

Introduction
Long QT syndrome (LQTS) is an inherited disorder with prolonged ventricular repolarization and an increased propensity to ventricular tachyarrhythmias of the torsade de pointes type that are responsible for arrhythmogenic syncope and sudden cardiac death.1 During the past 13 years, 10 different genotype forms of LQTS have been identified (LQT1-10), with the most frequent clinical types (LQT1-3) categorized as ion-channelopathies.2 The remaining 7 infrequently occurring forms of LQTS (LQT4-10) also affect myocellular ion-channel currents either directly or indirectly, but LQT4-10 make up less than 5% of the genotype-identified LQTS. To date, approximately 500 different LQTS mutations have been identified in the 10 LQTS genes, and cellular expression studies of these mutations have elucidated basic electrophysiologic mechanisms responsible for the delayed repolarization and the manifest QT prolongation. Different LQTS genes affect different ion-current mechanisms, and the clinical course of patients with LQT1, 2, and 3 genotypes have been shown to be quite different.3 In addition, different mutations on the same LQTS gene may produce different electrophysiologic effects. For example, mutations involving the LQT1 gene are all associated with reduction in the repolarizing IKs current, but the magnitude of the reduction in this current can vary considerably among the different LQT1 mutations.4 This variability in the electrophysiological effects of different mutations contributes to the variability in the risk of life-threatening cardiac events that are independent of the manifest QTc interval on the ECG. Thus, knowledge of the LQTS genotype and the associated specific mutations are useful in risk-stratifying individual patients for the selection of appropriate therapy for patient-specific risk-reduction.
Because of the extensive literature that currently exists in LQTS, we will focus on the 3 common forms of LQTS (LQT1, 2, and 3) and related mutations to make our point that this information is useful in managing patients with LQTS.
Risk by Genotype
By 1995, the three major LQT1, 2, and 3 genetic loci were identified, and shortly thereafter it was appreciated that the ECG manifestations and the clinical course of LQTS were different among the three common genotypes. Each of the three long QT syndrome genotypes was associated with somewhat distinctive ECG repolarization features.5 Among affected individuals, the QTonset-c was unusually prolonged in those individuals with LQT3 mutations (lead II QTonsetc : LQT1, 243 ±73 ms; LQT2, 290 ±56 ms; LQT3, 341 ±42 ms; p<0.001); Tamplitude was generally quite small in the LQT2 genotype (lead II Tamplitude, mV: LQT1, 0.37 ±0.17; LQT2, 0.13 ±0.07;LQT3, 0.36 ±0.14; p<0.001); and Tduration was particularly long in LQT1 genotype (lead II Tdurationc, ms: LQT1, 262 ±65; LQT2, 191 ±51; LQT3, 187 ±33; p<0.001).
Life-threatening cardiac events tend to occur under specific circumstances in a gene-specific manner (Fig. 1).6 LQT1 patients were shown to experience 68% of their lethal events during exercise, whereas most LQT2 and LQT3 patients experience lethal events during rest/sleep, respectively.6 The triggering role of sympathetic activation in LQT1 patients has important therapeutic implications, as it suggests that protection could be expected by the use of anti-adrenergic interventions. It should also be noted that although most events in LQT2 and LQT3 patients occur at rest or during sleep, the triggers associated with lethal events for LQT2 and LQT3 patients show a different pattern since LQT2 patients are particularly sensitive to startling and sudden noises, such as a telephone or alarm clock ring (Fig. 1).6
Figure 1.
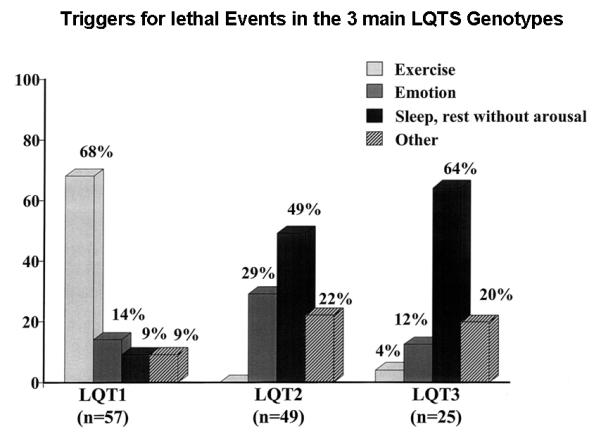
Triggers for lethal cardiac events according to 3 genotypes. Numbers in parentheses indicate number of triggers, not number of patients. (Reproduced with permission; Fig. 1 from Schwartz PJ, et al. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89-95.)6
In the 246 genotyped patients reported by Zareba, et al.3 the frequency of cardiac events was significantly higher among subjects with mutations at the LQT1 locus (63 percent) or the LQT2 locus (46 percent) than among subjects with mutations at the LQT3 locus (18 percent) (Fig. 2). However, the likelihood of dying during a cardiac event was significantly higher among families with mutations at the LQT3 locus (20 percent) than among those with mutations at the LQT1 locus (4 percent) or the LQT2 locus (4 percent).
Figure 2.
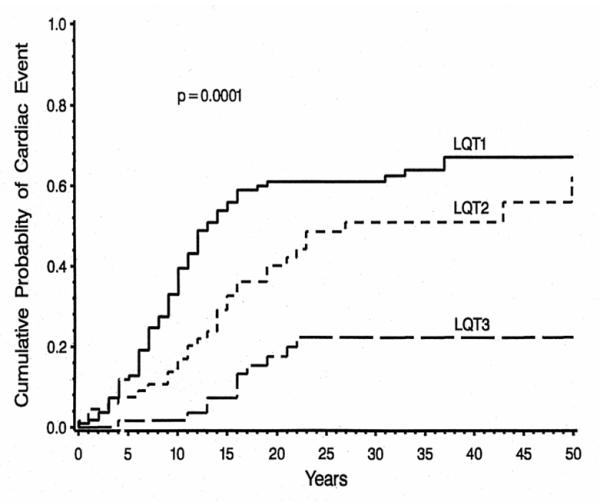
Kaplan-Meier estimate of the cumulative probability of an LQTS-related cardiac event in LQT1, LQT2, and LQT3 genotyped patients. Reproduced with permission; Fig. 2 from Zareba W, et al. Influence of genotype on the clinical course of the long-QT syndrome. International Long-QT Syndrome Registry Research Group. N Engl J Med. 1998;339:960-965).3
The genotypic risk is also influenced by age and sex of the patient. Priori, et al showed that the genotype of the causative mutation affects the clinical course of the long-QT syndrome and modulates the effects of the QTc and sex on clinical manifestations.7 Goldenberg, et al.8 reported in children age 1 to 12 years that the three major LQTS genotypes were associated with similar risks for life-threatening cardiac events after adjustment for clinical factors (LQT1 vs. LQT2: hazard ratio = 1.61, p=0.56; LQT3 vs. LQT2: hazard ratio = 2.37; p=0.49). Similar risk results were observed for the three genotypes during adolescence.9 However, in adults age 18 to 40 years, patients with the LQT2 genotype had a significantly higher cardiac event rate than patients with the other two genotypes, and this was especially so in women.10 The risk by genotype was also examined in female patients during their childbearing years.11 Compared with a time period before a woman’s first conception, the pregnancy time was associated with a reduced risk of cardiac events (hazard ratio 0.28, p=0.01), whereas the 9-month postpartum time had an increased risk (hazard ratio 2.7, p<0.001). Genotype analysis showed that women with the LQT2 genotype were more likely to experience a post-partum cardiac event than women with the LQT1 or LQT3 genotype (Fig. 3). In the only study looking at the clinical course of patients older than 40 years, Goldenberg, et al. showed that LQT3 genotype carriers exhibited the highest cumulative lethal event rate (35%) compared with LQT2, LQT1, and genotype negative subjects (24% and 14%, and 10%, respectively; p=0.001).12
Figure 3.
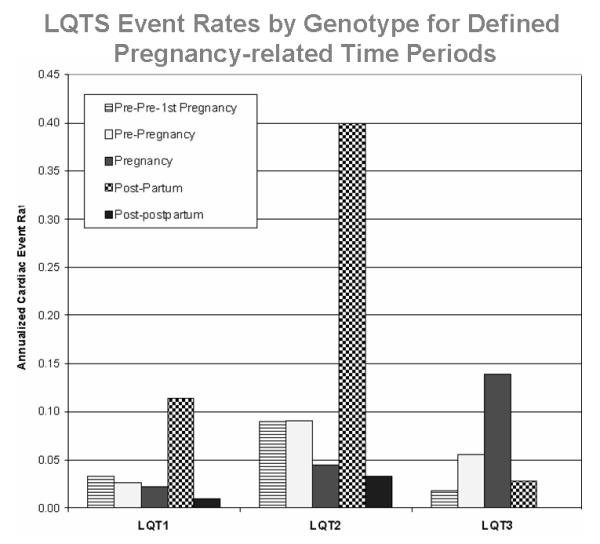
Annualized LQTS-related cardiac event rates by genotype in LQT1, LQT2, and LQT3 genotyped patients for defined pregnancy-related time periods. None of the 12 LQT3 women had a cardiac event during the post-postpartum period. (Reproduced and modified with permission; Fig. 4 from Seth R, et al. Long QT syndrome and pregnancy. J Am Coll Cardiol. 2007;49:1092-1098).11
A summary of the risk of cardiac events by the LQT1, 2, and 3 genotypes for four different age groups is presented in Table 1. The risk for cardiac events is augmented in female LQT2 patients in age 20-40 years and in LQT3 patients over age 40 years.
Table 1.
Risk for Aborted Cardiac Arrest or LQTS Death by Genotype and Age
| Age group | LQT1 | LQT2 | LQT3 |
|---|---|---|---|
| 1-12 years | ++ (M) | ++ (M) | ++ (M) |
| 12-20 years | +++ | +++ | +++ |
| 21-40 years | ++ | +++ (F) | ++ |
| 41-75 years | + | ++ | +++ |
The number of plus signs indicates the relative risk in ordinal mild (+), moderate (++), and severe (+++) categories, with M and F indicating the relative predominance of male and females, respectively.
Risk by Mutation Location and Function
There are 2 proposed molecular mechanisms that may account for reduced potassium currents in patients with KCNQ1 and HERG mutations:13,14 (1) coassembly or trafficking abnormalities, in which mutant subunits either do not coassemble with normal subunits or if they do, are not transported to the cell membrane, resulting in a net effect of ≤ 50% reduction in the number of functional channels (haplotype insufficiency); and (2) formation of defective channels involving mutant subunits, with the altered channel protein transported to the cell membrane, resulting in a > 50% reduction in channel function (dominant-negative effect). Knowledge of the functional effects of the mutation and its location have been demonstrated to provide incremental prognostic information to clinical risk factors and the genotype,4 that may be utilized for improved risk stratification and a more focused management of higher-risk LQTS patients.
Mutations in the KCNQ1 gene
Prior studies that assessed the functional role of LQT1 mutations have yielded conflicting results, possibly due to sample size limitations.15,16. However, a recent cooperative study comprising 600 LQT1 patients, derived from the from the US portion of the International LQTS Registry, the Netherlands’ LQTS Registry, and the Japanese LQTS Registry, has facilitated a comprehensive analysis of the clinical aspects of 77 different KCNQ1 mutations categorized by their location, coding type, and type of biophysical ion channel dysfunction.4 The study demonstrated that subjects with mutations having dominant-negative ion current effects had a longer QTc interval and a higher cumulative probability of cardiac events than subjects with mutations resulting in loss of function (haploinsufficiency) (Fig. 4A). After multivariable adjustment for clinical covariates, subjects with mutations having dominant-negative functional effects exhibited >2-fold increase in the risk for cardiac events (p<0.001) compared with those with haploinsufficiency mutations. The study further demonstrated that the frequency of cardiac events in LQT1 patients is also related to the location and type of the KCNQ1 mutation. Subjects with mutations located in the transmembrane region of the channel had a significantly higher rate of cardiac events than those with mutations located in the C-terminus regions (Fig. 4B), and those with missense mutations had a significantly higher event rate than those with non-missense mutations (Fig. 4C).4 It is possible that IKs channels with transmembrane mutations might have reduced responsiveness to the regulator β-adrenergic signaling of the ion-conduction pathway with more impairment of shortening of the QTc with exercise-related tachycardia than mutations in the C-terminus region.
Figure 4.
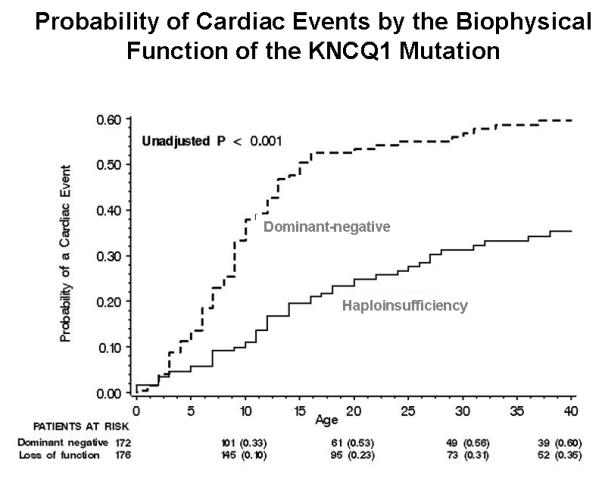
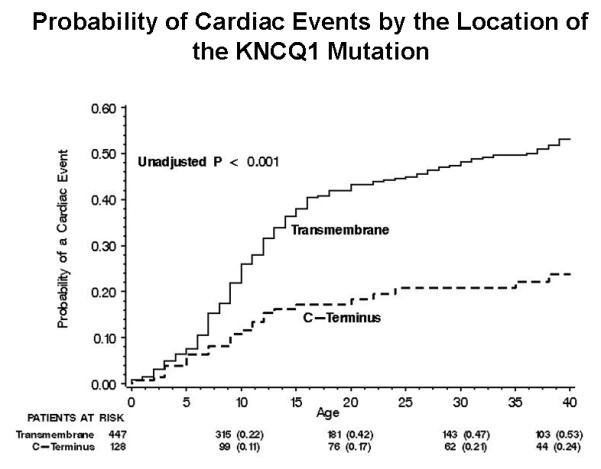
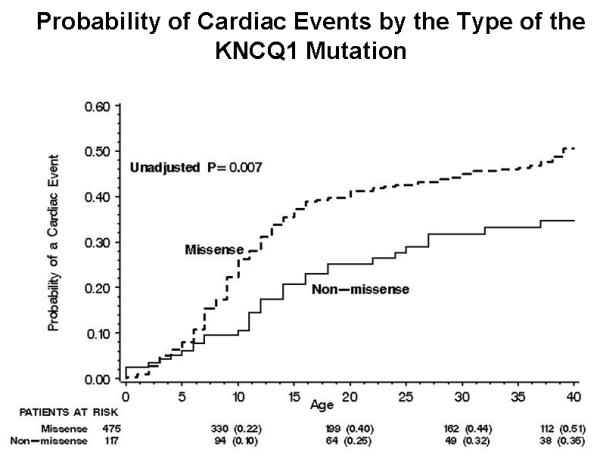
Kaplan-Meier estimate of the cumulative probability of a first cardiac event in KCNQ1 mutation carriers (LQT1 genotype) by (A) the biophysical function; (B) location; and (C) coding type of the mutation. (Reproduced with permission; Fig. 7 from Moss AJ, et al. Clinical aspects of type-1 long-QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene. Circulation. 2007;115:2481-2489).4
Mutations in the HERG gene
The pore region of the HERG channel provides the potassium conductance pathway. Most mutations involving this region are missense mutations with dominant-negative effects on IKr, whereas most mutations in the nonpore regions of HERG are associated with coassembly or trafficking abnormalities resulting in haplotype insufficiency.14 In a study of 201 subjects with a total of 44 different HERG mutations from the International LQTS Registry, subjects harboring pore mutations exhibited a more severe clinical course and experienced a higher frequency of arrhythmia-related cardiac events, occurring at earlier age, than did subjects with nonpore mutations (Fig. 5).17 Furthermore, pore mutations were shown to dominate the risk after multivariate adjustment for clinical factors, exhibiting an 11-fold increase in the risk for cardiac events in subjects with QTc at 500 ms, and a 16% increase in the pore hazard ratio for each 10-ms increase in QTc.17 Cellular expression studies also implicate the Per-Arnt-Sim (PAS) domain within the N-terminus and the cyclic nucleotide binding domain (cNBD) of the C-terminus as important regulators of the biophysical properties of HERG.18-20 However, the clinical risk associated with these mutation sites has not yet been assessed. The US, Japanese and Netherlands LQTS Registries are currently cooperating in the analysis of the risk associated with additional mutation sites in HERG gene.
Figure 5.

Kaplan-Meier cumulative probability of first cardiac events from birth through age 40 years for subjects with HERG mutations in pore (n=34), N-terminus (N-term; n=54), and C-terminus (C-term; n=91) regions (Reprinted with permission; Fig. 2 from Moss AJ, et al. Increased risk of arrhythmic events in long-QT syndrome with mutations in the pore region of the human ether-a-go-go-related gene potassium channel. Circulation. 2002;105:794-799).17
Mutations in the SCN5A gene
LQT3 mutations in the SCN5A gene produce a gain of function, through faster recovery from inactivation and/or increase in residual current. These mechanisms produce a small but functionally important enhancement of inward sodium plateau current sufficient to delay repolarization and increase vulnerability of the heart to arrhythmias.21. Preliminary data from the US portion of the International LQTS Registry indicate that the biophysical function and location of the LQT3 location are also important determinants of outcome in this population. These recent findings suggest that the ΔKPQ mutation, which is located in the intracellular loops and operates through both faster recovery from inactivation and an increase in residual sodium current, is associated with a significantly higher risk for cardiac events than the C-terminus D1790G mutation that has distinct biophysical function effects on steady state inactivation and intracellular calcium homeostasis.22.23 The US, Japanese and Netherlands LQTS Registries are currently cooperating in a combined assessment of the risk associated with the location, function, and coding type of LQT3 mutations.
Risk by Specific Mutation
Several individual mutations in the LQTS genotypes have been demonstrated to be associated with a more severe clinical course and reduced response to therapy that is not fully explained by the biophysical properties of the mutation. In a cohort study involving individuals from South Africa harboring the same A341V mutation in the KCNQ1 gene, mutation-carriers were shown to experience an unusual severe clinical course, independently of the origin of the families, and to be at higher risk for cardiac events compared with a more general LQT1 population. Importantly, patients who harbored the A341V mutation experienced a higher frequency of recurrent cardiac events despite β-blocker therapy.24 Furthermore, a recent study, comprising 78 A341V mutation carriers from 21 families and 8 countries,25 showed that A341V is associated with a significantly higher arrhythmic risk compared with non-A341V LQT1 mutations with a dominant-negative effect or other LQT1 transmembrane mutations despite the fact that A341V was shown to exert only a relatively mild dominant-negative effect. Individual mutations may also contribute independently to risk in LQT2 patients. Rossenbacker et al. have identified a novel nonpore missense mutation (K28E) in the PAS domain of the KCNH2 channel that is associated with a malignant phenotype,26 and Crotti et al.27 showed that the clinically latent non-pore KCNH2-A1116V mutation exhibits a severe clinical phenotype among patients who co-express the common K897T KCNH2 polymorphism. These findings suggest that cellular electrophysiological studies cannot always predict the clinical phenotype, and that data regarding individual LQTS mutations and associated polymorphisms in modifiers genes may be used to identify high risk patients with this genetic disorder who may have attenuated response to medical therapy.
Therapy by Genotype and Mutation
The main therapeutic modalities for the prevention of life-threatening cardiac events include β-blockers, left cervico-thoracic sympathetic denervation (LCSD), and the implantable cardioverter defibrillator (ICD). In non-genotyped patients, β-blockers comprise the mainstay therapy, whereas LCSD and implantation of an ICD are therapeutic options in high-risk LQTS patients who experience recurrent cardiac events despite β-blocker therapy. Genetic data can be used to provide an improved therapeutic management plan that is individualized by knowledge of genotype- and mutation-specific risk factors, prevention measures, and therapies. A proposed therapeutic regimen for each of the 3 major LQTS genotypes is considered separately below and summarized in Table 2.28
Table 2.
Risk mechanisms and genotype-specific therapy based on clinical and experim ental data in LQT1, LQT2, and LQT3 forms of long QT syndrome
| LQT1 | LQT2 | LQT3 | |
|---|---|---|---|
| Sensitivity to sympathetic stimulation |
Yes | To some degree | No |
| Torsade de pointes | Exercise related | Startle | Sleep/rest |
| Specific triggers/Occurrence |
Swimming | Telephone, alarm clock, postpartum |
Inactivity |
| Risk by mutation | Dominant neg., transmembrane location, A341V |
Pore location, K28E |
ΔKPQ |
| Exercise restriction | +++ | ++ | ? |
| Beta-blockers | +++ | ++ | ? |
| Potassium supplement |
+ | ++ | + |
| Mexiletine | + | + | ++ |
| Flecainide | No data | No data | +++ (ΔKPQ, D1790G) |
| Ranolazine | No data | No data | ++ (ΔKPQ) |
| LCSD in high -risk patients |
++ | ++ | ++ |
| ICD in high-risk patients |
+++ | +++ | +++ |
ICD = implantable cardioverter-defibrillator; LCSD = left cervicothoracic sympathetic denervation. The number of plussigns indicates the relative benefit of therapy in minimal (+), moderate (++), and marked (+++) effectiveness categories; ? = uncertain. (Reproduced and modified with permission. Table 1 from Shimizu W, et al. The long QT syndrome: therapeutic implications of a genetic diagnosis. Cardiovasc Res. 2005;67:347-356).29
LQT1 patients
Life-threatening events occur during sympathetic activation in patients with this genotype, therefore this patient subset benefit from appropriate lifestyle modifications and are effectively protected by the use of antiadrenergic interventions. LQT1 patients should not be allowed to participate in competitive sports. Swimming is particularly hazardous, as 99% of the arrhythmic episodes associated with swimming were shown to occur in patients with this genotype.29 Patients who are identified as carriers of the LQT1 genotype should be treated with β-blockers. Priori et al.30 observed in a group of 187 LQT1 patients who were treated with β-blockers a very low rate of life-threatening cardiac events (1.2%) during a median-term follow-up period of 4.7 years. Implantation of an ICD and/or LCSD should be considered in high-risk LQT1 patients, especially those harboring dominant negative and/or transmemebrane mutations who exhibit phenotypic QT prolongation and recent syncope despite β-blocker therapy. Since LQT1 patients are most sensitive to sympathetic stimulation, LCSD is expected to be most effective in this population. In a study 147 patients who underwent LCSD, Schwartz et al. 31 showed that the procedure was associated with a significant long-term reduction in the frequency of ACA and syncope, but was not entirely effective in preventing sudden death. However, LQT1 patients in the study were shown to experience a very low rate of life-threatening cardiac events after LCSD (1 event per 100 person-years). Recent data from the LQTS-ICD Registry demonstrate the efficacy and safety of the implantable defibrillator in combination with β-blockers in LQT1 patients: in a subset of 69 (36%) LQTS-ICD Registry who were genotyped, none of the LQT1 patients who received combined ICD and β-blocker therapy died or experienced appropriate ICD therapy during a median follow-up of 6 years, whereas the rate of appropriate ICD therapy among LQT2 patients during the same time period was 24%.33
LQT2 patients
Preventive measures in LQT2 patients include avoidance of unexpected auditory stimuli in the bedroom that can cause a startle reaction since these may be associated with lethal events, especially during rest or sleep. The efficacy of β-blocker therapy in LQT2 patients is lower than in patients with the LQT1 genotype. In the study of Priori et al.30 27 of 120 (23%) LQT2 patients experienced a cardiac event during follow-up, and the adjusted risk of cardiac events among LQT2 patients was significantly higher (HR=2.91; p=0.001) as compared with LQT1 patients. Similarly, the benefit of LCSD in this population may be more limited: Schwartz et al.32 reported that the combined incidence of aborted cardiac arrest of sudden cardiac death after LCSD was 3-fold higher among LQT2 patients as compared with LQT1 patients. Thus, high-risk LQT2 patients (e.g. those with pore mutations with concomitant phenotypic QT prolongation and recent syncope, and symptomatic adult females) should be considered for early ICD implantation.
LQT2 patients are especially vulnerable when their potassium levels are low since IKr is sensitive to extracellular potassium level. Experimental wedge studies suggested that an increase in extracellular potassium can limit the development of an arrhythmogenic substrate under long QT conditions.34 Furthermore, in clinical practice, exogenously administered potassium was reported to correct repolarization abnormalities in patients with IKr defects,35 and long-term oral potassium administration was recently shown to improve repolarization abnormalities in LQT2 patients.36
Therefore, efforts should be made to maintain a serum potassium level > 4 mEq/L in patients with this genotype.
LQT3 patients
Data regarding management of LQT3 patients are more limited. Patients harboring this genotype have excessive further prolongation of the QT intervals at slow heart rates,37 and the QTc was shown to prolong even further during the night when heart rate decreases,.38 Thus, a reduction in heart rate with β-blockers may pose a therapeutic problem in this population. Accordingly, β-blocker therapy in this population was shown to be associated with a relatively high rate of residual events, and the efficacy of this mode of medical therapy is lower in LQT3 patients compared with those with the other 2 main LQTS genotypes.30,39 In the study of Priori et al.30 9 of 28 (32%) LQT3 patients experienced a cardiac event while on β-blocker therapy and the adjusted risk for a cardiac event among LQT3 patients was 4-fold higher than among LQT1 patients.
Since most SCN5A mutations increase a late Na+ inward current, sodium channel blockers may shorten the QT interval in LQT3 patients. Administration of the sodium-channel blocker mexiletine was shown to shorten the QT interval by an average of 90 ms.40 However, the response to mexiletine was not consistent and was shown to be mutation-specific.41 Similarly, the effectiveness of flecainide, a class IC sodium channel blocker, in this population was also demonstrated to be mutation-specific.42,43 Benhorin et al.42 demonstrated a that administration of flecainide abbreviated the QT interval in LQT3 patients with the D1790G mutation in SCN5A, and Windle et al.43 showed that oral flecainide shortened the QTc interval and normalized the repolarization T-wave pattern patients with SCN5A-ΔKPQ mutation. The antianginal agent ranolazine reduces late sodium channel current, shortens the action potential duration, and suppress early afterdepolarization-triggered arrhythmias in animal models of LQT3 with sustained inward sodium current.44 In a recent study we have shown that therapeutic concentrations of ranolazine were associated with a dose-dependent shortening of QTc interval and improved diastolic relaxation in patients with the LQT3-ΔKPQ mutation.45 Thus, a possible management strategy in LQT3 patients may be to assess the degree of QT shortening produced by an oral sodium channel blocker or ranolazine, and to initiate medical therapy in combination with a beta-blocker in patients who respond with QTc shortening of ≥50 msec. It should be noted, however, that data regarding the clinical efficacy of sodium channel blockers and ranolazine in LQT3 patients are limited, and an electrocardiographic response may not correlate with clinical response in LQT3 carriers. Thus, due to limited long-term clinical data regarding the benefit of medical therapy in LQT3 patients, together with the fact that the lethality among carriers of this genotype is higher than in LQT1 and LQT2 patients,3 early ICD implantation should be encouraged more aggressively in symptomatic LQT3 patients than among patients with the LQT1 or LQT2 genotypes.
Conclusion
Patients with LQTS are at increased risk for sudden cardiac death, with the risk probability influenced by a number of phenotypic and genotypic factors. Optimal drug and/or device therapy to reduce the probability of a fatal event should be tailored to the magnitude of the perceived risk and to amelioration of the risk mechanisms involved in life-threatening ventricular tachyarrhythmias. During the past decade, various genetic and mutation-related risk mechanisms have been identified, and the available studies indicate that this genotype-specific information should be utilized in the selection of therapy to reduce morbidity and mortality in patients with LQTS.
Acknowledgments
Sources of Funding This work was supported in part by research grants HL-33843 and HL-51618 from the NIH, Bethesda, MD, and by research grants to the University of Rochester from Medtronic Inc., Cardiovascular Therapeutics, Inc., and BioReference Laboratories, Inc. Dr. Ilan Goldenberg received salary support from the Mirowski-Moss Career Development Award while a faculty member at the University of Rochester Medical Center.
Footnotes
Conflict of Interest Disclosures Dr. Moss received an honorarium when he served in the past year as member of an advisory group for Cardiovascular Therapeutics, Inc.
References
- 1.Moss AJ, Schwartz PJ, Crampton RS, Tzivoni D, Locati EH, MacCluer J, Hall WJ, Weitkamp L, Vincent GM, Garson A., Jr. The long QT syndrome. Prospective longitudinal study of 328 families. Circulation. 1991;84:1136–1144. doi: 10.1161/01.cir.84.3.1136. [DOI] [PubMed] [Google Scholar]
- 2.Roden DM. Clinical practice. Long-QT syndrome. N Engl J Med. 2008;358:169–176. doi: 10.1056/NEJMcp0706513. [DOI] [PubMed] [Google Scholar]
- 3.Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Robinson JL, Priori SG, Benhorin J, Locati EH, Towbin JA, Keating MT, Lehmann MH, Hall WJ. Influence of genotype on the clinical course of the long-QT syndrome. International Long-QT Syndrome Registry Research Group. N Engl J Med. 1998;339:960–965. doi: 10.1056/NEJM199810013391404. [DOI] [PubMed] [Google Scholar]
- 4.Moss AJ, Shimizu W, Wilde AA, Towbin JA, Zareba W, Robinson JL, Qi M, Vincent GM, Ackerman MJ, Kaufman ES, Hofman N, Seth R, Kamakura S, Miyamoto Y, Goldenberg I, Andrews ML, McNitt S. Clinical aspects of type-1 long-QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene. Circulation. 2007;115:2481–2489. doi: 10.1161/CIRCULATIONAHA.106.665406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moss AJ, Zareba W, Benhorin J, Locati EH, Hall WJ, Robinson JL, Schwartz PJ, Towbin JA, Vincent GM, Lehmann MH. ECG T-wave patterns in genetically distinct forms of the hereditary long QT syndrome. Circulation. 1995;92:2929–2934. doi: 10.1161/01.cir.92.10.2929. [DOI] [PubMed] [Google Scholar]
- 6.Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, Denjoy I, Guicheney P, Breithardt G, Keating MT, Towbin JA, Beggs AH, Brink P, Wilde AA, Toivonen L, Zareba W, Robinson JL, Timothy KW, Corfield V, Wattanasirichaigoon D, Corbett C, Haverkamp W, Schulze-Bahr E, Lehmann MH, Schwartz K, Coumel P, Bloise R. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89–95. doi: 10.1161/01.cir.103.1.89. [DOI] [PubMed] [Google Scholar]
- 7.Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, Vicentini A, Spazzolini C, Nastoli J, Bottelli G, Folli R, Cappelletti D. Risk stratification in the long-QT syndrome. N Engl J Med. 2003;348:1866–1874. doi: 10.1056/NEJMoa022147. [DOI] [PubMed] [Google Scholar]
- 8.Goldenberg I, Moss AJ, Peterson DR, McNitt S, Zareba W, Andrews ML, Robinson JL, Locati EH, Ackerman MJ, Benhorin J, Kaufman ES, Napolitano C, Priori SG, Qi M, Schwartz PJ, Towbin JA, Vincent M,G, Zhang L. Risk factors for aborted cardiac arrest and sudden cardiac death in children with the congenital long-QT syndrome. Circulation. 2008;117:2184–2191. doi: 10.1161/CIRCULATIONAHA.107.701243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hobbs JB, Peterson DR, Moss AJ, McNitt S, Zareba W, Goldenberg I, Qi M, Robinson JL, Sauer AJ, Ackerman MJ, Benhorin J, Kaufman ES, Locati EH, Napolitano C, Priori SG, Towbin JA, Vincent GM, Zhang L. Risk of aborted cardiac arrest or sudden cardiac death during adolescence in the long-QT syndrome. JAMA. 2006;296:1249–1254. doi: 10.1001/jama.296.10.1249. [DOI] [PubMed] [Google Scholar]
- 10.Sauer AJ, Moss AJ, McNitt S, Peterson DR, Zareba W, Robinson JL, Qi M, Goldenberg I, Hobbs JB, Ackerman MJ, Benhorin J, Hall WJ, Kaufman ES, Locati EH, Napolitano C, Priori SG, Schwartz PJ, Towbin JA, Vincent GM, Zhang L. Long QT syndrome in adults. J Am Coll Cardiol. 2007;49:329–337. doi: 10.1016/j.jacc.2006.08.057. [DOI] [PubMed] [Google Scholar]
- 11.Seth R, Moss AJ, McNitt S, Zareba W, Andrews ML, Qi M, Robinson JL, Goldenberg I, Ackerman MJ, Benhorin J, Kaufman ES, Locati EH, Napolitano C, Priori SG, Schwartz PJ, Towbin JA, Vincent GM, Zhang L. Long QT syndrome and pregnancy. J Am Coll Cardiol. 2007;49:1092–1098. doi: 10.1016/j.jacc.2006.09.054. [DOI] [PubMed] [Google Scholar]
- 12.Goldenberg I, Moss AJ, Peterson DR, Bradley J, Polonski S, McNitt S, Zareba W, Andrews ML, Robinson JL, Locati EH, Ackerman MJ, Benhorin J, Kaufman ES, Napolitano C, Priori SG, Qi M, Schwartz PJ, Towbin JA, Vincent M,G, Zhang L. Long-QT syndrome after age 40. Circulation. 2008;117:2192–2201. doi: 10.1161/CIRCULATIONAHA.107.729368. [DOI] [PubMed] [Google Scholar]
- 13.Shalaby FY, Levesque PC, Yang WP, Little WA, Conder ML, Jenkins-West T, Blanar MA. Dominant-negative KvLQT1 mutations underlie the LQT1 form of long QT syndrome. Circulation. 1997;96:1733–1736. doi: 10.1161/01.cir.96.6.1733. [DOI] [PubMed] [Google Scholar]
- 14.January CT, Gong Q, Zhou Z. Long-QT syndrome: cellular basis and arrhythmia mechanism in LQT2. J Cardiovasc Electrophysiol. 2000;11:1413–1418. doi: 10.1046/j.1540-8167.2000.01413.x. [DOI] [PubMed] [Google Scholar]
- 15.Zareba W, Moss AJ, Sheu G, Kaufman ES, Priori S, Vincent GM, Towbin JA, Benhorin J, Schwartz PJ, Napolitano C, Hall WJ, Keating MT, Qi M, Robinson JL, Andrews ML. Location of mutation in the KCNQ1 and phenotypic presentation of long QT syndrome. J Cardiovasc Electrophysiol. 2003;14:1149–1153. doi: 10.1046/j.1540-8167.2003.03177.x. [DOI] [PubMed] [Google Scholar]
- 16.Shimizu W, Horie M, Ohno S, Takenaka K, Yamaguchi M, Shimizu M, Washizuka T, Aizawa Y, Nakamura K, Ohe T, Aiba T, Miyamoto Y, Yoshimasa Y, Towbin JA, Priori SG, Kamakura S. Mutation site-specific differences in arrhythmic risk and sensitivity to sympathetic stimulation in the LQT1 form of congenital long QT syndrome: multicenter study in Japan. J Am Coll Cardiol. 2004;44:117–125. doi: 10.1016/j.jacc.2004.03.043. [DOI] [PubMed] [Google Scholar]
- 17.Moss AJ, Zareba W, Kaufman ES, Gartman E, Peterson DR, Benhorin J, Towbin JA, Keating MT, Priori SG, Schwartz PJ, Vincent GM, Robinson JL, Andrews ML, Feng C, Hall WJ, Medina A, Zhang L, Wang Z. Increased risk of arrhythmic events in long-QT syndrome with mutations in the pore region of the human ether-a-go-go-related gene potassium channel. Circulation. 2002;105:794–799. doi: 10.1161/hc0702.105124. [DOI] [PubMed] [Google Scholar]
- 18.Akhavan A, Atanasiu R, Noguchi T, Han W, Holder N, Shrier A. Identification of the cyclic-nucleotide-binding domain as a conserved determinant of ion-channel cell-surface localization. J Cell Sci. 2005;118:2803–2812. doi: 10.1242/jcs.02423. [DOI] [PubMed] [Google Scholar]
- 19.Cui J, Kagan A, Qin D, Mathew J, Melman YF, McDonald TV. Analysis of the cyclic nucleotide binding domain of the HERG potassium channel and interactions with KCNE2. J Biol Chem. 2001;276:17244–17251. doi: 10.1074/jbc.M010904200. [DOI] [PubMed] [Google Scholar]
- 20.Chen J, Zou A, Splawski I, Keating MT, Sanguinetti MC. Long QT syndrome-associated mutations in the Per-Arnt-Sim (PAS) domain of HERG potassium channels accelerate channel deactivation. J Biol Chem. 1999;274:10113–10118. doi: 10.1074/jbc.274.15.10113. [DOI] [PubMed] [Google Scholar]
- 21.Dumaine R, Wang Q, Keating MT, Hartmann HA, Schwartz PJ, Brown AM, Kirsch GE. Multiple mechanisms of Na+ channel-linked long-QT syndrome. Circ Res. 1996;78:916–924. doi: 10.1161/01.res.78.5.916. [DOI] [PubMed] [Google Scholar]
- 22.An RH, Wang XL, Kerem B, Benhorin J, Medina A, Goldmit M, Kass RS. Novel LQT-3 mutation affects Na+ channel activity through interactions between alpha-and beta1-subunits. Circ Res. 1998;83:141–146. doi: 10.1161/01.res.83.2.141. [DOI] [PubMed] [Google Scholar]
- 23.Wehrens XH, Abriel H, Cabo C, Benhorin J, Kass RS. Arrhythmogenic mechanism of an LQT-3 mutation of the human heart Na(+) channel alpha-subunit: A computational analysis. Circulation. 2000;102:584–590. doi: 10.1161/01.cir.102.5.584. [DOI] [PubMed] [Google Scholar]
- 24.Brink PA, Crotti L, Corfield V, Goosen A, Durrheim G, Hedley P, Heradien M, Geldenhuys G, Vanoli E, Bacchini S, Spazzolini C, Lundquist AL, Roden DM, George AL, Jr, Schwartz PJ. Phenotypic variability and unusual clinical severity of congenital long-QT syndrome in a founder population. Circulation. 2005;112:2602–2610. doi: 10.1161/CIRCULATIONAHA.105.572453. [DOI] [PubMed] [Google Scholar]
- 25.Crotti L, Spazzolini C, Schwartz PJ, Shimizu W, Denjoy I, Schulze-Bahr E, Zaklyazminskaya EV, Swan H, Ackerman MJ, Moss AJ, Wilde AA, Horie M, Brink PA, Insolia R, De Ferrari GM, Crimi G. The common long-QT syndrome mutation KCNQ1/A341V causes unusually severe clinical manifestations in patients with different ethnic backgrounds: toward a mutation-specific risk stratification. Circulation. 2007;116:2366–2375. doi: 10.1161/CIRCULATIONAHA.107.726950. [DOI] [PubMed] [Google Scholar]
- 26.Rossenbacker T, Mubagwa K, Jongbloed RJ, Vereecke J, Devriendt K, Gewillig M, Carmeliet E, Collen D, Heidbüchel H, Carmeliet P. Novel mutation in the Per-Arnt-Sim domain of KCNH2 causes a malignant form of long-QT syndrome. Circulation. 2005;111:961–968. doi: 10.1161/01.CIR.0000156327.35255.D8. [DOI] [PubMed] [Google Scholar]
- 27.Crotti L, Lundquist AL, Insolia R, Pedrazzini M, Ferrandi C, De Ferrari GM, Vicentini A, Yang P, Roden DM, George AL, Jr, Schwartz PJ. KCNH2-K897T is a genetic modifier of latent congenital long-QT syndrome. Circulation. 2005;112:1251–1258. doi: 10.1161/CIRCULATIONAHA.105.549071. [DOI] [PubMed] [Google Scholar]
- 28.Shimizu W. The long QT syndrome: therapeutic implications of a genetic diagnosis. Cardiovasc Res. 2005;67:347–56. doi: 10.1016/j.cardiores.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 29.Moss AJ, Robinson JL, Gessman L, Gillespie R, Zareba W, Schwartz PJ, Vincent GM, Benhorin J, Heilbron EL, Towbin JA, Priori SG, Napolitano C, Zhang L, Medina A, Andrews ML, Timothy K. Comparison of clinical and genetic variables of cardiac events associated with loud noise versus swimming among subjects with the long QT syndrome. Am J Cardiol. 1999;84:876–879. doi: 10.1016/s0002-9149(99)00458-0. [DOI] [PubMed] [Google Scholar]
- 30.Priori SG, Napolitano C, Schwartz PJ, Grillo M, Bloise R, Ronchetti E, Moncalvo C, Tulipani C, Veia A, Bottelli G, Nastoli J. Association of long QT syndrome loci and cardiac events among patients treated with β-blockers. JAMA. 2004;292:1341–1344. doi: 10.1001/jama.292.11.1341. [DOI] [PubMed] [Google Scholar]
- 31.Schwartz PJ, Priori SG, Cerrone M, Spazzolini C, Odero A, Napolitano C, Bloise R, De Ferrari GM, Klersy C, Moss AJ, Zareba W, Robinson JL, Hall WJ, Brink PA, Toivonen L, Epstein AE, Li C, Hu D. Left cardiac sympathetic denervation in the management of high-risk patients affected by the long-QT syndrome. Circulation. 2004;109:1826–1833. doi: 10.1161/01.CIR.0000125523.14403.1E. [DOI] [PubMed] [Google Scholar]
- 32.Zareba W, Moss AJ, Daubert JP, Hall WJ, Robinson JL, Andrews M. Implantable cardioverter defibrillator in high-risk long QT syndrome patients. J Cardiovasc Electrophysiol. 2003;14:337–341. doi: 10.1046/j.1540-8167.2003.02545.x. [DOI] [PubMed] [Google Scholar]
- 33.Zareba W, Goldenberg I, Moss AJ, Daubert J, McNitt S, Polonsky S, Mykins M. Efficacy and safety of implantable cardioverter-defibrillator therapy in long QT syndrome patients. Circulation (abstract) 2006;114(II):560–561. [Google Scholar]
- 34.Yan GX, Antzelevitch C. Cellular basis for the normal T wave and the electrocardiographic manifestations of the long QT syndrome. Circulation. 1998;98:1928–1936. doi: 10.1161/01.cir.98.18.1928. [DOI] [PubMed] [Google Scholar]
- 35.Compton SJ, Lux RL, Ramsey MR, Strelich KR, Sanguinetti MC, Green LS, Keating MT, Mason JW. Genetically defined therapy of inherited long-QT syndrome. Correction of abnormal repolarization by potassium. Circulation. 1996;94:1018–1022. doi: 10.1161/01.cir.94.5.1018. [DOI] [PubMed] [Google Scholar]
- 36.Etheridge SP, Compton SJ, Tristani-Firouzi M, Mason JW. A new oral therapy for long QT syndrome: long-term oral potassium improves repolarization in patients with HERG mutations. J Am Coll Cardiol. 2003;42:1777–1782. doi: 10.1016/j.jacc.2003.07.006. [DOI] [PubMed] [Google Scholar]
- 37.Schwartz PJ, Priori SG, Locati EH, Napolitano C, Cantù F, Towbin JA, Keating MT, Hammoude H, Brown AM, Chen LS. Long QT syndrome patients with mutations on the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate. Implications for gene-specific therapy. Circulation. 1995;92:3381–3386. doi: 10.1161/01.cir.92.12.3381. [DOI] [PubMed] [Google Scholar]
- 38.Stramba-Badiale M, Priori SG, Napolitano C, Locati EH, Viñolas X, Haverkamp W, Schulze-Bahr E, Goulene K, Schwartz PJ. Gene-specific differences in the circadian variation of ventricular repolarization in the long QT syndrome: a key to sudden death during sleep? Ital Heart J. 2000;1:323–328. [PubMed] [Google Scholar]
- 39.Moss AJ, Zareba W, Hall WJ, Schwartz PJ, Crampton RS, Benhorin J, Vincent GM, Locati EH, Priori SG, Napolitano C, Medina A, Zhang L, Robinson JL, Timothy K, Towbin JA, Andrews ML. Effectiveness and limitations of beta-blocker therapy in congenital long-QT syndrome. Circulation. 2000;101:616–623. doi: 10.1161/01.cir.101.6.616. [DOI] [PubMed] [Google Scholar]
- 40.Schwartz PJ, Priori SG, Locati EH, Napolitano C, Cantu F, Towbin JA, Keating MT, Hammoude H, Brown AM, Chen LS. Long QT syndrome patients with mutations on the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate. Implications for gene-specific therapy. Circulation. 1995;92:3381–3386. doi: 10.1161/01.cir.92.12.3381. [DOI] [PubMed] [Google Scholar]
- 41.Ruan Y, Liu N, Bloise R, Napolitano C, Priori SG. Gating properties of SCN5A mutations and the response to mexiletine in long-QT syndrome type 3 patients. Circulation. 2007;116(10):1137–1144. doi: 10.1161/CIRCULATIONAHA.107.707877. [DOI] [PubMed] [Google Scholar]
- 42.Benhorin J, Taub R, Goldmit M, Kerem B, Kass RS, Windman I, Medina A. Effects of flecainide in patients with new SCN5A mutation: mutation-specific therapy for long-QT syndrome? Circulation. 2000;101:1698–1706. doi: 10.1161/01.cir.101.14.1698. [DOI] [PubMed] [Google Scholar]
- 43.Windle R, Geletka RC, Moss AJ, Zareba W, Atkins DL. Normalization of ventricular repolarization with flecainide in long QT syndrome patients with SCN5A: DeltaKPQ mutation. Ann Noninvasive Electrocardiol. 2001;6:153–158. doi: 10.1111/j.1542-474X.2001.tb00100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Song Y, Shryock JC, Wu L, Belardinelli L. Antagonism by ranolazine of the proarrhythmic effects of increasing late INa in guinea pig ventricular myocytes. J Cardiovasc Pharmacol. 2004;44:192–199. doi: 10.1097/00005344-200408000-00008. [DOI] [PubMed] [Google Scholar]
- 45.Moss AJ, Zareba W, Schwarz KQ, Rosero S, McNitt S, Robinson JL. Ranolazine shortens repolarization in patients with sustained inward sodium current due to type-3 Long QT Syndrome. J Cardiovasc Eletrophysiol. 2008 doi: 10.1111/j.1540-8167.2008.01246.x. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]