

Abstract
The reaction of methyl indole-2-carboxylates and arynes affords a very efficient, high yielding synthesis of a novel indole-indolone ring system, which tolerates considerable functionality, is broad in scope and proceeds under mild reaction conditions.
Keywords: Aryne, Annulation, Indole
Nitrogen-containing heterocycles exhibit a diverse array of favorable biological and pharmacological properties. The indole core, in particular, is the foundation for many well known medicinally active compounds.1 Convenient methods for constructing fused indoles are of great value to medicinal chemists.1,2 In addition, indolone derivatives possess potent biological activities. Thus, new methodologies for their synthesis contribute significantly to the pursuit of new drugs.3 Of considerable current interest to medicinal chemists is the synthesis of new drug-like compounds in which two separate biologically interesting scaffolds, both independently known for their favorable activities, are fused.2,4 Since both indoles and indolones are relevent core structures for pharmaceuticals, a hybrid thereof could potentially lead to a series of biologically active compounds (Fig. 1).
Figure 1.
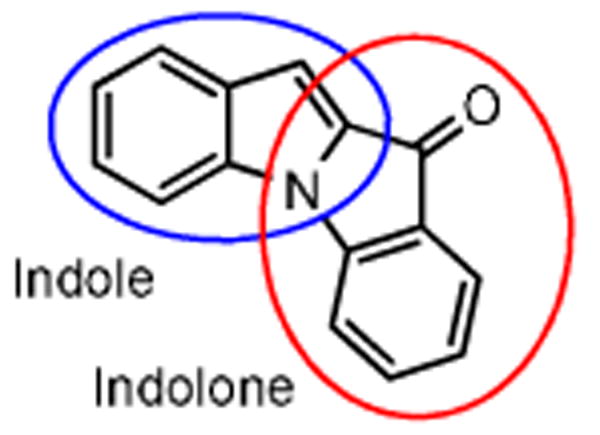
Indole-indolone scaffold
Arynes have been used extensively in recent years in the construction of many heteroaromatic structures.5 The highly electrophilic nature of benzyne is most suitable for cross coupling with various nucleophilic heteroatoms. If the substrate containing the nucleophilic heteroatom is tethered with a neighboring electrophile, such as a carbonyl group, intramolecular annulations often take place.6 Recently, our group has reported annulations between ortho-heteroatom (N, O, and S) benzoates with arynes, producing a diverse array of medicinally relevant acridones, xanthones, and thioxanthones in good to excellent yields.7 The proven success and utility of this novel methodology led us to envision a related [3+2] annulation involving the reaction of indole-2-carboxylate esters and arynes. Herein, we report the synthesis of a novel indole-indolone hybrid, a previously unexplored scaffold with respect to biological activity and materials applications, via the annulation of arynes by methyl indole-2-carboxylates (Scheme 1).
Scheme 1.
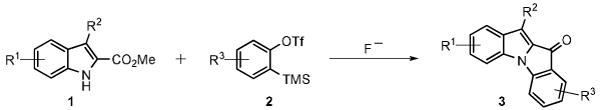
General reaction of arynes with methyl indole-2-carboxylates.
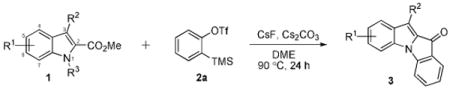
Initial experiments began with ethyl indole-2-carboxylate as our test substrate, but little success was realized using conditions similar to those of our previous annulation chemistry.7 Switching to the methyl ester, however, produced a slight increase in yield of the desired product (Table 1, entry 1). It was not until we switched to 1,2-dimethoxyethane (DME) as a solvent that decent results were obtained (entry 3). The addition of various bases was examined and a significant improvement in yield was realized when two equiv of Cs2CO3 were utilized (entry 6). The success of this base over others (entries 7-9) may be attributed to its superior solubility in DME. The success of aryne annulation chemistry often depends heavily on the relative rate of aryne formation versus bimolecular coupling of the substrate and the aryne. For that reason, both the quantity and the nature of the fluoride source, as well as the solvent and temperature, are often critical. CsF has generally been the most popular source of fluoride used in benzyne chemistry and, in fact, it seems to work best for our methodology. Other sources of fluoride, such as tetrabutylammonium reagents have been examined. However, the overall yields declined (entries 13 and 14), even though these fluorides are more soluble in DME.
Table 1.
Optimization studies of the reaction between methyl indole-2-carboxylate and o-(trimethylsilyl)phenyl triflate (Scheme 1, R1 = R2 = R3 = H).a
| Entry | Aryne Precursor (equivs) | Fluoride Source (equivs)b | Base (equivs) | Solvent | Temp. (°C) | % Yieldc |
|---|---|---|---|---|---|---|
| 1 | 1.2 | CsF (3) | - | THF | 65 | 22 |
| 2 | 1.2 | CsF (3) | - | MeCN | 80 | 34 |
| 3 | 1.2 | CsF (3) | - | DME | 90 | 44 |
| 4 | 1.2 | CsF (3) | Cs2CO3 (1.1) | DME | 90 | 52 |
| 5 | 1.5 | CsF (3) | Cs2CO3 (1.1) | DME | 90 | 55 |
| 6 | 1.5 | CsF (3) | Cs2CO3 (2.0) | DME | 90 | 70 |
| 7 | 1.5 | CsF (3) | Na2CO3 (2.0) | DME | 90 | 34 |
| 8 | 1.5 | CsF (3) | K2CO3 (2.0) | DME | 90 | 34 |
| 9 | 1.5 | CsF (3) | CsOPiv (2.0) | DME | 90 | 39 |
| 10 | 1.5 | CsF (3) | NaH (2.0) | DME | 90 | <10 |
| 11 | 1.5 | CsF (3) | Cs2CO3 (2.0) | DME | 120 | 62 |
| 12 | 2.0 | CsF (3) | Cs2CO3 (2.0) | DME | 90 | 62 |
| 13 | 1.5 | TBAT (2) | Cs2CO3 (2.0) | DME | rt | 33 |
| 14 | 1.5 | TBAF (2) | Cs2CO3 (2.0) | DME | rt | 19 |
Reaction conditions: 0.5 mmol of 1, 5 mL of solvent, 24 h.
Per one equivalent of benzyne precursor.
Isolated yields.
With optimized conditions in hand (Table 1, entry 6), we have explored the scope and limitations of our methodology. Suitable indole substrates were easily prepared by the esterification of commercially available 2-indolecarboxylic acids. It was found that indoles containing electron-donating groups generally resulted in lower yields than the unsubstituted indole (Table 1, entries 2-6). Possibly, the electron-donating groups decrease the acidity of the indole, resulting in a weaker nucleophile. In fact, for all substrates containing a methoxy group, it has been observed that a significant amount of the starting material remains after 24 h. Low yields are also obtained with indole 1g, a substrate containing a highly electron-withdrawing group. It is believed, however, that steric crowding of the nitro group around the nucleophilic nitrogen center is the main reason for the observed decrease in yield (entry 7). On the other hand, a variety of 5-haloindole-2-carboxylates have afforded good yields of the desired products (entries 8-10). Interestingly, even better results are observed when the halogen occupies the 3-position of the indole (entries 11-13). This trend is also observed in substrates 1n and 1o, whose 3-positions are substituted (entries 14 and 15) and near quantitative yields are observed. This may be attributed to steric effects, in that a substituent at the 3-position can better help direct the electrophilic ester moiety for intramolecular attack by the intermediate aryl carbanion. As expected, replacement of the N-H with a N-methyl group prevented any formation of the desired N-annulated product (entry 16).
After a number of methyl indolecarboxylates were examined, we turned our attention towards screening various benzyne precursors. Substrate 1l was chosen for these reactions due to its favorable performance with benzyne precursor 2a (Table 2, entry 12). A series of symmetrical benzyne precursors were examined first (Table 3, entries 1-4) and good yields were obtained. It was found that the overall yields depend greatly on the electronics of the aryne precursors. Thus, electron-rich aryne precursor 2c gave the highest yield (entry 2), whereas electron-poor aryne precursor 2d gave the lowest yield (entry 3). Unsymmetrical benzyne precursors present an issue of regioselectivity, since a nucleophile can attack either one of the two reactive aryne carbons. Some unsymmetrical benzyne precursors display higher regioselectivity than others. For example, it has been reported in the literature that benzyne precursor 2f undergoes nucleophilic attack at the meta-position for both steric and electronic reasons.9 As expected, our observations were consistent with the previous reports. Utilizing a 2D-NOESY experiment, the structure of product 3t was established as shown (entry 6). Benzyne precursor 2g also presents regioselectivity issues. Here a single product was formed. Although a 2D-NOESY experiment was not performed on the major isolated product, it is assumed that the nitrogen nucleophile would attack at the beta position based on steric hindrance. In addition, previous literature has shown that benzyne precursor 2g preferably undergoes attack at the beta position.10
Table 2.
[3+2] Annulations of methyl indole-2-carboxylates.a
 | |||
|---|---|---|---|
| Entry | Substrate | Product | % Yieldb |
| 1 | R1 = R2 = R3 = H 1a |
 |
70 |
| 2 | R1 = 5-Me R2 = R3 = H 1b |
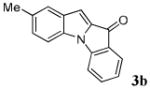 |
62 |
| 3 | R1 = 5-OMe R2 = R3 = H 1c |
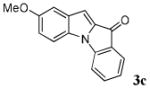 |
28 |
| 4 | R1 = 4-OMe R2 = R3 = H 1d |
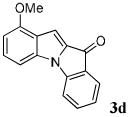 |
45 |
| 5 | R1 = 6-OMe R2 = R3 = H 1e |
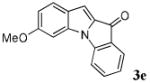 |
33 |
| 6 | R1 = 5,6-(OMe)2 R2 = R3 = H 1f |
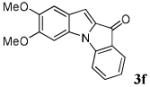 |
39 |
| 7 | R1 = 7-NO2 R2 = R3 = H 1g |
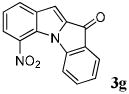 |
40 |
| 8 | R1 = 5-F R2 = R3 = H 1h |
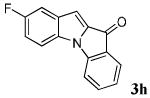 |
64 |
| 9 | R1 = 5-Cl R2 = R3 = H 1i |
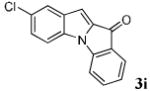 |
78 |
| 10 | R1 = 5-Br R2 = R3 = H 1j |
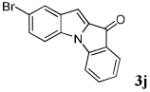 |
76 |
| 11 | R1 = R3 = H R2 = Cl 1k |
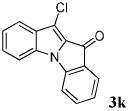 |
83 |
| 12 | R1 = R3 = H R2 = Br 1l |
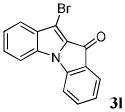 |
88 |
| 13 | R1 = H R2 = I R3 = H 1m |
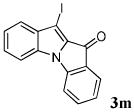 |
85 |
| 14 | R1 = R3 = H R2 = Ph 1n |
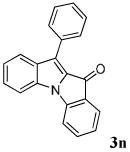 |
94 |
| 15 | R1 = R3 = H R2 = CO2Me 1o |
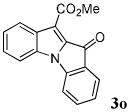 |
88 |
| 16 | R1 = R2 = H R3 = Me 1p |
- | 0 |
Reaction conditions: 0.50 mmol of 1, 0.75 mmol of 2, 2.25 mmol of CsF, 1.00 mmol of Cs2CO3, DME (5 mL) at rt for 1 d.
Isolated yields.
Table 3.
Reactions of indole 1l with various benzyne precursors.
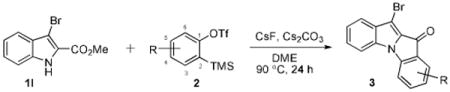 | |||
|---|---|---|---|
| Entry | Benzyne Precursor | Product | % Yielda |
| 1 | R = 4,5-Me2 2b |
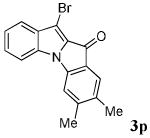 |
67 |
| 2 | R = 4,5-(OMe)2 2c |
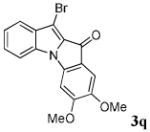 |
87 |
| 3 | R = 4,5-F2 2d |
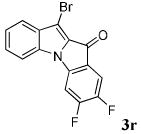 |
37 |
| 4 | R = 4,5-(CH)4- 2e |
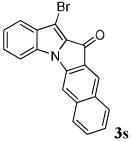 |
67 |
| 5 | R = 3-OMe 2f |
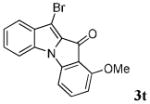 |
82 |
| 6 | R = 3,4-(CH)4- 2g |
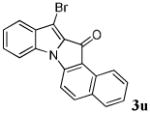 |
74 |
Isolated yields.
A possible mechanism for formation of the [3+2] annulated product parallels what has been proposed previously in the literature (Scheme 2).7 First, immediately following formation of the benzyne, a negatively charged indole nucleophilically attacks the aryne, resulting in intermediate A. The newly formed aryl carbanion then intramolecularly attacks the neighboring ester, displacing a methoxy group, which can also aid in deprotonation of the indole starting material.
Scheme 2.
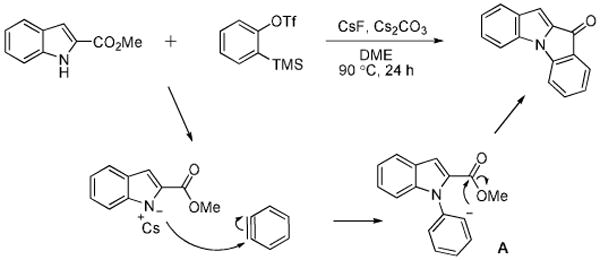
Proposed mechanism.
In conclusion, we have demonstrated a new and novel benzyne methodology for the synthesis of a previously unexplored indole-indolone scaffold, which provides good to excellent yields. The reaction involves a [3+2] annulation of arynes by methyl indole-2-carboxylates under mild conditions, producing a variety of functionally diverse compounds. In addition, a variety of substituted benzyne precursors, both symmetrical and unsymmetrical, undergo smooth annulations. Single regioisomers have been obtained in good yields for the unsymmetrical benzyne precursors studied. This novel indole-indolone scaffold holds great promise for biological activity. In addition, compounds 3j, 3l, 3m, and 3p-3u are suitable for further elaboration via palladium chemistry if a combinatorial library of these compounds is desired.
Acknowledgments
We thank the National Institute of General Medical Sciences (GM070620 and GM079593) and the National Institutes of Health Kansas University Center of Excellence in Chemical Methodology and Library Development (P50 GM069663) for their generous financial support. We also thank Dr. Feng Shi for his help in the preparation of some of the aryne precursors.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gil C, Brase S. J Comb Chem. 2009;11:175–197. doi: 10.1021/cc800102t. [DOI] [PubMed] [Google Scholar]
- 2.Horton DA, Bourne GT, Smythe ML. Chem Rev. 2003;103:893–930. doi: 10.1021/cr020033s. [DOI] [PubMed] [Google Scholar]
- 3.(a) Smith G, Glaser M, Perumal M, Nguyen QD, Shan B, Arstad E, Aboagye EO. J Med Chem. 2008;51:8057–8067. doi: 10.1021/jm801107u. [DOI] [PubMed] [Google Scholar]; (b) Manoury P, Binet J, Obitz D, Defosse G, Dewitte E, Veronique C. Indolone derivatives, their preparation and their application in therapy. U.S. Patent 5,010,079. 1991 April 23;; (c) Konkel M, Packiarajan M, Chen H. Aminoalkylphenyl indolone derivatives. PCT Int Appl. 2006 WO2006083538. [Google Scholar]
- 4.(a) Tietze LF, Bell HP, Chandrasekhar S. Angew Chem, Int Ed. 2003;42:3996–4028. doi: 10.1002/anie.200200553. [DOI] [PubMed] [Google Scholar]; (b) Zheng L, Xiang J, Dang Q, Guo S, Bai X. J Comb Chem. 2005;7:813–815. doi: 10.1021/cc0500809. [DOI] [PubMed] [Google Scholar]; (c) Meunier B. Acc Chem Res. 2008;41:69–77. doi: 10.1021/ar7000843. [DOI] [PubMed] [Google Scholar]; (d) Bajorath J. Mol Diversity. 2002;5:305–313. doi: 10.1023/a:1021321621406. [DOI] [PubMed] [Google Scholar]
- 5.Chen Y, Larock RC. In: Modern Arylation Methods. Ackerman J, editor. Wiley/VCH; New York: 2009. pp. 401–473. [Google Scholar]
- 6.(a) Liu Z, Larock RC.J Am Chem Soc 200612813112–13122.17017791 [Google Scholar]; (b) Yoshida H, Shirakawa E, Honda Y, Hiyama T. Angew Chem, Int Ed. 2002;41:3247–3249. doi: 10.1002/1521-3773(20020902)41:17<3247::AID-ANIE3247>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]; (c) Tambar UK, Stoltz BM. J Am Chem Soc. 2005;127:5340–5341. doi: 10.1021/ja050859m. [DOI] [PubMed] [Google Scholar]
- 7.(a) Zhao J, Larock RC. J Org Chem. 2007;72:583–588. doi: 10.1021/jo0620718. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhao J, Larock RC. Org Lett. 2005;7:4273–4275. doi: 10.1021/ol0517731. [DOI] [PubMed] [Google Scholar]
- 8.Representative procedure (3i, Table 2, entry 9): An oven dried 6-dram vial equipped with a stirrer bar was charged with 107.4 mg of 1i (0.50 mmol) and 223.8 mg of 2a (0.75 mmol), followed by 2.5 mL of dry DME. The mixture was briefly stirred and 325.8 mg of dry Cs2CO3 and 341.8 mg of dry CsF was added sequentially, followed by an additional 2.5 mL of DME, washing the sidewalls of the vial. The vial was sealed and placed in an oil bath of 90 °C and allowed to stir for 1 d. The resultant mixture was cooled and eluted through a plug of silica gel with ethyl acetate and DCM. The filtrate was dried with anhydrous Na2CO3, decanted, and evaporated. The residue was impregnated with silica gel and purified by column chromatography (35:1 to 10:1 hexane/ethyl acetate), affording 98.3 mg of the desired product 3i as a light orange solid; mp 203-204 °C; 1H NMR (400 MHz, CDCl3) δ 7.62 (d, J = 7.6 Hz, 1 H), 7.57 (s, 1 H), 7.50 (t, J = 7.6 Hz, 1 H), 7.31-7.39 (m, 2 H), 7.26 (d, J = 8.0 Hz, 1H), 7.09 (t, J = 7.6 Hz, 1 H), 6.99 (s, 1 H); 13C NMR (100 MHz CDCl3) δ 181.4, 145.3, 136.8, 135.8, 133.6, 132.5, 129.3, 128.5, 127.7, 125.5, 124.4, 124.4, 112.3, 111.4, 107.0; HRMS (EI): calcd for C15H8ClNO 253.0294, found 253.0301.
- 9.(a) Liu Z, Shi F, Martinez PDG, Raminelli C, Larock RC. J Org Chem. 2008;73:219–226. doi: 10.1021/jo702062n. [DOI] [PubMed] [Google Scholar]; (b) Blackburn T, Romtohul YK. Synlett. 2008:1159–1164. [Google Scholar]; (c) Jayanth TT, Cheng CH. Angew Chem, Int Ed. 2007;46:5921–5924. doi: 10.1002/anie.200701063. [DOI] [PubMed] [Google Scholar]
- 10.(a) Waldo JP, Zhang X, Shi F, Larock RC. J Org Chem. 2008;73:6679–6685. doi: 10.1021/jo8009215. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yoshida H, Shirakawa E, Honda Y, Hiyama T. Angew Chem, Int Ed. 2002;41:3247–3249. doi: 10.1002/1521-3773(20020902)41:17<3247::AID-ANIE3247>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]