxazolidine products and recrystallization to high enantiopurity.
In summary, we have developed the first enantioselective copper(II)-catalyzed intermolecular aminohydroxylation reaction of alkenes. This discovery is significant for a number of reasons. First, this method provides a convenient route to enantioenriched aminoalcohols from styrenes. The ability to perform this aminohydroxylation under osmium-free conditions and the very high levels of regioselectivity obtained make this method an attractive complement to the well-established Sharpless asymmetric aminohydroxylation. We anticipate, therefore, that this method could find significant utility in the syntheses of enantioenriched vicinal amino alcohols. In addition, these results provide strong evidence that the chiral copper(II) catalyst is intimately involved in the stereochemistry-determining step of the aminohydroxylation. Future efforts in our labs will focus on the discovery of ligands for copper that will improve the reactivity of this system and induce higher levels of enantioselectivity.
3. Experimental section
3.1. General methods
Oxaziridine 1 was prepared as described by Davis.10 (R)-Phenyl bis(oxazoline) was synthesized from (R)-phenylglycinol using the procedure reported by García and Pires.11 Product ratios were determined by supercritical fluid chromatography (SFC) on a Berger Analytix instrument equipped with an Agilent 1100 variable wavelength detector, using chiral columns and methods as noted. 1H and 13C NMR spectra were obtained using Bruker AC-300 and Varian Unity-300 spectrometers and were referenced to TMS (0.00 ppm) or CDCl3 (77.23 ppm), respectively. Mass spectrometry was performed with a Micromass LCT (electrospray ionization, time-of-flight analyzer). Optical rotations were obtained using a Rudolph Autopol III polarimeter. Isolated yields refer to the combined yields of both diastereomers obtained upon chromatography; characterization data were obtained from analytically pure samples isolated by further chromatography or selective crystallization.
3.2. Asymmetric aminohydroxylations
General procedure for enantioselective aminohydroxylation of styrenes (Table 2)
A flame-dried 2 dram vial containing (R,R)-2,2′-isopropylidenebis(4-phenyl-2-oxazoline) (0.15 mmol), copper(II) hexafluoroacetylacetonate (0.05 mmol), and a stirbar was charged with acetone (2.0 mL). The reaction vial was then capped with a septum and flushed with nitrogen, and the solution was stirred for 30 min. N-Benzensulfonyl-3-phenyloxaziridine (1, 2.5 mmol) was then added, followed by the styrene substrate (1.0 mmol). The reaction was stirred under positive pressure of nitrogen for 14–44 h and then quenched by the addition of 200 μL dimethylsulfide to destroy remaining excess oxaziridine. The solvent was removed by rotary evaporation, and the residue was dissolved in 2 mL toluene and loaded directly onto a silica gel column. Elution with 5:1 hexanes:ethyl acetate with 3% triethylamine co-solvent afforded the 1,3-oxazolidine products.
N-Benzensulfonyl-2,4-diphenyl-1,3-oxazolane (Table 2, entry 1)
Prepared according to the general procedure using 104 mg styrene. Yield 1: 298 mg, 80%, 1.6:1 cis:trans, 85/79% ee cis/trans (82% ee combined); yield 2: 293 mg, 79%, 1.44:1 cis:trans, 83/76% ee cis/trans (80 % ee combined). Spectral data were in agreement with previously reported values.8a Cis diastereomer: 85% ee [Daicel Chiracel OJ-H column, 10% MeOH 6 min, then 17% MeOH 10 min, 2.5 ml/min, 210 nm; t1=5.37 min (major), t2=6.38 min], [α]22D = +57.4 (c 0.93, CH2Cl2). Trans diastereomer: >99% ee [t1=12.62 min (major), t2=16.01 min], [α]22D = +114.2 (c 0.55, CH2Cl2).
N-Benzensulfonyl-2-phenyl-4-(4-methylphenyl)-1,3-oxazolane (Table 2, entry 2)
Yield 1: 0.264 g, 70%, 2.2:1 cis:trans, 86/65% ee cis/trans (82% ee combined); yield 2: 0.281 g, 74%, 2.2:1 cis:trans, 86/65% ee cis/trans (82% ee combined). Cis diastereomer: 86% ee [Daicel Chiracel OJ-H column, 3% MeOH 18 min, then 20% MeOH 6 min, 2.5 ml/min, 210 nm; t1=16.81 min (major), t2=18.41 min], [α]22D = +76.5 (c 2.00, H2Cl2); 1H NMR (300 MHz, CDCl3): δ 7.72–7.84 (−SO2PhH, m, 2H), 7.55–7.67 (ArH, m, 3H), 7.44–7.54 (ArH, m, 2H), 7.29–7.43 (ArH, m, 3H), 7.02–7.18 (ArH, m, 4H), 6.34 (Haminal, s, 1H), 4.82 (CH-CH2, t, J = 7.0 Hz, 1H), 4.15 (CH2, dd, J = 9.0, 7.1 Hz, 1H), 3.87 (CH2, dd, J = 9.0, 6.9 Hz, 1H), 2.30 (ArCH3, s, 3H); 13C NMR (75.4 MHz, CDCl3): δ 138.2, 137.9, 137.6, 135.6, 133.4, 129.5, 129.3, 129.0, 128.6, 128.2, 127.4, 127.3, 92.7, 73.4, 63.3, 21.3; HRMS (EI+) calc’d for [C22H21NO3SH]+ requires m/z 380.1315, found m/z 380.1313. Isolated as a white solid (mp = 178–180 °C). Trans diastereomer: 99% ee [t1=25.44 min (major), t2=26.52 min], [α]22D = +135.9 (c 0.69, CH2Cl2); 1H NMR (300 MHz, CDCl3): δ 7.48–7.56 (SO2PhH, m, 2H), 7.31–7.43 (ArH, m, 4H), 7.08–7.20 (ArH, m, 6H), 6.95 (ArH, d, J = 7.9 Hz, 2H), 6.35 (Haminal, s, 1H), 5.06 (CH-CH2, dd, J = 6.4, 2.2 Hz, 1H), 4.38 (CH2, dd, J = 8.8, 6.4 Hz, 1H), 3.98 (CH2, dd, J = 8.8, 2.2 Hz, 1H), 2.31 (ArCH3, s, 3H); 13C NMR (75.4 MHz, CDCl3): δ 140.7, 138.5, 138.1, 135.8, 131.9, 129.3, 128.5, 128.4, 128.0, 127.6, 127.1, 92.4, 73.2, 62.8, 21.3; HRMS (EI+) calc’d for [C22H21NO3SH]+ requires m/z 380.1315, found m/z 380.1315. Isolated as a colorless oil.
N-Benzensulfonyl-2-phenyl-4-(3-methylphenyl)-1,3-oxazolane (Table 2, entry 3)
Prepared according to the general procedure using 119 mg 3-methylstyrene. Yield 1: 290 mg, 76%, 1.6:1 cis:trans, 84/72% ee cis/trans (79% ee combined); yield 2: 296 mg, 78%, 1.6:1 cis:trans, 82/71% ee cis/trans (78 % ee combined). Spectral data were in agreement with previously reported values.8a Cis diastereomer: 84% ee [Daicel Chiracel OJ-H column, 12% MeOH, 2.5 ml/min, 210 nm; t1=3.88 min (major), t2=4.26 min], [α]22D = +61.6 (c 1.34, CH2Cl2). Trans diastereomer: 73% ee [t1=9.59 min, t2=11.05 min (major)], [α]22D = +93.1 (c 1.66, CH2Cl2).
N-Benzensulfonyl-2-phenyl-4-(3,5-dimethylphenyl)-1,3-oxazolane (Table 2, entry 4)
Prepared according to the general procedure using 140 mg 3,5-dimethylstyrene. Yield 1: 0.276 g, 66%, 1.5:1 cis:trans, 83/70% ee cis/trans (78% ee combined); yield 2: 0.253 g, 69%, 1.6:1 cis:trans, 85/70% ee cis/trans (79% ee combined). Cis diastereomer: 80% ee [Daicel Chiracel OJ-H column, 3% MeOH 12 min then 20% MeOH 7 min, 2.5 ml/min, 210 nm; t1=8.76 min (major), t2=11.52 min], [α]22D = +67.8 (c 1.19, CH2Cl2); 1H NMR (300 MHz, CDCl3): δ 7.74–7.82 (−SO2PhH, m, 2H), 7.56–7.66 (ArH, m, 3H), 7.44–7.54 (ArH, m, 2H), 7.31–7.42 (ArH, m, 3H), 6.85 (ArH, broad s, 1H), 6.79 (ArH, broad s, 2H), 6.37 (Haminal, s, 1H), 4.81 (CH-CH2, t, J = 6.9 Hz, 1H), 4.14 (CH2, dd, J = 8.9, 7.2 Hz, 1H), 3.86 (CH2, dd, J = 8.9, 6.7 Hz, 1H), 2.20 (ArCH3, s, 6H); 13C NMR (75.4 MHz, CDCl3): δ 138.5, 138.2, 138.2, 137.8, 133.4, 129.7, 129.3, 129.0, 128.6, 128.3, 127.4, 125.2, 92.6, 73.5, 63.4, 21.4; HRMS (EI+) calc’d for [C23H23NO3SNa]+ requires m/z 416.1291, found m/z 416.1308. Isolated as a white solid (mp = 121–124 °C). Trans Diastereomer: 70% ee [t1=21.90 min, t2=23.21 min (major)], [α]22D = +93.3 (c 1.20, CH2Cl2); 1H NMR (300 MHz, CDCl3): δ 7.51–7.62 (SO2PhH, m, 2H), 7.30–7.47 (ArH, m, 4H), 7.09–7.24 (ArH, m, 4H), 6.82 (ArH, broad s, 1H), 6.78 (ArH, broad s, 2H), 6.37 (Haminal, s, 1H), 5.01 (CH-CH2, dd, J = 6.4, 2.0 Hz, 1H), 4.34 (CH2, dd, J = 8.8, 6.4 Hz, 1H), 3.94 (CH2, dd, J = 8.8, 2.0 Hz, 1H), 2.15 (ArCH3, s, 6H); 13C NMR (75.4 MHz, CDCl3): δ 140.6, 138.8, 138.3, 138.2, 132.0, 129.9, 129.3, 128.6, 128.3, 127.5, 127.1, 126.0, 92.4, 73.0, 63.0, 21.3; HRMS (EI+) calc’d for [C23H23NO3SH]+ requires m/z 394.1472, found m/z 394.1469. Isolated as a white solid (mp = 94–95 °C).
N-Benzensulfonyl-2-phenyl-4-(2-methylphenyl)-1,3-oxazolane (Table 2, entry 5)
Prepared according to the general procedure using 59 mg 2-methylstyrene. Yield 1: 81 mg, 43%, 6.3:1 cis:trans, 11/50% ee cis/trans ( 16% ee combined); yield 2: 84 mg, 44%, 6.3:1 cis:trans, 12/47% ee cis/trans (17% ee combined). Spectral data were in agreement with previously reported values.8a Cis diastereomer: 48 % ee [Daicel Chiracel OJ-H column, 5% MeOH 15 min, then 20% MeOH 10 min, 2.0 ml/min, 210 nm; t1=7.61 min (major), t2=8.74 min], [α]22D = +23.2 (c 0.50, CH2Cl2). Trans diastereomer: 8% ee [t1=20.57 min (major), t2=22.35 min], [α]22D = +8.4 (c 2.25, CH2Cl2).
N-Benzensulfonyl-2-phenyl-4-(4-tert-butylphenyl)-1,3-oxazolane (Table 2, entry 6)
Prepared according to the general procedure using 161 mg 4-tert-butylstyrene. Yield 1: 0.283 g, 67%, 2.1:1 cis:trans, 87/73% ee cis/trans (83% ee combined); yield 2: 0.291 g, 67%, 2.4:1 cis:trans, 87/79% ee cis/trans (84% ee combined). Cis diastereomer: 85% ee [Daicel Chiralpak AD column, 10% MeOH ramp (0.75ml/min) to 25% MeOH, 2.0 ml/min, 210 nm; t1=5.01 min, t2=6.48 min (major)], [α]22D = +41.8 (c 1.10, CH2Cl2); 1H NMR (300 MHz, CDCl3): δ 7.69–7.80 (−SO2PhH, m, 2H), 7.53–7.67 (ArH, m, 3H), 7.31–7.52 (ArH, m, 5H), 7.22–7.30 (ArH, m, 2H), 7.08–7.19 (ArH, m, 2H), 6.37 (Haminal, s, 1H), 4.83 (CH-CH2, t, J = 7.1 Hz, 1H), 4.17 (CH2, dd, J = 9.0, 7.1 Hz, 1H), 3.87 (CH2, dd, J = 9.0, 7.1 Hz, 1H), 1.28 (tBuH, s, 9H); 13C NMR (75.4 MHz, CDCl3): δ 151.1, 138.3, 137.7, 135.4, 133.3, 129.3, 129.0, 128.6, 128.2, 127.4, 127.1, 125.7, 92.6, 73.4, 63.3, 34.7, 31.5; HRMS (EI+) calc’d for [C25H27NO3SH]+ requires m/z 422.1785, found m/z 422.1786. Isolated as a colorless oil. Trans Diastereomer: 66% ee [t1=8.97 min (major), t2=10.93 min], [α]22D = +33.0 (c 1.06, CH2Cl2); 1H NMR (300 MHz, CDCl3): δ 7.49–7.62 (−SO2PhH, m, 2H), 7.29–7.47 (ArH, m, 4H), 7.05–7.22 (ArH, m, 8H), 6.36 (Haminal, s, 1H), 5.09 (CH-CH2, dd, J = 6.3, 2.1 Hz, 1H), 4.37 (CH2, dd, J = 8.8, 6.3 Hz, 1H), 3.99 (CH2, dd, J = 8.8, 2.1 Hz, 1H), 1.30 (tBuH, s, 9H); 13C NMR (75.4 MHz, CDCl3): δ 151.3, 140.7, 138.8, 135.4, 132.0, 129.3, 128.6, 128.4, 127.9, 127.5, 127.1, 125.5, 92.2, 73.0, 62.7, 34.7, 31.6; HRMS (EI+) calc’d for [C25H27NO3SH]+ requires m/z 422.1785, found m/z 422.1789. Isolated as a white solid (mp = 158–160 °C).
N-Benzensulfonyl-2-phenyl-4-(4-biphenyl)-1,3-oxazolane (Table 2, entry 7)
Prepared according to the general procedure using 180 mg 4-vinylbiphenyl. Yield 1: 0.326 g, 71%, 1.9:1 cis:trans, 86/62% ee cis/trans (78% ee combined); yield 2: 0.311 g, 71%, 1.9:1 cis:trans, 87/62% ee cis/trans (78% ee combined). Cis diastereomer: 96% ee [Daicel Chiracel OD-H column, 5% MeOH 10 min then 15% MeOH 10 min, 3.0 ml/min, 210 nm; t1=20.25 min, t2=21.71 min (major)], [α]22D = +101.8 (c 1.07, CH2Cl2); 1H NMR (300 MHz, CDCl3): δ 7.73–7.85 (−SO2PhH, m, 2H), 7.27–7.68 (ArH, m, 17H), 6.38 (Haminal, s, 1H), 4.92 (CH-CH2, t, J = 6.9 Hz, 1H), 4.21 (CH2, dd, J = 9.0, 7.1 Hz, 1H), 3.94 (CH2, dd, J = 9.0, 6.8 Hz, 1H); 13C NMR (75.4 MHz, CDCl3): δ 141.1, 140.8, 138.1, 137.6, 133.5, 129.4, 129.1, 129.0, 128.7, 128.3, 127.8, 127.6, 127.5, 127.4, 127.3, 92.8, 73.3, 63.2; HRMS (EI+) calc’d for [C27H23NO3SNa]+ requires m/z 464.1291, found m/z 464.1292. Isolated as a white solid (mp = 131–133 °C). Trans Diastereomer: 98% ee [t1=25.43 min (major), t2=28.51 min], [α]22D = +59.2 (c 0.61, CH2Cl2); 1H NMR (300 MHz, CDCl3): δ 7.18–7.62 (ArH, m, 17H), 7.07–7.17 (ArH, m, 2H), 6.42 (Haminal, s, 1H), 5.14 (CH-CH2, dd, J = 6.2, 1.9 Hz, 1H), 4.42 (CH2, dd, J = 9.0, 6.2 Hz, 1H), 4.04 (CH2, dd, J = 9.0, 1.9 Hz, 1H); 13C NMR (75.4 MHz, CDCl3): δ 141.3, 140.9, 140.7, 138.6, 137.7, 132.0, 129.4, 129.1, 128.6, 128.5, 127.7, 127.5, 127.4, 127.2, 127.1, 92.5, 73.0, 62.7; HRMS (EI+) calc’d for [C27H23NO3SH]+ requires m/z 442.1472, found m/z 442.1461. Isolated as a white solid (mp = 195–197 °C).
N-Benzensulfonyl-2-phenyl-4-(4-bromophenyl)-1,3-oxazolane (Table 2, entry 8)
Prepared according to the general procedure using 184 mg 4-bromostyrene. Yield 1: 0.279 g, 63%, 1.0:1 cis:trans, 83/46% ee cis/trans (65% ee combined); yield 2: 0.275 g, 62%, 1.0:1 cis:trans, 83/47% ee cis/trans (65% ee combined). Cis diastereomer: >99% ee [Daicel Chiralpak AD column, 15% MeOH, 2.0 ml/min, 210 nm; t1=9.24 min, t2=13.11 min (major)], [α]22D = +80.2 (c 1.00, CH2Cl2); 1H NMR (300 MHz, CDCl3): δ 7.71–7.82 (−SO2PhH, m, 2H), 7.45–7.67 (ArH, m, 5H), 7.30–7.44 (ArH, m, 5H), 7.04–7.17 (ArH, m, 2H), 6.34 (Haminal, s, 1H), 4.83 (CH-CH2, t, J = 6.8 Hz, 1H), 4.17 (CH2, dd, J = 9.0, 6.8 Hz, 1H), 3.86 (CH2, dd, J = 9.0, 6.8 Hz, 1H); 13C NMR (75.4 MHz, CDCl3): δ 137.9, 137.8, 137.5, 133.6, 131.9, 129.4, 129.2, 129.1, 128.7, 128.2, 127.3, 122.2, 92.8, 73.1, 62.8; HRMS (EI+) calc’d for [C21H18BrNO3SNa]+ requires m/z 466.0083, found m/z 466.0084. Isolated as a white solid (mp = 149–150 °C). Trans Diastereomer: 99% ee [t1=11.21 min (major), t2=16.90 min], [α]22D = +94.0 (c 1.23, CH2Cl2); 1H NMR (300 MHz, CDCl3): δ 7.47–7.57 (SO2PhH, m, 2H), 7.34–7.45 (ArH, m, 4H), 7.06–7.30 (ArH, m, 8H), 6.37 (Haminal, s, 1H), 5.04 (CH-CH2, dd, J = 6.3, 1.8 Hz, 1H), 4.37 (CH2, dd, J = 9.0, 6.3 Hz, 1H), 3.94 (CH2, dd, J = 9.0, 1.8 Hz, 1H); 13C NMR (75.4 MHz, CDCl3): δ 140.6, 138.3, 137.9, 132.3, 131.8, 129.8, 129.4, 128.7, 128.6, 127.5, 127.0, 122.5, 92.5, 72.8, 62.3; HRMS (EI+) calc’d for [C21H18BrNO3SH]+ requires m/z 466.0083, found m/z 466.0090. Isolated as a white solid (mp = 163–164 °C).
N-Benzensulfonyl-2-phenyl-4-(4-chlorophenyl)-1,3-oxazolane (Table 2, entry 9)
Prepared according to the general procedure using 138 mg 4-chlorostyrene. Yield 1: 0.280 g, 70%, 0.9:1 cis:trans, 89/45% ee cis/trans (69% ee combined); yield 2: 0.280 g, 67%, 0.8:1 cis:trans, 89/45% ee cis/trans (69% ee combined). Cis diastereomer: >99% ee [Daicel Chiralpak AD column, 12% MeOH, 2.5 ml/min, 210 nm; t1=7.55 min, t2=9.36 min (major)], [α]22D = +77.9 (c 1.53, CH2Cl2); 1H NMR (300 MHz, CDCl3): δ 7.70–7.84 (−SO2PhH, m, 2H), 7.44–7.66 (ArH, m, 5H), 7.30–7.43 (ArH, 1, 3H), 7.10–7.28 (ArH, m, 4H), 6.34 (Haminal, m, 1H), 4.85 (CH-CH2, t, J = 6.8 Hz, 1H), 4.07 (CH2, dd, J = 9.1, 7.2 Hz, 1H), 3.78 (CH2, dd, J = 9.1, 6.5 Hz, 1H); 13C NMR (75.4 MHz, CDCl3): δ 137.8, 137.4, 137.3, 134.0, 133.6, 129.4, 129.2, 129.0, 128.7, 128.2, 127.3, 92.7, 73.2, 62.7; HRMS (EI+) calc’d for [C21H18ClNO3SNa]+ requires m/z 422.0589, found m/z 422.0592. Isolated as a white solid (mp = 141–143 °C). Trans Diastereomer: 87% ee [t1=8.42 min (major), t2=12.21 min], [α]22D = +98.1 (c 1.11, CH2Cl2); 1H NMR (300 MHz, CDCl3): δ 7.47–7.56 (−SO2PhH, m, 2H), 7.32–7.45 (ArH, m, 4H), 7.04–7.24 (ArH, m, 8H), 6.37 (Haminal, s, 1H), 5.06 (CH-CH2, dd, J = 6.3, 1.8 Hz, 1H), 4.37 (CH2, dd, J = 9.0, 6.3 Hz, 1H), 3.94 (CH2, dd, J = 9.0, 1.8 Hz, 1H); 13C NMR (75.4 MHz, CDCl3): δ 140.6, 138.3, 137.4, 134.3, 132.3, 129.4, 128.8, 128.6, 128.6, 127.5, 127.0, 92.5, 72.9, 62.2; HRMS (EI+) calc’d for [C21H18ClNO3SH]+ requires m/z 400.0769, found m/z 400.0767. Isolated as a white solid (mp = 144–148 °C).
N-Benzensulfonyl-2-phenyl-4-(4-fluorophenyl)-1,3-oxazolane (Table 2, entry 10)
Prepared according to the general procedure using 122 mg 4-fluorostyrene. Yield 1: 0.261 g, 68%, 1.6:1 cis:trans, 85/71% ee cis/trans (78% ee combined); yield 2: 0.265 g, 69%, 1.6:1 cis:trans, 85/71% ee cis/trans (78% ee combined). Cis diastereomer: 84% ee [Daicel Chiracel OJ-H column, 7% MeOH 15 min then 20% MeOH 10 min, 2.0 ml/min, 210 nm; t1=8.96 min (major), t2=13.71 min], [α]22D = +46.2 (c 4.42, CH2Cl2);1H NMR: (300 MHz, ) δ 7.70–7.81 (−SO2PhH, m, 2H), 7.43–7.65 (ArH, m, 5H), 7.29–7.43 (ArH, m, 3H), 7.14–7.27 (ArH, m, 2H), 6.87–7.01 (ArH, m, 2H), 6.34 (Haminal, s, 1H), 4.86 (CH-CH2, t, J = 6.8 Hz, 1H), 4.16 (CH2, dd, J = 9.0, 7.0 Hz, 1H), 3.87 (CH2, dd, J = 9.0, 6.6 Hz, 1H);13C NMR (75.4 MHz, CDCl3): δ 162.6, 161.0, 137.9, 137.5, 134.5, 133.6, 129.4, 129.2, 129.1, 129.1, 128.7, 128.2, 127.3, 115.7, 92.7, 73.3, 62.7; HRMS (EI+) calc’d for [C21H18FNO3SNa]+ requires m/z 406.0884, found m/z 406.0901. Isolated as a white solid (mp = 90–91 °C). Trans Diastereomer: 74% ee [t1=21.20 min, t2=22.30 min (major)], [α]22D = +96.7 (c 0.67, CH2Cl2); 1H NMR (300 MHz, CDCl3): δ 7.48–7.58 (−SO2PhH, m, 2H), 7.35–7.45 (ArH, m, 4H), 7.14–7.28 (ArH, m, 6H), 6.78–6.88 (ArH, m, 2H), 6.37 (Haminal, s, 1H), 5.08 (CH-CH2, dd, J = 6.2, 2.0 Hz, 1H), 4.38 (CH2, dd, J = 9.0, 6.2 Hz, 1H), 3.95 (CH2, dd, J = 9.0, 2.0 Hz, 1H); 13C NMR (75.4 MHz, CDCl3): δ 162.8, 140.7, 138.4, 134.7, 132.3, 129.9, 129.8, 129.4, 128.6, 127.5, 127.0, 115.5, 110.9, 92.4, 73.0, 62.2; HRMS (EI+) calc’d for [C21H18FNO3SNa]+ requires m/z 406.0884, found m/z 406.0877. Isolated as a white solid (mp = 140–144 °C).
Benzensulfonyl-2-phenyl-4-(4-methoxyphenyl)-1,3-oxazolane (Table 2, entry 11)
Prepared according to the general procedure using 67 mg 4-vinylanisole. Yield 1: 0.140 g, 71%, 0.4:1 cis:trans, 24/0% ee cis/trans (9% ee combined); yield 2: 0.133 g, 69%, 0.4:1 cis:trans, 25/0% ee cis/trans (9 % ee combined). Spectral data were in agreement with previously reported values.8a Cis diastereomer: 28% ee [Daicel Chiracel OJ-H column, 15% MeOH, 2.5 ml/min, 210 nm; t1= 5.26 min (major), t2=5.96 min], [α]22D = +15.8 (c 0.77, CH2Cl2). Trans diastereomer: 0% ee [t1=11.14 min, t2=12.29 min].
N-Benzensulfonyl-2-phenyl-4-(3-methoxyphenyl)-1,3-oxazolane (Table 2, entry 12)
Prepared according to the general procedure using 137 mg 4-vinylanisole. Yield 1: 0.306 g, 76%, 1.3:1 cis:trans, 87/65% ee cis/trans (77% ee combined); yield 2: 0.317 g, 78%, 1.3:1 cis:trans, 87/64% ee cis/trans (77 % ee combined). Cis diastereomer: 90% ee [Daicel Chiralpak AD column, 12% MeOH, 2.5 ml/min, 210 nm; t1=5.43 min, t2=6.08 min (major)], [α]22D = +69.6 (c 1.81, CH2Cl2); 1H NMR (300 MHz, CDCl3): δ 7.73–7.83 (−SO2PhH, m, 2H), 7.55–7.67 (ArH, m, 3H), 7.44–7.54 (ArH, m, 2H), 7.29–7.43 (ArH, m, 3H), 7.16 (ArH, t, J = 8.0 Hz, 1H), 6.64–8.85 (ArH, m, 3H), 6.40 (Haminal, s, 1H), 4.85 (CH-CH2, t, J = 7.0 Hz, 1H), 4.19 (CH2, dd, J = 9.0, 7.2 Hz, 1H), 3.88 (CH2, dd, J = 9.0, 6.8 Hz, 1H), 3.62 (Ar-OCH3, s, 3H); 13C NMR (75.4 MHz, CDCl3): δ 160.0, 140.3, 138.2, 137.7, 133.5, 129.7, 129.4, 129.0, 128.7, 128.3, 127.3, 119.6, 114.4, 112.2, 92.7, 73.5, 63.4, 55.3; HRMS (EI+) calc’d for [C22H21NO4SNa]+ requires m/z 418.1085, found m/z 418.1085. Isolated as a colorless oil. Trans Diastereomer: 53% ee [t1=7.79 min (major), t2=8.73 min], [α]22D = +56.3 (c 0.49, CH2Cl2); 1H NMR (300 MHz, CDCl3): δ7.51–7.57 (−SO2PhH, m, 2H), 7.36–7.43 (ArH, m, 4H), 7.07–7.24 (ArH, m, 5H), 6.85–6.90 (ArH, m, 1H), 6.73–6.78 (ArH, m, 1H), 6.67–6.70 (ArH, m, 1H), 6.36 (Haminal, s, 1H), 5.07 (CH-CH2, dd, J = 6.3, 2.0 Hz, 1H), 4.37 (CH2, dd, J = 8.9, 6.3 Hz, 1H), 3.98 (CH2, dd, J = 8.9, 2.0 Hz, 1H), 3.64 (ArOCH3, s, 3H); 13C NMR (75.4 MHz, CDCl3): δ 159.9, 140.6, 140.1, 138.6, 132.1, 129.6, 129.3, 128.6, 128.5, 127.5, 127.1, 120.8, 114.4, 112.8, 92.4, 73.0, 63.0, 55.3; HRMS (EI+) calc’d for [C22H21NO4SH]+ requires m/z 396.1265, found m/z 396.1268. Isolated as a colorless oil.
N-Benzensulfonyl-2-phenyl-4-(3-chlorophenyl)-1,3-oxazolane (Table 2, entry 13)
Prepared according to the general procedure using 139 mg 3-chlorostyrene. Yield 1: 0.202 g, 50%, 0.8:1 cis:trans, 39/83% ee cis/trans (60% ee combined); yield 2: 0.208 g, 52%, 0.8:1 cis:trans, 38/83% ee cis/trans (60% ee combined). Cis diastereomer: 85% ee [Daicel Chiracel OJ-H column, 18% MeOH, 2.5 ml/min, 210 nm; t1=3.58 min (major), t2=4.26 min], [α]22D = +53.5 (c 0.51, CH2Cl2); 1H NMR (300 MHz, CDCl3): δ 7.74–7.81 (−SO2PhH, m, 2H), 7.45–7.67 (ArH, m, 5H), 7.34–7.44 (ArH, m, 3H), 7.10–7.23 (ArH, m, 4H), 6.36 (Haminal, s, 1H), 4.85 (CH-CH2, t, J = 7.0 Hz, 1H), 4.18 (CH2, dd, J = 9.0, 7.2 Hz, 1H), 3.87 (CH2, dd, J = 9.0, 6.8 Hz, 1H); 13C NMR (75.4 MHz, CDCl3): δ 140.9, 137.7, 137.5, 134.6, 133.6, 130.1, 129.4, 129.2, 128.7, 128.3, 128.2, 127.6, 127.3, 125.4, 92.8, 73.2, 62.8; HRMS (EI+) calc’d for [C21H18ClNO3SH]+ requires m/z 400.0769, found m/z 400.0768. Isolated as a colorless oil. Trans diastereomer: 45% ee [t1=7.74 min, t2=11.88 min (major)], [α]22D = +69.9 (c 1.00, CH2Cl2); 1H NMR (300 MHz, CDCl3): δ 7.48–7.57 (m, 2H, SO2PhH), 7.34–7.45 (m, 4H, ArH), 7.08–7.28 (m, 8H, ArH), 6.39 (s, 1H, Haminal), 5.05 (dd, J = 6.2, 1.7 Hz, 1H, CH-CH2), 4.37 (dd, J = 9.0, 6.2 Hz, 1H, CH2), 3.94 (dd, J = 9.0, 1.7 Hz, 1H, CH2); 13C NMR (75.4 MHz, CDCl3): δ 140.7, 140.4, 138.3, 134.8, 132.5, 129.9, 129.4, 128.6, 128.5, 128.0, 127.5, 126.9, 126.5, 92.6, 72.8, 62.4; HRMS (EI+) calc’d for [C21H18ClNO3SNa]+ requires m/z 422.0589, found m/z 422.0569. Isolated as a white solid (mp = 126–128 °C).
3.3. Hydrolysis of 1,3-oxazolidines
N-Benzenesulfonyl-2-amino-2-(4-methylphenyl) ethanol
Into a oven dried 250 ml round bottom flask containing a stir bar was placed the N-benzensulfonyl-2-phenyl-4-(4-methylphenyl)-1,3-oxazolane (5.0 g, 13.2 mmol, 82% combined ee) and 48 ml 2:1 dioxane:H2O. Trifluoroacetic acid (4.52 g, 39.6 mmol) was then added dropwise while stirring at room temperature. The reaction was then heated to 80 °C and stirred under positive pressure of nitrogen for 4 hours. The mixture was then cooled to room temperature and 75 ml saturated NaHCO3 was added slowly. The mixture was then extracted with 3×100 ml CH2Cl2 and the combined organic dried over Na2SO4 before solvent removal. The product was purified by chromatography on silica gel with 1:1 EtOAc:hexanes as eluent. Yield 1: 3.36 g, 84% yield, 83% ee; yield 2: 3.38 g 88% yield, 82% ee. The product was the recrystalized from ethyl acetate:hexanes to afford 2.54 g, >99% ee. Data: 98% ee [Diacel Chiracel OJ-H column, 6% MeOH, 2.0 ml/min; t1=11.24 min, t2=14.98 min (major)], [α]22D = +109.52 (c 0.94, CH2Cl2). 1H NMR (300 MHz, CDCl3 ): δ 7.66–7.78 (−SO2PhH, m, 2H), 7.44–7.55 (ArH, m, 1H), 7.32–7.43 (ArH, m, 2H), 6.90–7.05 (ArH, m, 4H), 5.33 (NH, d, J = 6.1 Hz, 1H), 4.38 (CH-CH2, q, J = 6.1 Hz, 1H), 3.75 (CH2, t, J = 6.1 Hz, 2H), 2.27 (ArCH3, s, 3H), 1.98 (−OH, t, J = 6.1 Hz, 1H);13C NMR (75.4 MHz, CDCl3): δ 140.3, 138.1, 134.6, 132.7, 129.6, 129.1, 127.4, 126.9, 66.5, 59.5, 21.2; HRMS (EI+) calc’d for [C15H17NO3SNa]+ requires m/z 314.0822, found m/z 314.0811. Isolated as a white solid (mp = 151–153 °C).
N-Benzenesulfonyl-2-amino-2-(4-biphenyl)ethanol
Into a oven dried 250 ml round bottom flask containing a stir bar was placed the N-benzensulfonyl-2-phenyl-4-(4-biphenyl)-1,3-oxazolane (6.2 g, 14.0 mmol, 74% combined ee) and 52 ml 2:1 dioxane:H2O. Trifluoroacetic acid (4.81 g, 42.2 mmol) was then added dropwise while stirring at room temperature. The reaction was then heated to 80 °C and stirred under positive pressure of nitrogen for 4 hours. The mixture was then cooled to room temperature and 75 ml saturated NaHCO3 was added slowly. The mixture was then extracted with 3×100 ml CH2Cl2 and the combined organic dried over Na2SO4 before solvent removal. The product was purified by chromatography on silica gel with 1:1 EtOAc:hexanes as eluent. Yield 1: 4.35 g, 88% yield, 72% ee; yield 2: 4.00 g, 84% yield, 72% ee. The product was the recrystalized from ethyl acetate:hexanes to afford 3.40 g, 86% ee. Data: 86% ee [Diacel Chiralpak AD column, 12% MeOH, 3.0 ml/min; t1=28.61 min (major), t2=30.65 min], [α]22D = +129.6 (c 0.38, CH2Cl2). 1H NMR (300 MHz, CDCl3 ): δ 7.67–7.79 (−SO2PhH, m, 2H), 7.30–7.57 (ArH, m, 10H), 7.15 (ArH, m, 2H), 5.38 (−NH, d, J = 6.4 Hz, 1H), 4.45–4.55 (−CH-CH2, m, 1H), 3.74–3.87 (CH2, m, 2H), 1.93 (−OH, t, J = 6.1 Hz, 1H); 13C NMR (75.4 MHz, CDCl3): δ 141.3, 140.6, 140.4, 136.5, 132.8, 129.1, 129.0, 127.7, 127.6, 127.5, 127.4, 127.2, 66.4, 59.4; HRMS (EI+) calc’d for [C20H19NO3SNa]+ requires m/z 376.0978, found m/z 376.0996. Isolated as a white solid (mp = 173–174 °C).
N-Benzenesulfonyl-2-amino-2-(3,5-dimethylphenyl)ethanol
Into a oven dried 250 ml round bottom flask containing a stir bar was placed the N-benzensulfonyl-2-phenyl-4-(3,5-dimethylphenyl)-1,3-oxazolane (3.0 g, 7.9 mmol, 81% combined ee) and 29 ml 2:1 dioxane:H2O. Trifluoroacetic acid (2.7 g, 23.6 mmol) was then added dropwise while stirring at room temperature. The reaction was then heated to 80 °C and stirred under positive pressure of nitrogen for 4 hours. The mixture was then cooled to room temperature and 40 ml saturated NaHCO3 was added slowly. The mixture was then extracted with 3×100 ml CH2Cl2 and the combined organic dried over Na2SO4 before solvent removal. The product was purified by chromatography on silica gel with 1:1 EtOAc:hexanes as eluent. Yield 1: 2.06 g, 88% yield, 81% ee; yield 2: 1.99 g, 88% yield, 81% ee. The product was the recrystalized from ethyl acetate:hexanes to afford 1.33 g, 98% ee. Data: 95 % ee [Diacel Chiralpak AD column, 5% MeOH, 2.5 ml/min; t1=22.62 min (major), t2=25.46 min], [α]22D = +88.3 (c 1.28, CH2Cl2). 1H NMR: (300 MHz, CDCl3 ) δ 7.65–7.75 (−SO2PhH, m, 2H), 7.40–7.49 (−SO2PhH, m, 1H), 7.28–7.38 (−SO2PhH, m, 2H), 6.77 (ArH, s, 1H), 6.62 (ArH, s, 2H), 5.80 (−NH, d, J = 6.1 Hz, 1H), 4.40 (−CH-CH2, q, J = 6.1 Hz, 1H), 3.73 (−CH2, t, J = 6.1 Hz, 2H), 2.57 (−OH, t, J = 6.1 Hz, 1H), 2.14 (−ArCH3, s, 6H); 13C NMR: (75.4 MHz, CDCl3) δ 140.5, 138.3, 137.2, 132.6, 129.7, 128.8, 127.3, 124.9, 66.4, 59.9, 21.3; HRMS (EI+) calc’d for [C16H19NO3SH]+ requires m/z 306.1159, found m/z 306.1152. Isolated as a white solid (mp = 106–109 °C).
3.4. Determination of absolute stereochemistry
The absolute stereochemistry of N-benzensulfonyl-2,4-diphenyl-1,3-oxazolane (Table 2, entry 1), N-benzensulfonyl-2-phenyl-4-(4-methylphenyl)-1,3-oxazolane (Table 2, entry 2), and N-benzensulfonyl-2-phenyl-4-(4-tert-butylphenyl)-1,3-oxazolane (Table 2, entry 6) were determined as described below. The configuration of all other 1,3-oxazolidine products in Table 2 was assigned by analogy.
N-Benzensulfonyl-2,4-diphenyl-1,3-oxazolane (Table 2, entry 1)
(R)-N-Benzenesulfonyl-2-amino-2-phenylethanol was prepared from (R)-phenylglycinol using a procedure described by Quintard and coworkers.12 Spectral data were in agreement with previously reported values.8a 99.8 % ee [Daicel Chiracel OJ-H column, 10% MeOH, 2.5 ml/min, 210 nm; t1=4.74 min (major), t2=6.71 min], [α]22D = −96.6 (c 0.55, CH2Cl2). For comparison, N-benzensulfonyl-2,4-diphenyl-1,3-oxazolane synthesized according to the general procedure was subjected to hydrolysis using 3 equiv of trifuloroacetic acid in 2:1 dioxane:H2O at 80 °C. The optical rotation of the resulting (S)-N-benzensulfonyl amino alcohol was measured: 80% ee, [α]22D = +79.2 (c 0.72, CH2Cl2).
N-Benzensulfonyl-2-phenyl-4-(4-methylphenyl)-1,3-oxazolane (Table 2, entry 2)
The aminal was subjected to hydrolysis using 3 equiv of trifluoroacetic acid in 2:1 dioxane:H2O at 80 °C. The resulting N-sulfonyl amino alcohol was deprotected using the method of Martin.13 The N-sulfonyl amino alcohol (762 mg, 2.61 mmol) in a flame-dried 50 mL round-bottomed flask along with 4,4′-di-t-butylbiphenyl (69 mg, 0.26 mmol). The flask was charged with 24 mL THF, and to this solution was added 106 mg (15.3 mmol, 6 equiv) lithium wire cut into small pieces. After sealing the flask with a septum and flushing with argon, the reaction vessel was placed into a sonicating bath and stirred while sonicating over 4 h. After removal of the remaining lithium metal, the resulting orange mixture was quenched by the addition of 2 mL ethanol and 1.0 g solid NH4Cl. This mixture was stirred for 5 min before the solids were removed by filtration and the filtrate concentrated by rotary evaporation. The resulting thick oil was dissolved in EtOH (3 mL) and passed through a short plug of silica gel using EtOH as eluent. The solvent was removed by rotary evaporation, and the residue was diluted with 25 mL CH2Cl2. The precipitate was isolated by vacuum filtration, affording 0.44 g (2.34 mmol, 90% yield) of (S)-2-amino-2-(4-methylphenyl)ethanol hydrochloride salt. An analytically pure sample of the free base was generated by treatment with 6 N KOH, and the optical rotation was measured. [α]22D = +22.9 (c 0.68, abs. EtOH) [lit. [α]22D = +26 (c 1.0, abs. EtOH)].14
N-Benzensulfonyl-2-phenyl-4-(4-t-butylphenyl)-1,3-oxazolane (Table 2, entry 6)
The 1,3-oxazolidine (.223 g, .67 mmol) was converted to 2-amino-2-(4-t-butylphenyl)ethanol using the procedure described for N-benzensulfonyl-2-phenyl-4-(4-methylphenyl)-1,3-oxazolane. Yield: .088g, 68%. [α]22D = +22.8 (c 1.85, CHCl3) [lit. [α]22D = +37.5 (c 0.47, CHCl3)].15
Acknowledgments
This paper is dedicated to Prof. Michael Krische, recipient of this year’s Tetrahedron Young Investigator Award, in recognition of his many creative contributions to synthetic methodology. Funding for this research has been provided by the NIH (National Institute of General Medical Sciences: R01-GM084022). D.J.M. was supported by fellowships from the NIH (CBI Training Grant GM08505) and the American Chemial Society. The NMR spectroscopy facility at UW–Madison is funded by the NIH (S10-RR04981-01) and NSF (CHE-9629688).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.For a recent review of methods for the construction of 1,2-aminoalcohols, see: Bergmeier SC. Tetrahedron. 2000;56:2561–2571.
- 2.Li G, Chang HT, Sharpless KB. Angew Chem Int Ed Engl. 1996;35:451–454. [Google Scholar]
- 3.For reviews of the scope of the Sharpless asymmetric aminohydroxylation and its use in synthesis, see: (a) O’Brien P. Angew Chem Int Ed. 1999;38:326–329. doi: 10.1002/(SICI)1521-3773(19990201)38:3<326::AID-ANIE326>3.0.CO;2-T.Nilov D, Reiser O. Adv Synth Catal. 2002;344:1169–1173.Bodkin JA, McLeod MD. J Chem Soc, Perkin Trans. 1;2002:2733–2746.
- 4.For selected examples, see: (a) Nicolaou KC, Natarajan S, Li H, Jain NF, Hughes R, Solomon ME, Ramanjulu JM, Boddy CNC, Takayanagi M. Angew Chem Int Ed Engl. 1998;37:2708–2714. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2708::AID-ANIE2708>3.0.CO;2-E.Boger DL, Miyazaki S, Kim SH, Wu JH, Castle SL, Loiseleur O, Jin Q. J Am Chem Soc. 1999;121:10004–10011.Cao B, Park H, Jouillé MM. J Am Chem Soc. 2002;124:520–521. doi: 10.1021/ja017277z.Crowley BM, Mori Y, McComas CC, Tang D, Boger DL. J Am Chem Soc. 2004;126:4310–4317. doi: 10.1021/ja039795a.
- 5.(a) Noack M, Göttlich R. Chem Commun. 2002:536–537. doi: 10.1039/b111656h. [DOI] [PubMed] [Google Scholar]; (b) Chikkanna D, Han H. Synlett. 2004:2311–2314. [Google Scholar]; (c) Mahoney JM, Smith CR, Johnston JN. J Am Chem Soc. 2005;127:1354–1355. doi: 10.1021/ja045608c. [DOI] [PubMed] [Google Scholar]; (d) Alexanian EJ, Lee C, Sorensen EJ. J Am Chem Soc. 2005;127:7690–7691. doi: 10.1021/ja051406k. [DOI] [PubMed] [Google Scholar]; (e) Szolcsányi P, Gracza T. Chem Commun. 2005:3948–3950. doi: 10.1039/b506731f. [DOI] [PubMed] [Google Scholar]; (f) Correa A, Tellitu I, Domínguez E, SanMartin R. J Org Chem. 2006;71:8316–8319. doi: 10.1021/jo061486q. [DOI] [PubMed] [Google Scholar]; (g) Liu G, Stahl SS. J Am Chem Soc. 2006;128:7179–7181. doi: 10.1021/ja061706h. [DOI] [PubMed] [Google Scholar]; (h) Schultz MJ, Sigman MS. J Am Chem Soc. 2006;128:1460–1461. doi: 10.1021/ja0579053. [DOI] [PubMed] [Google Scholar]; (i) Desai LV, Sanford MS. Angew Chem Int Ed. 2007;46:5737–5740. doi: 10.1002/anie.200701454. [DOI] [PubMed] [Google Scholar]; (j) Cochran BM, Michael FE. Org Lett. 2008;10:5039–5042. doi: 10.1021/ol8022165. [DOI] [PubMed] [Google Scholar]
- 6.Fuller PH, Kim JW, Chemler SR. J Am Chem Soc. 2008;130:17638–17639. doi: 10.1021/ja806585m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Davis FA, Nadir UK, Kluger EW. J Chem Soc Chem Commun. 1977:25–26. [Google Scholar]; (b) Davis FA, Jenkins R, Yocklovich SG. Tetrahedron Lett. 1978;52:5171–5174. [Google Scholar]
- 8.(a) Michaelis DJ, Shaffer CJ, Yoon TP. J Am Chem Soc. 2007;129:1866–1867. doi: 10.1021/ja067894t. [DOI] [PubMed] [Google Scholar]; (b) Partridge KM, Anzovino ME, Yoon TP. J Am Chem Soc. 2008;130:2920–2921. doi: 10.1021/ja711335d. [DOI] [PubMed] [Google Scholar]; (c) Michaelis DJ, Ischay MA, Yoon TP. J Am Chem Soc. 2008;130:6610–6615. doi: 10.1021/ja800495r. [DOI] [PubMed] [Google Scholar]
- 9.(a) Ghosh AK, Mathivanan P, Cappiello J. Tetrahedron: Asymmetry. 1998;9:1–45. doi: 10.1016/S0957-4166(97)00593-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Johnson JS, Evans DA. Acc Chem Res. 2000;33:325–335. doi: 10.1021/ar960062n. [DOI] [PubMed] [Google Scholar]; (d) McManus HA, Guiry PJ. Chem Rev. 2004;104:4151–4202. doi: 10.1021/cr040642v. [DOI] [PubMed] [Google Scholar]; (e) Desimoni G, Faita G, Jørgensen KA. Chem Rev. 2006;106:3561–3651. doi: 10.1021/cr0505324. [DOI] [PubMed] [Google Scholar]; (f) Rasappan R, Laventine D, Reiser O. Coord Chem Rev. 2008;252:702–714. [Google Scholar]
- 10.Vishwakarma LC, Stringer OD, Davis FA. Org Syn Col Vol 8. 1993:546–550. [Google Scholar]
- 11.Cornejo A, Fraile JM, García JI, Gil MJ, Martínez-Merino V, Mayoral JA, Pires E, Villalba I. Synlett. 2005:2321–2324. [Google Scholar]
- 12.Coeffard V, Thobie-Gautier C, Beaudet I, Le Grognec E, Quintard JP. Eur J Org Chem. 2008:383–391. [Google Scholar]
- 13.Neipp CE, Humphrey JM, Martin SF. J Org Chem. 2001;66:531–537. doi: 10.1021/jo001386z. [DOI] [PubMed] [Google Scholar]
- 14.Endeshaw MM, Bayer A, Hansen LK, Gautun OR. Eur J Org Chem. 2006:5249–5259. [Google Scholar]
- 15.Kawasaki KI, Katsuki T. Tetrahedron. 1997;53:6337–6350. [Google Scholar]