

Abstract
The fluorescence signal of single organic fluorophores is characterized by random blinking and irreversible photobleaching. Photoinduced blinking of Cy5 has posed various limitations of this popular near-infrared (NIR) probe in biological applications. Here we show that fluorophore–metal nanoparticle (NP) complexes greatly suppress Cy5 blinking and noticeably reduce photobleaching events. The blinking behavior of single Cy5 molecules was investigated and compared in the absence and the presence of silver nanostructures. A power-law distribution of off time population was observed for single Cy5 molecules. Average off times were compared to evaluate the plasmonic effect of silver nanoparticles on the triplet decay rates. We furthermore demonstrate enhanced photostability in the presence of silver NPs. The results show that plasmonic-controlled fluorescence can lead to a novel physical mechanism to enhance fluorescence intensity, reduce blinking, and increase photostability.
Introduction
Single molecule detection (SMD)1–4 and fluorescence correlation spectroscopy (FCS)5–7 experiments are usually performed on organic fluorophores with high extinction coefficients and high quantum yields. Fluorescence emission of such single fluorophores is characteristically intermittent (blinking). Thus, the continuous stream of emitted photons observed on long time scales is interrupted by so-called dark, or off, periods, i.e., a state which does not fluoresce, either because it does not adsorb efficiently or because its fluorescence yield is too low. As a result, the single emitter is found to be intermittent: bright and dim/dark intervals randomly follow each other. In ensemble measurements, this behavior is always hidden because the fluctuations of individual objects are not synchronized. For single-molecule studies both in vitro and in vivo, fluorophores with high emission rates and excellent photostability must be identified that are nearly devoid of blinking on all related time scales, while maintaining small overall sizes.
Several possible mechanisms have been discussed to explain the fluorescence intermittency and the nature of long-lasting states in organic fluorophores.8–14 One well-studied source of on–off intermittency in single fluorophores is intersystem crossing from the single excited state (on state) to the triplet state (off state).11 The fluorescence yield of the molecule in the triplet state is usually low. This suggests that the triplet state is dark. The fluorescence trace presents dark periods lasting for the triplet lifetime on average. Besides fast triplet blinking, particularly long-lived dark states with typical off times longer than triplet lifetime have been reported.15,16 Depending on the molecule, this time can be as short as microseconds and as long as tens of milliseconds for normal organic dyes. The second possibility is charge-transfer.10,12 The molecule may react with its environment and accept an electron from its environment or donate one. The remaining radical ion has a very low fluorescence yield. Another blinking mechanism could be conformational changes.17 A typical example is cis–trans isomerization, which is commonly found in cyanine dyes.13,18 These photochemical reactions often require significant activation relating to large atom rearrangements. Unlike triplet state blinking, dark states due to photoisomers can last from milliseconds to seconds. Some recent studies have shown that local environments play a key role in this blinking phenomenon and can affect the emission properties of organic fluorophores.14,19 Single fluorophores show various blinking behaviors in different polymer matrixes.14 Surrounding the fluorophores with the thiol groups can also suppress the blinking.19 The suppression is most probably due to the passivation of surface trap by ligand groups.
Additionally, the observability of single organic fluorophores is limited by their photostability and brightness. The typical observation times for single fluorophores only last several seconds under continuous illumination, followed by permanent photobleaching. The molecule lost fluorescence permanently due to the irreversible photochemical destruction occurring at the excited state. All organic fluorophores photobleach at room temperature and there is a need to screen and design probes which are able to reduce photobleaching.
The use of fluorophore–metal interactions has the potential to dramatically increase the detectability of single fluorophores for both SMD and FCS experiments.20–22 It is now agreed that the metal-enhanced fluorescence occurs via through space near-field interaction of fluorophore with the metal substrate, which can be described as localizing a dipole fluorophore in the electric field near a metal particle.23–25 Our previous measurements have shown increased quantum yields, decreased lifetimes, and increased photostabilities.23,26,27 Decreased lifetimes should result in higher photostability because there is less time for chemical reactions to occur in the excited state. These effects will provide longer observation times prior to photobleaching. This approach to obtain stronger fluorescence signals promises significant progress for experimental schemes where weak fluorescence signals need to be retrieved with high signal-to-noise ratios such as single-molecule methods, fluorescence correlation spectroscopy, and biosensors.
In the present paper, we discussed the effects of nearby silver particles on the blinking and photostability of a Cy5-labeled DNA oligomer. These studies would increase our understanding of fluorophore–metal interaction which will allow conjugates of fluorophore and metallic nanostructure to be utilized in a variety of biophysical studies or high-sensitivity assay applications.28 Novel probes can also be generated on the basis of these effects.
Experimental Section
All reagents and spectroscopic grade solvents were used as received. The oligonucleotides labeled with biotin, Cy5 (Biotin-3′-AGG-TGT-ATG-ACC-GGT-AGA-AG-5′-Cy5) were obtained from the Biopolymer Shared Service at the University of Maryland, School of Medicine. Nanopure water (>18.0 MΩ), purified using the Millipore Milli-Q gradient system, was used for all experiments. All buffer components were purchased from Sigma-Aldrich (St. Louis, MO) and used as received. The coverslips (18 × 18 μm2, Corning) used in the experiments were first soaked in a 10:1 (v/v) mixture of concentrated H2SO4 and 30% H2O2 overnight, extensively rinsed with water, sonicated in absolute ethanol for 2 min, and dried with air stream. The purity was checked by fluorescence measurements at single molecule levels.
Immobilization of Cy5-Labeled Oligonucleotides on Silver Island Films (SIFs)
SIFs were deposited on cleaned glass coverslips by reduction of silver nitrate as reported previously.22,29 Only one side of each slide was coated with SIF. The particles are typically 100–500 nm across and 70 nm high, covering about 20% of the surface. The immobilization process was described elsewhere. Briefly, the cy5-labeled oligonucleotide was hybridized to the complementary thiolated oligonucleotide. After incubation, labeled DNA molecules were tethered to SIFs on an aminated glass substrate.
Preparation of Fluorophore/Nanoparticle Complex (Cy5–SNP)
N-(2-Mercapto-propionyl)glycine (abbreviated as tiopronin)-coated silver nanoparticles were prepared by chemical reduction of silver nitrate using ascorbic acid as previously described.30 The size-controlled silver particles were succinimidylated by ligand exchange. Aminated oligonucleotides were chemically attached to the succinimidylated silver particles by condensation between the amino moieties on the oligonucleotides and the terminal succinimidyl ester moieties on the silver particles. The fluorophore-labeled complementary oligonucleotides were bound to the metal particles by hybridization with the particle-bound oligonucleotides (molar ratio 1:1). The unbound oligo left in the solution was removed by centrifugation and washing. Transmission electron micrographs of nanoparticles were taken using a side-entry Philips electron microscope at 120 keV. Samples were cast from water solutions onto standard carbon-coated (200–300 Å) Formvar films on copper grids (200 mesh) by placing a droplet of a 1 mg/mL aqueous sample solution on grids. The size distribution of metal core was analyzed with Scion Image. Transmission electron microscopy (TEM) images depict the average diameter of metal cores as ca. 50 nm on the basis of at least 50 image counts. In order to avoid multiple labeling on the single metal particle, the molar ratio of succinimidyl ligand and metal particle was controlled to be 0.5. It assures that most of single nanoparticles are conjugated to a single fluorophore.
Single-Molecule Experiments
All single-molecule studies were performed using time-resolved confocal microscopy (MicroTime 200, PicoQuant). Immobilization of nanoparticles on glass coverslips was achieved by casting 20 μL of a 100× diluted Cy5 nanoparticle suspension onto an amino-silanized coverslip following by spin-drying at 4000 rpm. A single-mode pulsed laser diode (635 nm, 100ps, 40 MHz) (PDL800, PicoQuant) was used as the excitation light. The collimated laser beam was spectrally filtered by an excitation filter (D637/10, Chroma) before directing into an inverted microscope (Olympus, IX 71). An oil immersion objective (Olympus, 100×, 1.3NA) was used both for focusing laser light onto the sample and collecting fluorescence emission from the sample. The fluorescence that passed a dichroic mirror (Q655LP, Chroma) was focused onto a 75 μm pinhole for spatial filtering to reject out-of-focus signals and then reached the single-photon avalanche diode (SPAD) (SPCM-AQR-14, Perkin-Elmer, Inc). Images were recorded by raster scanning (in a bidirectional fashion) the sample over the focused spot of the incident laser with a pixel integration of 0.6 ms. The excitation power into the microscope was maintained around 1.4 kW/cm2. Time-dependent fluorescence data were collected with a dwell time of 50 ms. The surface immobilized samples were air-dried before use. All measurements were performed in a dark compartment at room temperature. The off periods can be directly measured by placing an appropriate threshold in an intensity histogram of the fluorescence emission.
Results and Discussion
Single-Molecule Blinking
Figure 1 displays representative fluorescence images recorded from this experiment. The well-separated bright spots are attributed to fluorescence from single Cy5 molecules, as deduced from the discrete single-step photobleaching events observed in the time-dependent studies. As depicted in Figure 1a, some blinking effects of free Cy5 probes can already be identified from the image (circles in Figure 1a). Some spots show dark stripes, demonstrating a discrete on–off–on switching process. This blinking is a characteristic behavior of single cyanine dyes. The intensity variations among the molecules may be due to diverse molecular orientations and environments. In contrast, the typical images of single fluorophores coupling to SIFs or SNPs show well-defined spots as displayed in Figure 1b,c. Such spots are ascribed to single fluorophores bound to silver nanoparticles, which is confirmed by several criteria:22,31,32 (1) The spot size is about the diffraction-limited size of the laser focus in this experiment. (2) The signal intensity is consistent with that expected for single-molecule emission considering the detection efficiency. (3) The spot density in the image is proportional to the immobilized dye concentration. (4) The photobleaching process occurs in a single step with an abrupt drop to the background level. We occasionally observe intense intensity fluctuations and “nondestructive” blinking, which has been ascribed to silver nanoclusters under continuous illumination.22 Only those spots that displayed a single-step photobleaching were assumed to the single molecules in this experiment. Obviously, silver nanoparticles gave rise to a strong fluorescence enhancement, resulting in much brighter spots over the confocal image, which show consistent emission rates during the illumination.
Figure 1.

Fluorescence images (10 μm × 10 μm) of (a) single Cy5 labeled DNA oligo molecules spin-coated on a glass coverslip (white circles indicate some blinking effect), (b) single Cy5 labeled DNA oligo molecules tethered to SIFs, and (c) single Cy5 labeled DNA oligo molecules coupled to single silver nanoparticles. Illumination intensity at 633 nm, 1.4 kW/cm2.
Images shown above were recorded principally to locate individual single molecules for additional time-dependent analysis. Typical emission intensity time transients of single Cy5 molecules are presented in Figure 2. The first example corresponds to the characteristic behavior of a free Cy5 fluorophore on the glass substrate. The free Cy5 trace clearly displays fast emission intensity fluctuations with several long dark durations. The right-hand panel in Figure 2a shows an intensity histogram before photobleaching occurred. Periods of background fluorescence are clearly visible, and two intensity levels are clearly noticeable. Similar blinking events with a period lasting from milliseconds to a few seconds have been observed for other Cy5 molecules on glass substrates in the experiment. In contrast, in the presence of silver nanostructure, the off states appear rather infrequently, as presented in Figure 1b. In Figure 2c, fast fluorescence blinking appears to be completely eliminated for Cy5 bound to silver colloids. Fluorescence intensity histograms right next to time traces in Figure 2b,c show narrow distributions and can be fitted well with Gaussian.
Figure 2.

Typical time traces of (a) single Cy5 labeled DNA oligo molecules spin-cast on a glass coverslip, (b) single Cy5 labeled DNA oligo molecules tethered to SIFs, and (c) single Cy5 labeled DNA oligo molecules coupled to single silver nanoparticles. Right panels show intensity frequency histograms from single time transients before photobleaching.
The blinking behavior of single fluorophores can provide useful information on the photophysics underlying by analyzing the distribution or duration of on/off times. To examine blinking on the large time scale of many seconds, a threshold was set at a level of 2-fold standard deviation with respect to the mean background during the off time histogram analysis to distinguish between the states as described previously.11,31–33 To characterize the fluorescence blinking of fluorophore probes on various substrates, the off-time histogram was constructed from an emission time transient with 1 ms bin time. An insufficient number of an off-time histogram from a single molecule could render the statistical analysis, and the combined off-time distribution for more than 50 molecules is used herein. The log–log distribution can be fitted to a straight line. (Figure 3a) We can described this off-time distribution using a power law function p(τoff) = p0τoff−αoff, with αoff being the power law exponent. The single molecule power exponent for the off-time statistics is sensitive to the molecule environment and/or the molecule structure.10 It is obvious from the linear log–log plot that the observed off times cover a time range and is described using a power law distribution. The αoff value of 1.74 is in agreement with other organic fluorophores obtained on glass substrates.9
Figure 3.

(a) Log–log off-time distribution compiled from single Cy5 molecules on glass substrate. The solid line is the best fit with a power law exponent αoff = 1.74. Histograms of average off times of (b) single fluorophores on bare-glass substrates, (c) single fluorophores immobilized on silvered surfaces and (d) single fluorophores coupled to silver nanoparticles.
Several processes have been discussed to explain the occurrence of power-law kinetics in single fluorophore intermittency.9,15,16 Cy5 dye molecules tend to be strictly immobilized on glass surfaces, thus hampering any molecular orientation. Rotational motion therefore plays a trivial role in effecting fluorescence intensity fluctuations. The broad distribution of off-time durations can be explained using a charge separation model recently employed in dye molecules.11 In this case, an electron is postulated to be transferred from Cy5 to a nearby acceptor (i.e., charge traps) on the glass matrix is involved in switching fluorescence off. Subsequent charge recombination by back-charge tunneling from the trap to the Cy5 cation restores the fluorescence. After expulsion of an electron or hole from the singlet excited or the triplet state, the remaining photoinduced radical ion either cannot be excited by the incident photons or returns to its ground state via a nonradiative pathway.
In the presence of silver nanostructures, a large fraction of molecule studies (>70%) did not display any long off times of interest. As showed in Figure 1c, we do not observe dark states during the 60-s observation time. The fluorescence intensity for this particular fluorophore–SNP complex in this example is well above the noise level at all times. We want to address the long-life dark states are rare events in the cases of fluorophore coupling to nanoparticles. A large number of the fluorophore–SNP complexes rarely blink. It is not easy to obtain off-time distributions for individual molecules and therefore a more appropriate description of the blinking statistics is to calculate the average duration of off times. Average off times were calculated as the sum of all detected off events by their number from the beginning of a time trace to the last detected on period in order to avoid contributions from irreversible photobleaching. Figure 3 shows histograms of the average off times compiled from 50 to 70 molecules for Cy5/SIF (gray bars) and Cy5/SNP (black bars) compared with those from Cy5 immobilized on glass substrates (white bars). With an exposure time of 60 s, free Cy5 molecules exhibit average off times sparsely in the 0–10 ms range. This behavior differs significantly from those observed from samples in the presence of metallic nanostructures. For Cy5 tethered to SIFs, ~50% of single molecules shows an average off time less than 1 ms, while more than 70% of a single Cy5–SNP complex shows an average off time in this regime. The difference in the distribution of average times is clearly evident.
The large disparity of average off-time distributions we observed in Figure 3 is likely caused by the interaction with the surrounding metallic nanostructure. The metallic structure with the sub-wavelength size usually displays an energy resonance arising from the collective oscillation of migrated electrons on its surfaces, which is defined as plasmon resonance.34,35 The radiating energy from the fluorophore is dramatically altered through coupling with the metal plasmon resonance to cause a change of the emission properties, which is referred to as radiative decay engineering (RDE).20 These energy resonances express as absorbance, excitation, and scattering and induce strongly enhanced local electromagnetic fields in a near-field region spatially overlapping with fluorophores. The highly enhanced scattering fields can be absorbed by the fluorophore. We assume the plasmonic interactions could hamper the charge-separation process.36 Such interactions affect transitions coupled to the electromagnetic field and introduce additional electromagnetic nonradiative channels. This would eliminate the photoinduced electron-transfer process, leading to reducing the frequency of blinking. However, suppression of the electron transfer may not be complete, for instance, due to heterogeneous distributions of plasmon field (ascribed to assorted shapes and sizes of nanostructures), accounting for the remaining blinking events.
Decreasing of Photobleaching
Photobleaching is an irreversible process that is caused by light-induced chemical reactions, which transform the fluorophore into a molecule with significantly decreased emission probability. In single-molecule fluorescence detection, the one-step photobleaching of the observed molecule is a characteristic difference compared to ensemble observation of an apparent decrease of the emission signal as a function of illumination time. Thus, the photobleaching imposes a limit to the useful observation time of a fluorophore. Both a decrease in lifetime and an increase in quantum yield lead to a significant decrease of photobleaching. In Figure 2b,c, the molecule displays nearly constant emission intensity before undergoing irreversible photobleaching after 30 s. The survival time (photobleaching) time histograms of single Cy5 molecules at various configurations are displayed in Figure 4 for consistent excitation intensities in an observation window of 60 s, and only those showed single-step photobleaching within 60 s were counted. For comparison purposes, bin widths of 5 s were used to build the histograms. It is obvious that the bleaching times decay exponentially for free Cy5 molecules immobilized on a glass substrate with an appropriate average bleaching time of 3.7 s by fitting the histogram with a single-exponential function (Figure 4a). As expected, long-surviving molecules can be seen in the presence of metallic nanostructures. The survival time distributions presented in Figure 4b,c reflect considerable differences and frequency characteristics can be categorized into two different distributions. As depicted in Figure 4b, the Gaussian distribution is centered at around 18 s for the SIF samples. The wide distribution obtained on silvered film most clearly illustrates changes in fluorophore spectra properties induced by the varied strength of metallic electromagnetic field, which could be the result of assorted probe proximities to metallic surfaces. It can also be assumed that the spread of surface features accounts for lifetime and intensity variation from molecule to molecule. As for Cy5–SNP complexes, there are two clear populations of survival times, with a predominant survival time of 50 s under continuous illumination. Unfortunately, we are not able to determine the exact origins of this distribution at single-molecule species arising from diverse electromagnetic interactions. TEM images (data not showed) indicate some distribution in particle shapes and sizes, which can play a role in field variations.
Figure 4.
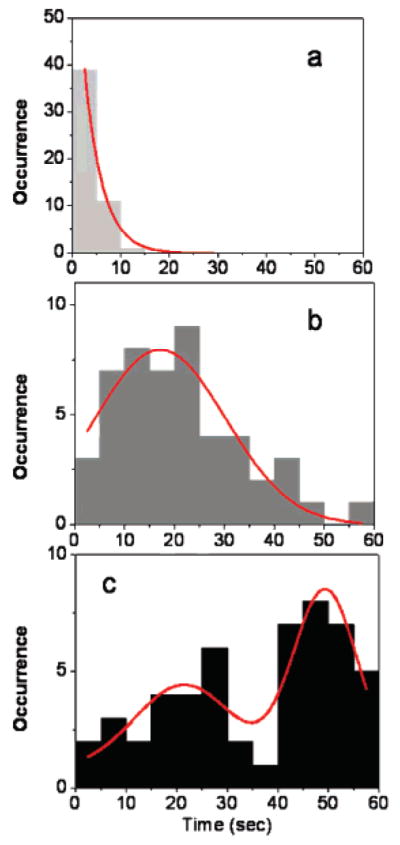
Histograms of survival times for (a) single fluorophores on a glass substrate, (b) single fluorophore immobilized on silvered surfaces, and (c) single fluorophores coupled to silver nanoparticles (red lines are approximate Gaussian fits).
These results show that the suppression of photobleaching occurs in the presence of metallic nanostructures. The longest survival time occurs on a high-frequency basis for Cy5–SNP complexes. The interaction of the dye with silver nanostructures can occur from the excited state (S1). These processes would lead to an increase in the total radiative decay rate of the dye from S1, with a consequent reduction of the excited-state lifetime and increase in emission rate. The rate of photon emission by an excited molecule can be modified by changing the density of possible electromagnetic decay channels. As previously reported, for a given fluorophore–matrix system, a longer stay in the off state leads to a higher probability of photochemistry, i.e., photobleaching.
In conclusion, we demonstrated reduced blinking and decreased photobleaching of fluorophores proximity to SIFs and silver nanoparticles. We ascribe the effects to the modification of spectral properties by the surface plasmon resonance from the metallic nanostructure. In this report, it is important to note that single-molecule measurements may be biased toward measuring those molecules show comparatively longer on times. The relatively low average off time is accompanied by a stronger fluorescence signal. The residence time in the dark state is reduced due to its enhanced decay. This finding may lead to highly photostable fluorophores, which are important for sensitive optical detections.
Acknowledgments
This work was supported by the NCRR, RR-08119 and NHGRI, HG-002655, EB000682, EB0065211 and NIBIB.
References
- 1.Shera EB, Seitzinger NK, Davis LM, Keller RA, Soper SA. Chem Phys Lett. 1990;174(6):553–557. [Google Scholar]
- 2.Molski A, Hofkens J, Gensch T, Boens N, DeSchryver F. Chem Phys Lett. 2000;318:325–332. [Google Scholar]
- 3.Ambrose WP, Goodwin PM, Jett JH, Van Order A, Werner JH, Keller RA. Chem Rev. 1999;99(10):2929–2956. doi: 10.1021/cr980132z. [DOI] [PubMed] [Google Scholar]
- 4.Xie XS, Trautman JK. Annu Rev Phys Chem. 1998;49:441–480. doi: 10.1146/annurev.physchem.49.1.441. [DOI] [PubMed] [Google Scholar]
- 5.Schwille P. Theory and applications. Springer; Berlin: 2001. Fluorescence correlation spectroscopy. [Google Scholar]
- 6.Webb WW. Appl Opt. 2001;40(24):3969–3983. doi: 10.1364/ao.40.003969. [DOI] [PubMed] [Google Scholar]
- 7.Edman L. J Phys Chem A. 2000;104:6165–6170. [Google Scholar]
- 8.Gensch T, Bohmer M, Aramendia PF. J Phys Chem A. 2005;109:6652–6658. doi: 10.1021/jp0510847. [DOI] [PubMed] [Google Scholar]
- 9.Haase M, Hubner CG, Reuther E, Herrmann A, Mullen K, Basche T. J Phys Chem A. 2004;108:10445–10450. [Google Scholar]
- 10.Schuster J, Cichos F, Borczyskowski CV. Optics Spectrosc. 2005;98(5):712–717. [Google Scholar]
- 11.Yeow E, Melnikov S, Bell T, Schryver FCD, Hofkens J. J Phys Chem A. 2006;110:1726–1734. doi: 10.1021/jp055496r. [DOI] [PubMed] [Google Scholar]
- 12.Schuster J, Cichos F, von Borczyskowski C. Appl Phys Lett. 2005;87:051915. [Google Scholar]
- 13.Huang Z, Ji D, Xia A. Colloids Surf, A. 2005;257–258:203–209. [Google Scholar]
- 14.Clifford J, Bell T, Tinnefeld P, Heilemann M, Melnikov S, Yeow E. J Phys Chem B. 2007;111(25):6987–6991. doi: 10.1021/jp072864d. [DOI] [PubMed] [Google Scholar]
- 15.Hoogenboom J, Hernando J, Dijk Ev, Hulst Nv, Garcia-Parajo M. ChemPhysChem. 2007;8:823–833. doi: 10.1002/cphc.200600783. [DOI] [PubMed] [Google Scholar]
- 16.Hoogenboom JP, Dijk EMv, Hernando J, Hulst NFv, Garcia-Parajo MF. Phys Rev Lett. 2005;95:097401. doi: 10.1103/PhysRevLett.95.097401. [DOI] [PubMed] [Google Scholar]
- 17.Huang Z, Ji D, Xia A, Koberling F, Patting M, Erdmann R. J Am Chem Soc. 2005;127:8064–8066. doi: 10.1021/ja050050+. [DOI] [PubMed] [Google Scholar]
- 18.Huang ZJD, Wang S, Xia A, Koberling F, Pattings M, Erdmann R. J Phys Chem A. 2006;110:45–5. doi: 10.1021/jp0562936. [DOI] [PubMed] [Google Scholar]
- 19.Rasnik I, McKinney SA, Ha T. Nat Methods. 2006;3(11):891–893. doi: 10.1038/nmeth934. [DOI] [PubMed] [Google Scholar]
- 20.Lakowicz JR, Malicka J, Gryczynski I, Gryczynski Z, Geddes CD. J Phys D: Appl Phys. 2003;36:R240–249. doi: 10.1088/0022-3727/36/14/203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fu Y, Lakowicz JR. J Phys Chem B. 2006;110:22557–22562. doi: 10.1021/jp060402e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fu Y, Lakowicz JR. Anal Chem. 2006;78:6238–6245. doi: 10.1021/ac060586t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lakowicz JR, Malika J, Gryczynski I, Gryczynski Z, Geddes CD. J Phys D: Appl Phys. 2003;36:R240–R249. doi: 10.1088/0022-3727/36/14/203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Enderlein J. Chem Phys. 1999;247:1–9. [Google Scholar]
- 25.Enderlein J. Biophys J. 2000;78:2151–2158. doi: 10.1016/S0006-3495(00)76761-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malicka J, Gryczynski I, Fang J, Kusba J, Lakowciz JR. J Fluoresc. 2002;12:439–447. doi: 10.1023/A:1021370111590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lakowicz JR, Shen Y, D’ Auria S, Malicka J, Fang J, Gryczynski Z, Gryczynski I. Anal Biochem. 2002;301:111–116. doi: 10.1006/abio.2001.5503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang J, Fu Y, Lakowicz JR. J Phys Chem C. 2007;111:50–56. doi: 10.1021/jp062665e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lukomska JMJ, Gryczynski I, Leonenko Z, Lakowicz JR. Biopolymers. 2005;77:31–37. doi: 10.1002/bip.20179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang J, Fu Y, Chowdlhury M, Lakowicz JR. Nano Lett. 2007;7(7):2101–2107. doi: 10.1021/nl071084d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hou Y, Higgins DA. J Phys Chem B. 2002;106:10306–10315. [Google Scholar]
- 32.Weston KD, Carson PJ, Metiu H, Buratto SK. J Chem Phys. 1998;109(17):7474–7485. [Google Scholar]
- 33.Yip WT, Hu D, Yu J, Bout DAV, Barbara PF. J Phys Chem A. 1998;102:7564–7575. [Google Scholar]
- 34.Antunes PA, Constantino CJL, Aroca RF, Duff J. Langmuir. 2001;17:2958–2964. [Google Scholar]
- 35.Kumbhar AS, Kinnan MK, Chumanov G. J Am Chem Soc. 2005;127:12444–12445. doi: 10.1021/ja053242d. [DOI] [PubMed] [Google Scholar]
- 36.Okamoto K, Vyawahare S, Scherer A. J Opt Soc Am B. 2006;23(8):1674–1677. [Google Scholar]