

Abstract
A series of seven 2-amino-4-oxo-6-substituted thieno[2,3-d]pyrimidines, with bridge length variations (from 2-8 carbon atoms) were synthesized as selective folate receptor (FR) α and β substrates and as antitumor agents. The syntheses were accomplished from appropriate allylalcohols and 4-iodobenzoate to afford the aldehydes which were converted to the appropriate 2-amino-4-carbethoxy-5-substituted thiophenes 23-29. Cyclization with chlorformamidine afforded the thieno[2,3-d]pyrimidines 30-36 which were hydrolyzed and coupled with diethyl-L-glutamate, followed by saponification to give the target compounds 2-8. Compounds 3-6 were potent growth inhibitors (IC50 4.7 to 334 nM) of human tumor cells (KB and IGROV1) that express FRs. In addition, compounds 3-6 inhibited the growth of Chinese hamster ovary (CHO) cells that expressed FRs but not the reduced folate carrier (RFC) or proton-coupled folate transporter (PCFT). However, the compounds were inactive toward CHO cells that lacked FRs but contained either the RFC or PCFT. By nucleoside and 5-amino-4-imidazole carboxamide (AICA) protection studies, along with in vitro and in situ enzyme activity assays, the mechanism of antitumor activity was identified as the dual inhibition of glycinamide ribonucleotide formyltransferase and, likely, AICA ribonucleotide formyltransferase. The dual inhibitory activity of the active thieno[2,3-d]pyrimidine antifolates and the FR specificity represent unique mechanistic features for these compounds distinct from all other known antifolates. The potent inhibitory effects of compounds 3-6 toward cells expressing FRs but not PCFT provide direct evidence that cellular uptake of this series of compounds by FRs does not depend on the presence of PCFT and argues that direct coupling between these transporters is not obligatory.
INTRODUCTION
The biological importance of reduced folates derives from their essential roles in one-carbon transfer leading to thymidylate, purine nucleotides, serine, and methionine, and in biological methylation reactions from S-adenosylmethionine.1 Since mammalian cells are unable to synthesize folates de novo, internalization of extracellular folates is essential. Three major folate uptake systems have been described.2 The ubiquitously expressed reduced folate carrier (RFC) is an anion exchanger and is the primary transport mechanism of folates at physiologic pH.3 The folate receptors (FRs) α and β are glycosylphosphoinositol-anchored proteins that transport folates by receptor-mediated endocytosis.4 The proton-coupled folate transporter (PCFT) is a proton-folate symporter that functions optimally at low pH.5 It has been reported that cellular uptake of (anti)folates by FR involves a functional “coupling” in which (anti)folates bind to surface FRs, followed by internalization and vesicle (endosome) formation, followed by extravesicular export by PCFT.6
Antifolates, typified by methotrexate (MTX), pemetrexed (PMX) and raltitrexed (RTX) (Figure 1) are structurally similar to folic acid and typically bind to folate-dependent enzymes to inhibit folate-dependent pathways.7 MTX continues to be an integral component of the chemotherapeutic arsenal for several cancers including pediatric acute lymphoblastic leukemia, osteogenic sarcoma, lymphoma, and breast cancer.8 RTX is used outside of the US for advanced colorectal cancer.3 PMX was approved in 2004 for pleural mesothelioma in the US,9 and subsequently, as a second line treatment for non-small cell lung cancer.10
Figure 1. Folate and antifolate structures.

Structures are shown for folic acid, depicting the pteridine, p-aminobenzoate, and glutamate motifs, along with structures for classical antifolates, MTX, PMX, RTX, LMX, 1a and 1b and the non-classical antifolate, trimetrexate (TMQ).
MTX is a potent inhibitor of dihydrofolate reductase, whereas PMX and RTX derive their primary cytotoxic effects by inhibiting thymidylate synthase. Lometrexol (LMX) (Figure 1) was introduced in 198511 as a targeted antipurine antifolate. The notion of selectively targeting de novo purine nucleotide biosynthesis has roots in early studies of azaserine12 or thiopurines13 and is based on the assumption that depletion of purine nucleotide pools can limit nucleotides for DNA synthesis and repair, while also impacting ATP and GTP stores important for cellular energetics.14 This effect could be even more acute in several cancer cells that lack enzymes involved in purine salvage.15 Although there have been reports that antipurine antifolates such as LMX were cytostatic rather than cytotoxic,16 in other reports, antipurine antifolates were distinctly cytotoxic.17, 18
While the aforementioned antifolates can be envisaged to possess limited selectivity for tumor cells over normal proliferative tissues such as bone marrow and gastrointestinal mucosa, interest in the development of antifolates remains particularly high for agents that target high affinity folate receptors (FRs).19 This reflects the restricted patterns of tissue expression for FRs, including the vast majority of ovarian and endometrial cancers for FRα and myeloid leukemias for FRβ.20 There are other factors that account for tumor selectivity of FR-targeted therapies, including the apical localization for FRα in normal epithelia such as renal tubules or choroid plexus where it is inaccessible to the circulation, and synthesis of non-functional FRβ in normal hematopoietic cells.4
Ample literature documents applications of FRs for tumor targeting with folic acid as the targeting agent. For instance, cytotoxins (e.g., mitomycin C), liposome-encapsulated drugs (e.g., doxorubicin), or radionuclides have been conjugated to folic acid for targeting FR-expressing tumors.21-23 There are at least two potential complications of this approach. These include: (i) instability in plasma versus that within tumor cells such that the folate conjugate may be prematurely cleaved and release the cytotoxic agent prior to reaching the tumor, resulting in toxicity to normal cells, thus precluding selectivity; and (ii) the possibility that free folic acid released upon cleavage within the tumor could provide a growth-sustaining nutrient detrimental to tumor inhibition. Another approach involves a targeting ligand which itself is cytotoxic. Unfortunately, most folate-based therapeutics such as classical antifolates (including RTX, PMX, and LMX) that are substrates for FRs are also substrates for the ubiquitously expressed RFC resulting in decreased tumor selectivity with these agents.24 Indeed, the lack of continued clinical development of LMX can be directly traced to the severe myelosuppression encountered in a Phase 1 clinical trial,25 at least partly due to its excellent substrate activity for RFC uptake and polyglutamylation by normal tissues. Although classical antifolates can be transported by PCFT5, the role of this transporter in chemotherapy is still emerging.
Clearly, a specific FR-targeted agent that also possesses cytotoxic activity without transport by RFC would circumvent many of the drawbacks of targeting FRs with folic acid-conjugated cytotoxic agents and the lack of selectivity often associated with clinically used antifolates. Indeed, FR selective cytotoxic agents can be envisaged to provide highly selective antitumor agents against tumors expressing FR with little or no host toxicity. Ideally, analogs could be identified that are selective substrates for FRs over RFC. One possibility, 1a, (Figure 1), was reported nearly 30 years ago,26, 27 however, its modest cell growth inhibitory potency and significant toxicity profile limited its further clinical development and led to introduction of RTX.28, 29 More recently, Jackman and colleagues described novel cyclopenta[g]quinazoline antifolates as FR-targeted agents,30,31 which like 1a and RTX are potent inhibitors of human thymidylate synthase. One of these cyclopenta[g]quinazolines was evaluated in mice and showed no systemic toxicity, as reflected in no weight loss or macroscopic signs of toxicity to major organs, consistent with its selective targeting of FRs.
Most recently, we18 described a novel series of 6-substituted classical pyrrolo[2,3-d]pyrimidine antifolates, differing only in the lengths of the carbon bridge (3- to 6-carbons), characterized by potent and selective substrate activities for FRα and FRβ, and negligible substrate activity for RFC. The intracellular enzyme target of the 6-substituted pyrrolo[2,3-d]pyrimidines was identified as glycinamide ribonucleotide formyltransferase (GARFTase), the first folate-dependent reaction in the de novo purine synthesis pathway.
In this study, we extend this focus to a novel isosteric series of 6-substituted thieno[2,3-d]pyrimidine antifolates [compounds 2-8 (Figure 2)], with a 2- to 8-carbon bridge between the thieno[2,3-d]pyrimidine and the benzoyl moiety, respectively. Isosteric replacement of the pyrrolo ring with a thieno ring for this series provides an increase in ring size that more closely approximates the pteridine, 6-6 fused ring system of the natural cofactor. In addition, replacement of the NH of the pyrrole with a S also allows for comparison of the relative importance of a hydrogen bond donor (NH) with a hydrogen bond acceptor (S). Our biological results establish the cytotoxic effects of the active compounds directly reflect potent inhibitory activities against GARFTase and cellular uptake capacities by FRs over RFC and PCFT. From nucleoside protection studies, lesser inhibitory effects of a second enzyme target, most likely 5-amino-4-imidazole carboxamide ribonucleotide formyltransferase (AICARFTase), was strongly implied. The potent cytotoxicity in cells expressing FRs but not PCFT provides direct evidence that cellular uptake of this series of compounds by FRs does not depend on the presence of PCFT and argues that direct coupling between these transporters is not obligatory.
Figure 2. Structures of novel 6-substituted thieno[2,3-d]pyrimidine antifolates.

CHEMISTRY
Target compounds 2-8 were synthesized as shown in Scheme 1. The appropriate allyl alcohols 9-15 were treated with palladium diacetate, ethyl 4-iodobenzoate, LiCl, LiOAc and Bu4NCl in DMF to afford the aldehydes 16-22 (80-85% yield).32 A modified reaction temperature of 80 °C from that reported in the literature improved the yield. Aldehydes 16-22 were then reacted with sulfur, ethyl cyanoactetate and morpholine in EtOH for 24 h at room temperature under Gewald reaction conditions to afford 23-29.33 Cyclization of 23-29 with chlorformamidine hydrochloride afforded the thieno[2,3-d]pyrimidines 30-36 in 55-80% yield. Hydrolysis of the ethyl esters of 30-36 with 1 N NaOH in ethanol followed by acid workup gave the corresponding acids 37-43. With 2-Cl-4,6-dimethoxy-1,3,5-triazine and 4-methylmorpholine as the coupling reagents, acids 37-43 were coupled with diethyl-L-glutamate hydrochloride to afford compounds 44-50 (69-77%). Hydrolysis of 44-50 in 1 N NaOH, followed by acid workup gave target compounds 2-8 in 96-98% yield.
Scheme 1.

Reagents and conditions:
(a) 10%Pd(AcO)2, Bu4NCl,LiCl, LiOAc, DMF 80 °C 12 h.; (b)S, CN-CH2-COOEt,Morpholine R.T. 24 h; (c) Chlorformamidine hydrochloride,DMSO2, 140 °C 4 h; (d) 4-Methylmorpholine, 2-Cl-4,6-dimethoxy-1,3,5-triazine,DMF,diethyl L- glutamate hydrochloride,10 h;
BIOLOGICAL EVALUATION AND DISCUSSION
Confirmation of the lack of PCFT in Chinese hamster ovary sublines and generation of PCFT stable transfectant
We previously described a panel of Chinese hamster ovary (CHO) cell lines engineered from RFC- and FR-null MTXRIIOuaR2-4 (R2) cells to express human FRs (α and β; designated RT16 and D4, respectively) or RFC (PC43-10).18 To confirm the lack of PCFT in the hamster sublines, we screened CHO sublines derived from R2 cells by RT-PCR, using PCR primers to regions in hamster PCFT completely homologous to the human PCFT. While PCFT transcripts were detected in RNA from MDA-MB-231 and HepG2 human cells, endogenous PCFT transcripts were not detected in any of the CHO sublines, in spite of 98% identical sequence to human PCFT (100% for the primer binding sites) (Figure 1S, Supplement).
We used R2 CHO cells as the parental subline for transfection with human PCFT cDNA. Following isolation of a stable clonal subline, R2/PCFT4, cells were assayed for transport and PCFT expression by RT-PCR and results compared to those for R2 cells transfected with empty pCDNA3.1 plasmid (R2/VC) (negative control) (Supplement). While transport of [3H]MTX in R2/VC cells was insignificantly different at pH 5.5 (transport by PCFT is maximal at pH 5.5)5 from that at pH 7.2 (PCFT is essentially inactive at this pH)5 [0.56±0.061 (SEM) and 0.466 ± 0.076 pmol/mg/2 min, respectively; p=0.4130, n=8], transport was markedly elevated in R2/PCFT4 cells [6.96 ± 0.43 pmol/mg/2 min (p=0.0001; n=8) (Figure 3).
Figure 3. PCFT-mediated transport of [3H]methotrexate at pH 5.5.
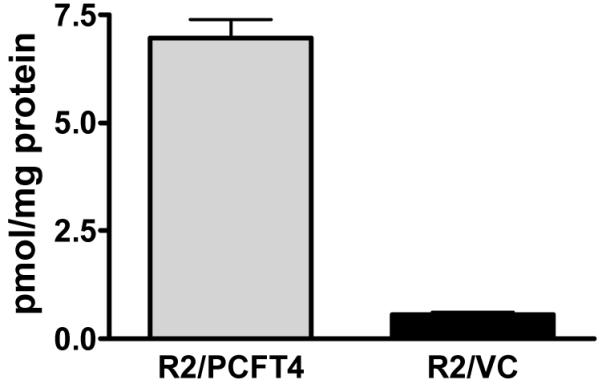
R2/PCFT4 and R2/VC cells were assayed for uptake of [3H]MTX (0.5 μM) at pH 5.5 in MES-buffered saline at 37° C over 2 min. Results are presented as mean values ± SEM (n=8).
Identification of 6-substituted thieno[2,3-d]pyrimidine antifolates as potent growth inhibitors for cells expressing FRs, but not for cells expressing exclusively RFC or PCFT
Compounds 2-8, containing 2- to 8-methylenes in the bridge region, were initially evaluated for their growth inhibitory effects during a 4 day exposure against the series of isogenic CHO sublines expressing the invidual folate transporters including FRα (RT16), FRβ (D4), RFC (PC43-10), or PCFT (R2/PCFT4). Growth inhibitory effects were compared to those for the classical antifolate inhibitors MTX, LMX, PMX, and RTX. For PC43-10 and R2/PCFT4 cells, results were compared to those for R2 or R2/VC cells, respectively, with identical results, whereas FR-effects in RT16 and D4 cells were established from parallel incubations in the presence of excess (200 nM) folic acid which blocked FR-mediated cellular uptake18. An identical series of experiments was performed with RFC-, PCFT-, and FRα-expressing KB and IGROV1 human tumor cells. The FR- binding levels and RFC transport activities for all these models were previously reported.18 The PCFT levels in KB and IGROV1 human solid tumor cell lines are significant and will be reported elsewhere (S. Kugel Desmoulin, Y. Wang, A. Gangjee, and L.H. Matherly manuscript in preparation).
All of the classical antifolates (MTX, LMX, PMX and RTX) were active against CHO sublines expressing RFC-, PCFT-, or FRs (Table 1). In RT16 and D4 cells, growth inhibitory effects were abolished by excess folic acid. For KB and IGROV1 cells, folic acid had at most a marginal effect on drug activity since the antifolates could still enter cells via RFC which is not blocked by folic acid.
Table 1. IC50s (nM) for thienopyrimidine compounds 2 to 8 in cell proliferation inhibition of RFC- and FR-expressing cell lines.
| Antifolate | RFC | PCFT | FRα | FRβ | RFC/FRα | RFC/FRα | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PC43-10 | R2 | R2/PCFT4 | R2/VC | RT16 | RT16 (+FA) |
D4 | D4 (+FA) |
KB | KB (+FA) |
IGROV1 | IGROV1 (+FA) |
|
| 2 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 |
| 3 | >1000 | >1000 | >1000 | >1000 | 13(3.4) | >1000 | 112(12) | >1000 | 23(5.5) | >1000 | 4.7(1.9) | >1000 |
| 4 | >1000 | >1000 | >1000 | >1000 | 9(2.9) | >1000 | 20(3.9) | >1000 | 4.9(1.3) | >1000 | 5.9(1.9) | >1000 |
| 5 | >1000 | >1000 | >1000 | >1000 | 56(9.8) | >1000 | 56(13) | >1000 | 132(23) | >1000 | 25(1.7) | >1000 |
| 6 | >1000 | >1000 | >1000 | >1000 | 108(17) | >1000 | 364(46) | >1000 | 310(54) | >1000 | 334(59) | >1000 |
| 7 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 |
| 8 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 |
| Methotrexate | 12(1.1) | 216(8.7) | 120.5(16.8) | >1000 | 114(31) | 461(62) | 106(11) | 211(43) | 6.0(0.6) | 20(2.4) | 21(3.4) | 22(2.1) |
| Pemetrexed | 138(13) | 894(93) | 13.2(2.4) | 974.0(18.1) | 42(9) | 388(68) | 60(8) | 254(78) | 68(12) | 327(103) | 102(25) | 200(18) |
| Raltitrexed | 6.3(1.3) | >1000 | 99.5(11.4) | >1000 | 15(5) | >1000 | 22(10) | 746(138) | 5.9(2.2) | 22(5) | 12.6(3.3) | 20(4.3) |
| Lometrexol | 12(2.3) | >1000 | 248.0(18.2) | >1000 | 12(8) | 188(41) | 2.6(1.0) | 275(101) | 1.2(0.6) | 31(7) | 3.1(0.9) | 16(6) |
| 1b | 11(3.3) | >1000 | >1000 | >1000 | 277(81) | >1000 | 52(12) | >1000 | 5.8(3.5) | 32(15) | 5.2(1.7) | 6.9(1.6) |
Growth inhibition assays were performed as described in the Experimental Section .For the FR experiments, growth inhibition assays were performed in the absence and presence of 200 nM folic acid (FA). The data shown are mean values from three experiments (plus/minus SEM in parentheses). For RFC- and FR-expressing cells, IC50 data of classical antifolate compounds, methotrexate, pemetrexed, raltitrexed, lometrexol, and 1b, were previously published from our laboratory.18 ND, not determined.
Compound 2 (2-carbon bridge) was uniformly inactive (IC50 > 1000 nM) in our assays. Compounds 3-6 (3- to 6-carbon bridge) were inhibitors of proliferation for CHO cells expressing either FRα or FRβ, but not with RFC-expressing PC43-10, PCFT-expressing R2/PCFT4 cells, or R2 or R2/VC cells without detectable FR, RFC, or PCFT (Table 1). The most potent analogs of the series, compounds 3 and 4, with 3- and 4-methylene groups in the bridge region, respectively, showed activities similar to those for the most active classical inhibitors, LMX and RTX, toward FR-expressing cells but were somewhat more active than either PMX or MTX. There was a progressive diminution of the growth inhibitory effects for the 5-, 6-, 7- and 8-methylene analogs (compounds 5 to 8, respectively). Analogous results were obtained with KB and IGROV1 human tumor cells (Table 1). For compounds 3-6, growth inhibitory effects were abolished with 200 nM folic acid indicating a requirement for FR uptake for these compounds.
Similar, albeit slightly less potent effects of compounds 3-5 and MTX were detected by colony-forming assays with KB cells over long term (10 day) continuous exposures to drug (Figure 4). For the thieno[2,3-d]pyrimidine antifolates (but not MTX), the effects were completely reversed by an excess of folic acid, establishing a requirement of FRs for transport of the thieno[2,3-d]pyrimidines. These results establish that compounds 3-5 are cytotoxic toward KB human tumor cells.
Figure 4. Colony-forming inhibition assay.

KB cells were plated into 60 mm dishes with a density of 100 cells per dish, in the presence or absence of different concentrations of antifolates (compounds 3-5 and MTX) from 1 to 1000 nM. The highest concentration (1000 nM) of inhibitors was also evaluated in the presence or absence of folic acid (100 μM). Colonies were enumerated after 10 days using a cut-off of 50 cells/colony. Data were calculated as percent of controls treated in an identical fashion but without drugs.
Protection from the growth inhibitory effects of thieno[2,3-d]pyridimidine antifolates with nucleosides
To localize the probable enzyme target(s) for the thieno[2,3-d]pyrimidine series that results in suppression of cell proliferation, we tested the growth inhibitory effects of compound 4, the most potent of our analogs, against KB cells in the presence of adenosine (60 μM) or thymidine (10 μM). Thymidine (10 μM) did not alter the growth inhibitory effects of compound 4, whereas adenosine (60 μM) was completely protective (Figure 5). This establishes the de novo purine biosynthetic pathway as the primary metabolic target.
Figure 5. Protection of cell growth inhibition by nucleosides, AICA, and folic acid.

Cell proliferation inhibition by thieno[2,3-d]pyrimidine compound 4 was measured on 96-well plates with 4000 KB cells per well initially and a range of antifolate concentrations (1-1000 nM), in the presence or absence of folic acid (200 nM), adenosine (60 μM); thymidine (10 μM), or AICA (320 μM). Cell densities were measured with CellTiter Blue fluorescence dye and a fluorescence plate reader. Results were normalized to cell density in the absence of drug. Results shown are representative data of experiments performed in triplicate.
Since there are two folate-dependent enzymes in the de novo purine synthesis pathway, GARFTase and AICARFTase, we tested the protection by 5-amino-4-imidazole (AICA) (320 μM), which can be converted to 5-amino-4-imidazolecarboxamide ribonucleotide (AICAR), an intermediate metabolite between GARFTase and AICARFTase which circumvents the GARFTase step.18, 34 AICA (320 μM) almost completely protected KB cells against the toxicity of compound 4 at low and medium concentrations (≤ 50 nM), whereas growth was still significantly inhibited at higher concentrations of compound 4 (above 100 nM) (Figure 5) even in the presence of AICA.
Thus, the pattern of nucleoside protection indicates a potent inhibition by the thieno[2,3-d]pyrimidine antifolate 4 on de novo purine nucleotide biosynthesis. GARFTase appears to be the major enzyme target leading to growth inhibition of FR-expressing KB cells, although a secondary target, most likely AICARFTase, also seems likely at higher doses of drug. Analogous results were obtained with compound 3. These results suggest a unique mechanistic feature of the thieno[2,3-d]pyrimidines 3 and 4 distinguishing this series from the pyrrolo[2,3-d]pyrimidines reported previously18 which were only GARFTase inhibitors.
Molecular mechanisms of 6-substituted thieno[2,3-d]pyrimidine antifolates
The results of the above experiments strongly indicate that the 6-substituted thieno[2,3-d]pyrimidine antifolates 3-8 are (i) substrates for FR-mediated cellular uptake but not for RFC or PCFT (Table 1), and (ii) inhibitors of de novo purine biosynthesis, reflecting primary inhibition of GARFTase (Figure 5). Additional mechanistic experiments were performed to validate these conclusions.
For FRs, substrate binding is a good reflection of FR-mediated uptake.24 Accordingly, competition by compounds 2-8 with [3H]folic acid for binding to FRs α and β (in RT16 and D4 cells, respectively) was measured and the results compared to those with the classical antifolates MTX, LMX, and PMX, and the folate cofactor leucovorin (LCV). Cells were washed at pH 3.5 to remove bound folate, then treated with 50 nM [3H]folic acid in the presence of a range of inhibitor concentrations. After additional washing (at neutral pH), binding of [3H]folic acid was measured. Relative affinities were determined over a range of concentrations of unlabeled ligands and were calculated as the inverse molar ratios of unlabeled ligands required to inhibit 50% of [3H]folic acid binding.
Using relative binding of folic acid set to a value of 1, MTX showed negligible binding (<0.05), whereas LMX showed high (∼0.8) and comparable affinities toward both FRα and β (Figure 6). Similarly, relative binding affinities of compounds 3-8 were high (above 0.8) to both FRα and FRβ. Compound 2 resembled PMX and LCV in its moderate binding affinity for FRα (∼0.25-0.45) and poor binding affinity for FRβ (<0.01). This establishes a clear chain length preference for binding FRα and FRβ.
Figure 6. Competitive inhibition of FR binding activities by 6-substituted thieno[2,3-d]pyrimidines.

Data are shown for the effects of the thieno[2,3-d]pyrimidine antifolates 2-8 with FRα-expressing RT16 CHO cells and FRβ-expressing D4 CHO cells. Relative binding affinities for assorted folate/antifolate substrates were determined over a range of ligand concentrations and were calculated as the inverse molar ratio of unlabeled ligands required to inhibit [3H]folic acid binding by 50%. By definition, the relative affinity of folic acid (FA) is 1. The data for MTX, PMX, LMX LCV and folic acid (FA) were previously published.18
The most potent of the 6-substituted thieno[2,3-d]pyrimidine series, compounds 2-6, were tested for their inhibition of GARFTase using an in vitro assay with recombinant mouse GARFTase, which followed the absorbance changes at 295 nm accompanying one-carbon transfer from 10-formyl-5,8-dideazafolic acid to β-glycinamideribonucleotide (βGAR), forming 5,8-dideazafolic and formyl GAR (FGAR). Table 2 summarizes the IC50 values for in vitro GARFTase inhibitions by compounds 2-6, for comparison with those for LMX and PMX. Similar to our cytotoxicity results, compounds 3 and 4 were the most potent of the series, with IC50s less than 10 μM. However, by in vitro assay, compounds 3 and 4 were less inhibitory than was LMX (Table 2). Compounds 2, 5 and 6, along with PMX, all showed IC50s above 20 μM, the upper limit of the in vitro activity assay (Table 2).
Table 2. IC50s for thieno[2,3-d]pyrimidines compound 2-6 in in vitro and in situ GARFTase inhibition assays.
| Antifolate | IC50 | |
|---|---|---|
| In vitro (μM) | In situ (nM) | |
| 2 | >20 | 32.3 (9.5) |
| 3 | 8.52 (2.71) | 13.8 (9.5) |
| 4 | 5.51 (0.82) | 13.3 (5.5) |
| 5 | >20 | 23.6 (6.2) |
| 6 | >20 | 26.6 (7.5) |
| Pemetrexed | >20 | 30 (7.7) |
| Lometrexol | 0.78 (0.08) | 14 (5.6) |
GARFTase inhibition assays, both in vitro and in situ, were performed as described in the Experimental Section. The IC50 data shown are mean values from 3 (in vitro; SEM in parentheses) or 2 (in situ; range in parentheses) experiments. IC50 data of classical antifolate compounds, pemetrexed and lometrexol, were previously published.18
To assess the inhibitory effects of compounds 2-6 in cells, in situ experiments were performed in which KB cells were metabolically labeled with [14C]glycine in the presence of azaserine, and the accumulations of [14C]FGAR were measured following ion exchange fractionation. Again, compounds 3 and 4 showed the lowest IC50s of the series (Table 2) and were equipotent with LMX but slightly more active than PMX and compounds 2, 5 and 6. While for compounds 3 to 6 (but not compound 2), the results of the in situ GARFTase assays generally paralleled their relative in vitro growth inhibitions (Table 1), this was less-than-complete and likely further reflects at least a partial effect of the thieno[2,3-d]pyrimidines at an intracellular target other than GARFTase.
Conclusions
Taken together, these results establish that the thieno[2,3-d]pyrimidines are potent and selective inhibitors of cell proliferation in cells that express FRs α and β, and that, with the exception of compound 2, there is no appreciable difference in affinity for the FR isoforms for these analogs. In contrast, by growth inhibition assays, the thieno[2,3-d]pyrimidine antifolates 2-8 are all exceedingly poor substrates for both RFC and PCFT. Compounds 3 and 4 were consistently the most potent inhibitors of proliferation in cells expressing FRs α and β and this was significantly associated with inhibition of GARFTase. By both in vitro and in situ GARFTase assays, the bridge length with 3- and 4-methylenes is optimal for GARFTase binding and inhibition, analogous to our previous findings with the 6-pyrrolo[2,3-d]pyrimidine antifolates.18 The dramatic differences in inhibition potencies by in situ (nM) versus in vitro (μM) assays of GARFTase (Table 2) likely reflect increased enzyme affinities by drug polyglutamates within cells. The possibility of a secondary enzyme target, most likely AICARFTase, suggested by our protection studies with AICA and in situ GARFTase assays, raises the intriguing possibility of a dual inhibition at high concentrations of these novel agents. The presence of two molecular targets would likely reduce the acquisition of resistance due to alterations in enzyme affinities for the cytotoxic drugs.
The thieno[2,3-d]pyrimidine antifolates 2-8 are unique from all the other classical antifolates evaluated, including pyrrolo[2,3-d]pyrimidine (PMX), quinazoline (RTX) and pteridine (MTX) antifolates, in that they are neither substrates for RFC nor PCFT. This characteristic results in selective targeting of FR-expressing cells, while exerting no cytotoxic effects toward cells that express these other transporters in the absence of FRs. Given that PCFT has been suggested to function in tandem with FRs in mediating folate uptake,6 our finding of exclusive FR-selective activity in FR-expressing CHO cells that do not express detectable PCFT is of further interest in that they establish that that the presence of PCFT is not obligatory to FR-mediated cellular uptake, at least for the thieno[2,3-d]pyrimidine antifolates described herein.
To summarize, this report documents our continued efforts to identify novel antifolates with potential selectivity for tumors expressing high affinity FRs over normal cells that express RFC and PCFT. We describe a novel series of 6-substituted thieno[2,3-d]pyrimidine antifolates 2-8 that differ in the lengths of the methylene bridge from 2- to 8-carbons. These analogs, like our previous series of 6-substituted pyrrolo[2,3-d]pyrimidine antifolates, are excellent substrates for FRs and potently inhibit de novo purine nucleotide biosynthesis rather than thymidylate synthesis. Further, as with the pyrrolo[2,3-d]pyrimidine antifolates, GARFTase appeared to be primary enzyme target for the 6-substituted thieno[2,3-d]pyrimidine analogs, although our results strongly suggested a secondary enzyme target, most likely AICARFTase, may also be important. Thus, unlike the pyrrolo[2,3-d]pyrimidines, the thieno[2,3-d]pyrimidines appear to be dual acting agents and likely inhibit both GARFTase and AICARFTase in the de novo purine nucleotide biosynthetic pathway at higher drug concentrations. In addition, the thieno[2,3-d]pyrimidines afford structural specificity for transport via FRs and are not stubstrates for RFC or PCFT. Thus, the thieno[2,3-d]pyrimidines provide a scaffold that allows selectivity of uptake by FRs over RFC and PCFT. This is the only example in the literature of antifolates that afford this exquisite selectivity for cellular uptake by FRs over RFC and PCFT that can be exploited for selective inhibition of tumors that express FRs.
EXPERIMENTAL SECTION
All evaporations were carried out in vacuo with a rotary evaporator. Analytical samples were dried in vacuo (0.2 mmHg) in a CHEM-DRY drying apparatus over P2O5 at 55 °C. Melting points were determined on a MEL-TEMP II melting point apparatus with FLUKE 51 K/J electronic thermometer and are uncorrected. Nuclear magnetic resonance spectra for proton (1H NMR) were recorded on a Bruker WH-300 (300 MHz) or a Bruker 400MHz/52 MM (400 MHz) spectrometer. The chemical shift values are expressed in ppm (parts per million) relative to tetramethylsilane as internal standard: s ) singlet, d ) doublet, t ) triplet, q ) quartet, m ) mutiplet, br ) broad singlet. The relative integrals of peak areas agreed with those expected for the assigned structures. High-resolution mass spectra (HRMS), using Electron Impact (EI), were recorded on a VG AUTOSPEC (Fisons Instruments) micromass (EBE Geometry) double focusing mass spectrometer. Thin-layer chromatography (TLC) was performed on Whatman Sil G/UV254 silica gel plates with fluorescent indicator, and the spots were visualized under 254 and 366 nm illumination. Proportions of solvents used for TLC are by volume. Column chromatography was performed on a 230-400 mesh silica gel (Fisher, Somerville, NJ) column. Elemental analyses were performed by Atlantic Microlab, Inc. Norcross, GA. Element compositions are within ±0.4% of the calculated values. Fractional moles of water or organic solvents frequently found in some analytical samples of antifolates could not be prevented despite 24-48 h of drying in vacuo and were confirmed where possible by their presence in the 1H NMR spectra. All solvents and chemicals were purchased from Aldrich Chemical Co. or from Fisher Scientific and were used as received.
General Procedure for the Synthesis of Compounds 16-22
To a 250 mL round bottom flask, fitted with a magnetic stir bar, were placed palladium diacetate (0.269 g, 1.2 mmol), the appropriate allyl alcohol (20 mmol), ethyl 4-iodobenzoate (5.52 g, 20 mmol), LiCl (0.848 g,20 mmol), LiOAc (3.3 g, 50 mmol) and Bu4NCl (11.12 g, 40 mmol) and DMF (40 mL). The mixture was stirred vigorously at 70 °C for 24 h. The reaction mixture was cooled to room temperature and 80 mL of water was added and then 100 mL of ethyl acetate. The ethyl acetate layer was separated, washed with brine (30 mL × 3), dried over anhydrous sodium sulfate, and concentrated under reduced pressure to afford a brown oil. The residue was loaded on a silica gel column and eluted with 5% ethyl acetate in hexane. The fractions containing the desired product (TLC) were pooled and evaporated to afford the product.
Ethyl 4- (4-oxobutyl)benzoate (16)
Using the general procedure 16 was obtained as a colorless liquid (4.11 g, 87.2 %); Rf 0.70 (hexane/EtOAc 3:1); 1H NMR (CDCl3): δ1.37-1.41 (3 H, t, COOCH2CH3), 1.96-2.01 (2 H, m, CH2CH2CH2CHO), 2.36-2.49 (2 H, q, CH2CH2CH2CHO), 2.69-2.76 (2 H, t, CH2CH2CH2CHO), 4.36-4.38 (2 H, q, COOCH2CH3), 7.23-7.98 (4 H, dd, C6H4 ), 9.77 (1 H, s, CHO). This compound was unstable and could not be analyzed by HRMS and was used directly for the next step.
Ethyl 4- (5-oxopentyl)benzoate (17)
Using the general procedure 17 was obtained as a colorless liquid (4.36 g, 87.9 %); Rf 0.71 (hexane/EtOAc 3:1); 1H NMR (CDCl3): δ 1.33-1.36 (3 H, t, COOCH2CH3), 1.62-1.64 (4 H, m, CH2CH2CH2CH2CHO), 2.38-2.42 (2 H, m, CH2CH2CH2CH2CHO), 2.66-2.71 (2 H, m, CH2CH2CH2CH2CHO), 4.32-4.37 (2 H, q, COOCH2CH3), 7.21-7.95 (4 H, dd, C6H4 ), 9.79 (1 H, s, CHO). HRMS (ESI, pos mode) m/z [M + Na]+ calcd for C14H18O3, 257.1154; found, 257.1144.
Ethyl 4- (6-oxohexyl)benzoate (18)
Using the general procedure 18 was obtained as a colorless liquid (3.25 g, 65.5 %); Rf 0.73 (hexane/EtOAc 3:1); 1H NMR (CDCl3): δ 1.22-1.27 (5 H, t, COOCH2CH3 and CH2CH2CH2CH2CH2CHO), 1.47-1.57 (4 H, m, CH2CH2CH2CH2CH2CHO), 2.26-2.31 (2 H, m, CH2CH2CH2CH2CH2CHO), 2.51-2.56 (2 H, m, CH2CH2CH2CH2CH2CHO), 4.19-4.26 (2 H, q, COOCH2CH3), 7.11-7.84 (4 H, dd, C6H4 ), 9.62 (1 H, s, CHO). This compound was unstable and could not be analyzed by HRMS and was used directly for the next step.
Ethyl 4- (7-oxoheptyl)benzoate (19)
Using the general procedure 19 was obtained as a colorless liquid (4.3 g, 82.06 %); Rf 0.74 (hexane/EtOAc 3:1); 1H NMR (CDCl3): δ 1.17-1.27 (7 H, m, COOCH2CH3 and CH2CH2CH2CH2CH2CH2CHO), 1.44-1.55 (4 H, m,CH2CH2CH2CH2CH2CH2CHO), 2.25-2.31 (2 H, m,CH2CH2CH2CH2CH2CH2CHO), 2.49-2.54 (2 H, t, CH2CH2CH2CH2CH2CH2CHO), 4.19-4.26 (2 H, q, COOCH2CH3), 7.08-7.85 (4 H, dd, C6H4 ), 9.61 (1 H, s, CHO). This compound was unstable and could not be analyzed by HRMS and was used directly for the next step.
Ethyl 4- (8-oxooctyl)benzoate (20)
Using the general procedure 20 was obtained as a colorless liquid (4.63 g, 88.3 %); Rf 0.76 (hexane/EtOAc 3:1); 1H NMR (CDCl3): δ 1.32-1.42 (9 H, m, COOCH2CH3 and CH2CH2CH2CH2CH2CH2CH2CHO), 1.50-1.68 (4 H, m,CH2CH2CH2CH2CH2CH2CH2CHO),2.13-2.45 (2 H, m,CH2CH2CH2CH2CH2CH2CH2CHO), 2.59-2.68 (2 H, m, CH2CH2CH2CH2CH2CH2CH2CHO), 4.34-4.41 (2H, q, COOCH2CH3), 7.22-7.98 (4H, dd, C6H4 ), 9.36 (1H, s, CHO). This compound was unstable and could not be analyzed by HRMS and was used directly for the next step.
Ethyl 4- (9-oxononyl)benzoate (21)
Using the general procedure 21 was obtained as a colorless liquid (4.53 g, 78.1 %); Rf 0.76 (hexane/EtOAc 3:1); 1H NMR (CDCl3): δ 1.34-1.53 (11 H, m, COOCH2CH3 and CH2CH2CH2 CH2CH2CH2CH2CH2CHO), 1.60-1.63 (4 H, m, CH2CH2CH2CH2CH2CH2CH2CH2CHO),2.33-2.41 (2 H, m,CH2CH2CH2CH2CH2CH2CH2CH2CHO), 2.62-2.65 (2 H, t, CH2CH2CH2CH2CH2CH2 CH2CH2CHO), 4.33-4.37 (2 H, q, COOCH2CH3), 7.21-7.96 (4 H, dd, C6H4), 9.35 (1 H, s, CHO). HRMS (ESI, pos mode) m/z [M + K]+ calcd for C18H26O3, 329.1519; found, 329.1498.
Ethyl 4- (10-oxodecyl)benzoate (22)
Using the general procedure 22 was obtained as a colorless liquid (5.05 g, 83.5 %); Rf 0.77 (hexane/EtOAc 3:1); 1H NMR (CDCl3): δ 1.29-1.38 (13 H, m, COOCH2CH3 and CH2CH2CH2CH2CH2CH2CH2CH2CH2CHO), 1.52-1.56 (4 H, m, CH2CH2CH2CH2CH2CH2CH2CH2CH2CHO), 2.23-2.30 (2 H, m, CH2CH2CH2CH2CH2CH2CH2CH2CH2CHO), 2.52-2.55 (2 H, t, CH2CH2CH2CH2CH2CH2CH2CH2CH2CHO), 4.23-4.27 (2 H, q, COOCH2CH3), 7.12-7.85 (4 H, dd, C6H4), 9.56 (1 H, s, CHO). HRMS (ESI, pos mode) m/z [M + Na]+ calcd for C19H28O3, 327.1936; found, 327.1950.
General Procedure for the Synthesis of Compounds 23-29
A mixture of sulfur (1 mmol), the appropriate aldehyde (1 mmol), ethyl cyanoactetate (1 mmol) and EtOH (5 mL) were placed in a round bottom flask and warmed to 45 °C and treated dropwise with morpholine (1 mmol) over 15 min. The mixture was stirred for 5 h at 45 °C and 24 h at room temperature. Unreacted sulfur was removed by filtration, and the filtrate was concentrated under reduced pressure to afford an orange oil. The residue was loaded on a silica gel column and eluted with 10% ethyl acetate in hexane. The fractions containing the desired product (TLC) were pooled and evaporated to afford the products.
Ethyl 2-amino-5-{2-[4- (ethoxycarbonyl)phenyl]ethyl}thiophene-3-carboxylate (23)
Compound 23 (3.68 g, 67.65 %) was obtained as an orange liquid by reacting 16 (3.7 g, 15.67 mmol) with sulfur (0.5 g, 15.67 mmol) and ethyl cyanoactetate (1.78 g, 15.67 mmol) as described in the General Procedure above; Rf 0.68 (hexane/EtOAc 3:1); 1 H NMR (CDCl3): δ 1.30-1.34 (3 H, t, COOCH2CH3), 1.36-1.41 (3 H, t, COOCH2CH3), 2.88-2.97 (4 H, m, C6H4-CH2CH2), 4.21-4.27 (2 H, q, COOCH2CH3), 4.33-4.40 (2 H, q, COOCH2CH3), 5.79 (2 H, s, NH2exch), 6.63 (1 H, s, 4-H), 7.23-7.97 (4 H, dd, C6H4). HRMS (ESI, pos mode) m/z [M + Na]+ calcd for C18H21NO4S, 370.1089; found, 370.1112.
Ethyl 2-amino-5-{3-[4- (ethoxycarbonyl) phenyl]propyl}thiophene-3-carboxylate (24)
Compound 24 (1.49 g, 65.32 %) was obtained as an orange liquid by reacting 17 (1.58 g, 6.31 mmol), sulfur (0.20 g, 6.31 mmol) and ethyl cyanoactetate (0.71 g, 6.31 mmol) as described in the General Procedure above; Rf 0.69 (hexane/EtOAc 3:1); 1H NMR (CDCl3): δ 1.32-1.36 (3 H, t, COOCH2CH3), 1.37-1.42 (3 H, t, COOCH2CH3) 1.88-1.98 (2 H, p, C6H4-CH2CH2CH2), 2.59-2.64 (2 H, t, C6H4-CH2CH2CH2), 2.69-2.74 (2 H, t, C6H4-CH2CH2CH2), 4.23-4.30 (2 H, q, COOCH2CH3), 4.34-4.41 (2 H, q, COOCH2CH3), 5.80 (2 H, s, NH2 exch), 6.65 (1 H, s, 4-H), 7.22-8.00 (4 H, dd, C6H4 ). HRMS (ESI, pos mode) m/z [M + Na]+ calcd for C19H23NO4S, 384.1245; found, 384.1281.
Ethyl 2-amino-5-{4- [4-(ethoxycarbonyl)phenyl]butyl}thiophene-3-carboxylate (25)
Compound 25 (2.99 g, 66.73 %) was obtained as an orange liquid by reacting 18 (2.96 g, 11.92 mmol), sulfur (0.38 g, 11.92 mmol) and ethyl cyanoactetate (1.35 g, 11.92 mmol) as described in the General Procedure above; Rf 0.71 (hexane/EtOAc 3:1); 1 H NMR (CDCl3): δ 1.27-1.32 (3 H, t, COOCH2CH3), 1.33-1.37 (3 H, t, COOCH2CH3) 1.57-1.69 (4 H, m,C6H4-CH2CH2CH2CH2), 2.54-2.59 (2 H, t,C6H4-CH2CH2CH2CH2), 2.62-2.67 (2 H, t, C6H4-CH2CH2CH2CH2) 4.18-4.25 (2 H, q, COOCH2CH3), 4.29-4.36 (2 H, q, COOCH2CH3), 5.73 (2 H, s, NH2 exch), 6.58 (1 H, s, 4-H), 7.18-7.93 (4 H, dd, C6H4 ). HRMS (ESI, pos mode) m/z [M + Na]+ calcd for C20H25NO4S, 398.1402; found, 398.1392.
Ethyl 2-amino-5-{5-[4- (ethoxycarbonyl) phenyl]pentyl}thiophene-3-carboxylate (26)
Compound 26 (2.62 g, 63.17 %) was obtained as an orange liquid by reacting 19 (2.8 g, 10.67 mmol), sulfur (0.34 g, 10.67 mmol) and ethyl cyanoactetate (1.20 g, 10.67 mmol) as described in the General Procedure above; Rf 0.71 (hexane/EtOAc 3:1); 1H NMR (CDCl3): δ 1.37-1.47 (8 H, m, 2COOCH2CH3 and C6H4-CH2CH2CH2CH2 CH2 ),1.62-1.76 (4 H, m, C6H4-CH2CH2CH2CH2CH2), 2.60-2.65 (2 H, t, C6H4-CH2CH2CH2CH2CH2), 2.69-2.74 (2 H, t, C6H4-CH2CH2CH2CH2CH2), 4.28-4.35 (2 H, q, COOCH2CH3), 4.39-4.46 (2 H, q, COOCH2CH3), 5.85 (2 H, s, NH2 exch), 6.66 (1 H, s, 4-H), 7.28-8.03 (4 H, dd, C6H4 ). HRMS (ESI, pos mode) m/z [M + Na]+ calcd for C21H27NO4S, 412.1559; found, 412.1537.
Ethyl 2-amino-5-{6-[4- (ethoxycarbonyl)phenyl]hexyl}thiophene-3-carboxylate (27)
Compound 27 (3.62 g, 68.86 %) was obtained as an orange liquid by reacting 20 (3.6 g, 13.03 mmol), sulfur (0.42 g,13.03 mmol) and ethyl cyanoactetate (1.47 g,13.03 mmol) as described in the General Procedure above; Rf 0.73 (hexane/EtOAc 3:1); 1H NMR (CDCl3): δ 1.31-1.41 (10 H, m, 2COOCH2CH3 and C6H4-CH2CH2CH2CH2 CH2CH2),1.46-1.66 (4 H, m,C6H4-CH2CH2CH2CH2CH2CH2), 2.53-2.58(2 H, t,C6H4-CH2CH2CH2CH2CH2CH2), 2.62-2.69 (2 H, t, C6H4- CH2CH2CH2CH2CH2CH2), 4.22-4.29 (2 H, q, COOCH2CH3), 4.33-4.40 (2 H, q, COOCH2CH3), 5.77 (2 H, s, NH2 exch), 6.62 (1 H, s, 4-H), 7.22-7.97 (4 H, dd, C6H4 ). HRMS (ESI, pos mode) m/z [M + Na]+ calcd for C22H29NO4S, 426.1715; found, 426.1688.
Ethyl 2-amino-5-{7-[4- (ethoxycarbonyl) phenyl]heptyl}thiophene-3-carboxylate (28)
Compound 28 (1.94 g, 63.5 %) was obtained as an orange liquid by reacting 21 (2.1 g, 7.2 mmol), sulfur (0.23 g, 7.2 mmol) and ethyl cyanoactetate (0.81 g, 7.2 mmol) as described in the General Procedure above; Rf 0.72 (hexane/EtOAc 3:1); 1H NMR (CDCl3): δ 1.29-1.41 (12 H, m, 2COOCH2CH3_and C6H4-CH2CH2CH2CH2CH2 CH2CH2),1.46-1.66 (4 H, m,C6H4-CH2CH2CH2CH2CH2CH2CH2),2.51-2.58 (2 H, t,C6H4-CH2CH2CH2CH2CH2CH2CH2), 2.60-2.71 (2 H, t, C6H4-CH2CH2CH2CH2CH2CH2CH2), 4.20-4.27 (2 H, q, COOCH2CH3), 4.31-4.38 (2 H, q, COOCH2CH3), 5.86 (2 H, s, NH2 exch), 6.60 (1 H, s, 4-H), 7.16-8.01 (4 H, dd, C6H4 ). HRMS (ESI, pos mode) m/z [M + Na]+ calcd for C23H31NO4S, 440.1872; found, 440.1837.
Ethyl 2-amino-5-{8-[4- (ethoxycarbonyl)phenyl]octyl}thiophene-3-carboxylate (29)
Compound 29 (4.77 g, 62.4 %) was obtained as an orange liquid by reacting 22 (5.4 g, 17.7 mmol), sulfur (0.57 g, 17.7 mmol) and ethyl cyanoactetate (2.0 g, 17.7 mmol) as described in the General Procedure above; Rf 0.74 (hexane/EtOAc 3:1); 1H NMR (CDCl3): δ 1.30-1.43 (14 H, m, 2COOCH2CH3 and C6H4-CH2CH2CH2CH2CH2CH2 CH2CH2), 1.47-1.66 (4 H, m,C6H4-CH2CH2CH2CH2CH2CH2CH2CH2), 2.53-2.58 (2H, t,C6H4-CH2CH2CH2CH2CH2CH2CH2CH2), 2.62-2.67 (2 H, t, C6H4-CH2CH2CH2CH2CH2CH2CH2CH2), 4.21-4.28 (2 H, q, COOCH2CH3), 4.32-4.39 (2 H, q, COOCH2CH3), 5.77 (2 H, s, NH2 exch), 6.61 (1 H, s, 4-H), 7.20-7.96 (4 H, dd, C6H4 ). HRMS (ESI, pos mode) m/z [M + Na]+ calcd for C24H33NO4S, 454.2028; found, 454.2016.
General Procedure for the Synthesis of Compounds 30-36
A mixture of appropriate thiophene and chloroformamidine hydrochloride (1:4) in DMSO2 was heated at 140° C for 4 h. The mixture was cooled to room temperature and 15 mL of water was added and ammonium hydroxide was used to neutralize the suspension. The brown solid, obtained by filtration, was washed with water and dried over P2O5 vacuum. The solid was dissolved in methanol and silica gel was added. A dry silica gel plug was obtained after evaporation of the solvent. The plug was loaded on to a silica gel column and eluted with 5% methanol in chloroform. The fractions containing the desired product (TLC) were pooled and evaporated to afford the products.
Ethyl 4-[2- (2-amino-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidin-6-yl)ethyl]benzoate (30)
Using the General Procedure above compound 30 (0.73 g, 71.51 %) was obtained as a yellow solid by reacting 23 (0.7 g, 2.96 mmol) and chloroformamidine hydrochloride (1.30 g, 11.85 mmol); mp 274.6-275.9 °C; Rf 0.53 (MeOH/CHCl3, 1:6); 1H NMR (DMSO-d6): δ 1.28-1.33 (3 H, t, COOCH2CH3), 2.98-3.04 (4 H, m, C6H4-CH2CH2), 4.25-4.32 (2 H, q, COOCH2CH3), 6.46 (2 H, s, 2-NH2 exch), 6.77 (1 H, s, 5-H), 7.37-7.88 (4 H, dd, C6H4 ), 10.81 (1 H, s, 3-NH exch). Anal. (C17H17N3O3S2) C, H, N, S.
Ethyl 4-[3- (2-amino-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidin-6-yl)propyl]benzoate (31)
Using the General Procedure above compound 31 (0.43 g, 81.13 %) was obtained as a yellow solid by reacting 24 (0.54 g, 1.49 mmol) and chloroformamidine hydrochloride (0.68 g, 5.9 mmol); mp 224.4-225.3 °C; Rf 0.53 (MeOH/CHCl3, 1:6); 1H NMR (DMSO-d6): δ 1.28-1.33 (3 H, t, COOCH2CH3), 1.86-1.96 (2 H, p, C6H4-CH2CH2CH2), 2.68-2.74 (4 H, m, C6H4-CH2CH2CH2) 4.26-4.33 (2 H, q, COOCH2CH3), 6.46 (2 H, s, 2-NH2 exch), 6.82 (1 H, s, 5-H), 7.35-7.89 (4 H, dd, C6H4 ), 10.81 (1 H, s, 3-NH exch). Anal. (C18H19N3O3S·0.4CH3OH) C, H, N, S.
Ethyl 4-[4- (2-amino-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidin-6-yl)butyl]benzoate (32)
Using the General Procedure above compound 32 (1.05 g, 76.51 %) was obtained as a yellow solid by reacting 25 (1.40 g, 3.71 mmol) and chloroformamidine hydrochloride (1.70 g, 14.84 mmol); mp 273.5-275 °C; Rf 0.54 (MeOH/CHCl3, 1:6); 1H NMR (DMSO-d6): δ 1.27-1.32 (3 H, t, COOCH2CH3), 2.47-2.52 (4 H, m, C6H4-CH2CH2CH2CH2), 2.65-2.73 (4 H, m, C6H4-CH2CH2CH2CH2), 4.24-4.32 (2 H, q, COOCH2CH3), 6.44 (2 H, s, 2-NH2 exch), 6.78 (1 H, s, 5-H), 7.32-7.87 (4 H, dd, C6H4 ), 10.84 (1 H, s, 3-NH exch). Anal. (C19H21N3O3S·0.2H2O) C, H, N, S.
Ethyl 4-[5- (2-amino-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidin-6-yl)pentyl]benzoate (33)
Using the General Procedure above compound 33 (0.56 g, 54.37 %) was obtained as a yellow solid by reacting 26 (1.08 g, 2.76 mmol) and chloroformamidine hydrochloride (1.58 g, 13.8 mmol); mp 219.3-220.1 °C; Rf 0.55 (MeOH/CHCl3, 1:6); 1H NMR (DMSO-d6):δ 1.27-1.31 (5 H, t,COOCH2CH3 and C6H4-CH2CH2CH2CH2CH2), 1.54-1.64 (4 H, m,C6H4-CH2CH2CH2CH2CH2), 2.59-2.69 (4 H, m, C6H4-CH2CH2CH2CH2CH2), 4.24-4.31 (2 H, q, COOCH2CH3), 6.45 (2 H, s, 2-NH2 exch), 6.78 (1 H, s, 5-H), 7.32-7.87 (4 H, dd, C6H4 ), 10.86 (1 H, s, 3-NH exch). Anal. (C20H23N3O3S) C, H, N, S.
Ethyl 4-[6- (2-amino-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidin-6-yl)hexyl]benzoate (34)
Using the General Procedure above compound 34 (0.78 g, 67.24 %) was obtained as a yellow solid by reacting 27 (1.18 g, 2.91 mmol) and chloroformamidine hydrochloride (1.30 g, 11.64 mmol); mp 196.8-197.4 °C; Rf 0.53 (MeOH/CHCl3, 1:6); 1H NMR (DMSO-d6):δ 1.27-1.32 (7 H, m, COOCH2CH3 and C6H4-CH2CH2CH2CH2CH2CH2), 1.50-1.61 (4 H, m,C6H4-CH2CH2CH2CH2CH2CH2), 2.50-2.71 (4 H, m,C6H4-CH2CH2CH2 CH2CH2CH2), 4.25-4.32 (2 H, q, COOCH2CH3), 6.53 (2 H, s, 2-NH2 exch), 6.85 (1 H, s, 5-H), 7.31-7.87 (4 H, dd, C6H4 ), 10.82 (1 H, s, 3-NH exch). Anal. (C21H25N3O3S·0.2H2O) C, H, N, S.
Ethyl 4-[7- (2-amino-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidin-6-yl)heptyl]benzoate (35)
Using the General Procedure above compound 35 (0.67 g, 67.7 %) was obtained as a yellow solid by reacting 28 (1 g, 2.4 mmol) and chloroformamidine hydrochloride (1.4 g, 12 mmol); mp 190.2-192.1 °C; Rf 0.55 (MeOH/CHCl3, 1:6); 1H NMR (DMSO-d6):δ 1.16-1.33 (9 H, m, COOCH2CH3 and C6H4-CH2CH2CH2 CH2CH2CH2CH2), 1.56 (4 H, m,C6H4-CH2CH2CH2CH2CH2CH2CH2), 2.50-2.69 (4 H, m,C6H4-CH2CH2CH2CH2CH2CH2CH2CH2), 4.25-4.32 (2 H, q, COOCH2CH3), 6.46 (2 H, s, 2-NH2 exch), 6.76 (1 H, s, 5-H), 7.30-7.87 (4 H, dd, C6H4 ), 10.83 (1 H, s, 3-NH exch). HRMS (ESI, pos mode) m/z [M + Na]+ calcd for C22H27N3O3S, 436.5266; found, 436.5239.
Ethyl 4-[8- (2-amino-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidin-6-yl)octyl]benzoate (36)
Using the General Procedure above compound 36 (1.52 g, 70.3 %) was obtained as a yellow solid by reacting 29 (2.18 g, 5.05 mmol) and chloroformamidine hydrochloride (2.3 g, 20.2 mmol); 274.5-276.2 °C; Rf 0.54 (MeOH/CHCl3, 1:6); 1H NMR (DMSO-d6):δ 1.13-1.33 (11H, m, COOCH2CH3 and C6H4-CH2CH2CH2 CH2CH2CH2CH2CH2), 1.55 (4 H, m,C6H4-CH2CH2CH2CH2CH2CH2CH2CH2), 2.50-2.72 (4 H, m,C6H4-CH2CH2CH2CH2CH2CH2CH2), 4.25-4.32 (2 H, q, COOCH2CH3), 6.46 (2 H, s, 2-NH2 exch), 6.78 (1 H, s, 5-H), 7.31-7.87 (4 H, dd, C6H4 ), 10.84 (1 H, s, 3-NH exch). HRMS (ESI, pos mode) m/z [M + Na]+ calcd for C23H29N3O3S, 450.1827; found, 450.1790.
General Procedure for the Synthesis of Compounds 37-43
To a solution of 30-36 in ethanol (10-50 mL) was added aqueous 1 N NaOH and the reaction mixture stirred at room temperature for 12 h. The ethanol was evaporated under reduced pressure and the residue was dissolved in water (5-10 mL). The solution was carefully acidified to pH 3 with the drop wise addition of 1 N HCl. The resulting suspension was left at 0 °C for an hour and then the residue was collected by filtration, washed with water (5 mL) and dried over P2O5/vacuum at 50 °C to afford the free acids 37-43.
4-[2- (2-amino-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidin-6-yl)ethyl]benzoic acid (37)
Using the General Procedure described above compound 37 (0.529 g, 95.0 %) was obtained as a white solid from 30 (0.61 g, 1.77 mmol) by hydrolysis in ethanol (50 mL) and 1 N NaOH (25 mL); mp >300 °C; Rf 0.55 (MeOH/CHCl3, 1:6 + 1drop of gl. HOAc); 1H NMR (DMSO-d6): δ 2.97-2.99 (2 H, t, C6H4-CH2CH2), 3.02-3.41 (2 H, t, C6H4-CH2CH2), 6.50 (2 H, s, 2-NH2 exch), 6.78 (1 H, s, 5-H), 7.35-7.86 (4 H, dd, C6H4 ), 10.85 (1 H, s, 3-NH exch), 12.76 (1 H, s, -COOH exch). Anal. (C15H13N3O3S·0.6HCl) C, H, N, S, Cl.
4-[3- (2-amino-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidin-6-yl)propyl]benzoic acid (38)
Using the General Procedure described above compound 38 (0.18 g, 92.6 %) was obtained as a white solid from 31 (0.22 g, 0.60 mmol) under hydrolysis in ethanol (20 mL) and 1 N NaOH (10 mL); mp 292.7-293.4 °C; Rf 0.52 (MeOH/CHCl3, 1:6 + 1drop of gl. HOAc); 1H NMR (DMSO-d6): δ 1.85-1.95 (2 H, m, C6H4-CH2CH2CH2), 2.66-2.73 (4 H, m, C6H4-CH2CH2CH2), 6.58 (2 H, s, 2-NH2 exch), 6.82 (1 H, s, 5-H), 7.32-7.90 (4 H, dd, C6H4 ), 10.94 (1 H, s, 3-NH exch), 12.79 (1H, s, -COOH exch). Anal. (C16H15N3O3S·0.7 CH3OH) C, H, N, S.
4-[4- (2-amino-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidin-6-yl)butyl]benzoic acid (39)
Using the General Procedure described above compound 39 (0.138 g, 93.6 %) was obtained as a yellow solid 32 (0.16 g, 0.43 mmol) under hydrolysis in ethanol (15 mL) and 1 N NaOH (7 mL); mp >300°C; Rf 0.52 (MeOH/CHCl3, 1:6 + 1drop of gl. HOAc); 1H NMR (DMSO-d6): δ 1.59-1.62 (4 H, m, C6H4-CH2CH2CH2CH2), 2.65-2.75 (4 H, m, C6H4-CH2CH2 CH2CH2), 6.46 (2 H, s, 2-NH2 exch), 6.79 (1 H, s, 5-H), 7.29-7.86 (4 H, dd, C6H4 ), 10.83 (1 H, s, 3-NH exch), 12.77 (1 H, s, -COOH exch). Anal.(C17H17N3O3S·0.5H2O) C, H, N, S.
4-[5- (2-amino-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidin-6-yl)pentyl]benzoic acid (40)
Using the General Procedure described above compound 40 (0.199 g, 93.3 %) was obtained as a white solid from 33 (0.23 g, 0.60 mmol) under hydrolysis in ethanol (20 mL) and 1 N NaOH (10 mL); mp 259.4-260 °C; Rf 0.52 (MeOH/CHCl3, 1:6 + 1drop of gl. HOAc); 1H NMR (DMSO-d6): δ 1.28-1.38 (2 H, m, C6H4-CH2CH2CH2CH2CH2), 1.56-1.66 (4 H, m, C6H4-CH2CH2 CH2CH2CH2), 2.61-2.71 (4 H, p, C6H4-CH2CH2CH2CH2CH2), 6.45 (2 H, s, 2-NH2 exch), 6.79 (1 H, s, 5-H), 7.29-7.85 (4 H, dd, C6H4 ), 10.82 (1 H, s, 3-NH exch), 12.77 (1 H, s, -COOH exch). Anal. (C18H19N3O3S·0.4H2O) C, H, N, S.
4-[6- (2-amino-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidin-6-yl)hexyl]benzoic acid (41)
Using the General Procedure described above compound 41 (0.141 g, 95.2 %) was obtained as a yellow solid from 34 (0.16 g, 0.40 mmol) and ethanol (15 mL) and 1 N NaOH (7 mL); mp 282-283.8 °C; Rf 0.52 (MeOH/CHCl3, 1:6 + 1drop of gl. HOAc); 1H NMR (DMSO-d6): δ 1.31-1.32 (4 H, m, C6H4-CH2CH2CH2CH2CH2CH2), 1.51-1.60 (4 H, m, C6H4-CH2CH2CH2CH2CH2CH2), 2.59-2.69 (4 H, p, C6H4-CH2CH2CH2CH2CH2CH2), 6.45 (2 H, s, 2-NH2 exch), 6.78 (1 H, s, 5-H), 7.28-7.85 (4 H, dd, C6H4 ), 10.82 (1 H, s, 3-NH exch), 12.76 (1 H, s, -COOH exch). Anal. (C19H21N3O3S) C, H, N, S.
4-[7- (2-amino-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidin-6-yl)heptyl]benzoic acid (42)
Using the General Procedure described above compound 42 (0.22 g, 94.9 %) was obtained as a white solid from 35 (0.25 g, 0.60 mmol) and ethanol (20 mL) and 1 N NaOH (10 mL); mp 233.8-235 °C; Rf 0.51 (MeOH/CHCl3, 1:6 + 1drop of gl. HOAc); 1H NMR (DMSO-d6): δ 1.29 (6 H, m, C6H4-CH2CH2CH2CH2CH2CH2CH2), 1.53-1.58 (4 H, m, C6H4-CH2CH2CH2CH2CH2CH2CH2), 2.60-2.69 (4 H, m, C6H4-CH2CH2CH2CH2CH2CH2CH2), 6.46 (2 H, s, 2-NH2 exch), 6.78 (1 H, s, 5-H), 7.29-7.85 (4 H, dd, C6H4 ), 10.83 (1 H, s, 3-NH exch), 12.74 (1H, s, -COOH exch). HRMS (ESI, pos mode) m/z [M + Na]+ calcd for C20H23N3O3S, 408.1358; found, 408.1380.
4-[8- (2-amino-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidin-6-yl)octyl]benzoic acid (43)
Using the General Procedure described above compound 43 (0.22 g, 92.8 %) was obtained as a yellow solid from 36 (0.26 g, 0.60 mmol) and ethanol (20 mL) and 1 N NaOH (9 mL); Rf 0.53 (MeOH/CHCl3, 1:6 + 1drop of gl. HOAc); 1H NMR (DMSO-d6): δ 1.29 (8 H, m, C6H4-CH2CH2CH2CH2CH2CH2CH2CH2), 1.56 (4 H, m, C6H4-CH2CH2CH2CH2CH2CH2CH2CH2), 2.60-2.69 (4 H, m, C6H4-CH2CH2CH2CH2 CH2CH2CH2CH2), 6.46 (2 H, s, 2-NH2 exch), 6.78 (1 H, s, 5-H), 7.30-7.87 (4 H, dd, C6H4 ), 10.83 (1 H, s, 3-NH exch), 12.75 (1 H, s, -COOH exch). HRMS (ESI, pos mode) m/z [M + Na]+ calcd for C21H25N3O3S, 422.1514; found, 422.1553.
General Procedure for the Synthesis of Compound 44-50
To a solution of 37-43 (0.1 mmol) in anhydrous DMF (5-10 mL) was added N-methylmorpholine (0.12 mmol) and 2-chloro-4,6-dimethoxy-1,3,5-triazine (0.12 mmol). The resulting mixture was stirred at room temperature for 2 h. N-methylmorpholine (0.12 mmol) and diethyl-L- glutamate hydrochloride (0.1 mmol) were added to the mixture. The reaction mixture was stirred for an additional 3 h at room temperature and silica gel was added to this solution and the suspension evaporated under reduced pressure. The plug obtained was loaded on a silica gel column and eluted with 2% methanol in chloroform. The fractions containing the desired product (TLC) were pooled and evaporated to afford the products.
Diethyl N-{4-[2- (2-amino-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidin-6-yl)ethyl]benzoy l}-L-glutamate (44)
Using the General Procedure described above compound 44 (0.114 g, 71.5 %) was obtained as a white solid from 37 (0.126 g, 0.40 mmol); mp 153.4-154 °C; Rf 0.63 (MeOH/CHCl3, 1:6); 1H NMR (DMSO-d6): δ 1.14-1.21 (6 H, m, 2COOCH2CH3),1.95-2.23 (2 H, m, Gluβ-CH2), 2.41-2.46 (2 H, t, Gluγ-CH2), 2.97-3.05 (4 H, m, C6H4-CH2CH2), 4.00-4.06 (2 H, q, 2COOCH2CH3 ), 4.08-4.14 (2 H, q, COOCH2CH3 ), 4.38-4.45 (1 H, m, Gluα-CH), 6.45 (2 H, s, 2-NH2 exch), 6.79 (1 H, s, 5-H), 7.33, 7.36-7.77, 7.80 (4 H, dd, C6H4 ), 8.63-8.66 (1 H, d, CONH exch), 10.81 (1 H, s, 3-NH exch). Anal. (C24H28N4O6S) C, H, N, S.
Diethyl N-{4-[3- (2-amino-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidin-6-yl)pentyl]benzoyl}-L-glutamate (45)
Using the General Procedure described above compound 45 (0.106 g, 73.1 %) was obtained as a yellow solid from 38 (0.10 g, 0.30 mmol); mp 122.5-123.2 °C; Rf 0.63 (MeOH/CHCl3, 1:6); 1H NMR (DMSO-d6): δ 1.15-1.21 (6 H, m, 2COOCH2CH3), 1.89-1.93 (2 H, m, C6H4-CH2CH2CH2), 1.99-2.11 (2 H, m, Gluβ-CH2), 2.41-2.45 (2 H, t, Gluγ-CH2), 2.68-2.71 (4 H, m, C6H4-CH2CH2CH2), 4.00-4.13 (4 H, m, 2COOCH2CH3 ), 4.41-4.45 (1 H, m, Gluα-CH), 6.47 (2 H, s, 2-NH2 exch), 6.82 (1 H, s, 5-H), 7.30, 7.32-7.79, 7.82 (4 H, dd, C6H4 ), 8.64-8.66 (1 H, d, CONH exch), 10.86 (1 H, s, 3-NH exch). Anal. (C25H30N4O6S·0.2H2O) C, H, N, S.
Diethyl N-{4-[4- (2-amino-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidin-6-yl)butyl]benzoyl}-L-glutamate (46)
Using the General Procedure described above compound 46 (0.134 g, 67.0 %) was obtained as a white solid from 39 (0.13 g, 0.38 mmol); mp 166.8-167.3 °C; Rf 0.64 (MeOH/CHCl3, 1:6); 1H NMR (DMSO-d6): δ 1.13-1.20 (6 H, m, 2COOCH2CH3), 1.57-1.62 (4 H, m, C6H4-CH2CH2CH2CH2), 1.95-2.12 (2 H, m, Gluβ-CH2), 2.40-2.45 (2 H, t, Gluβ-CH2 ), 2.65-2.75 (4 H, m, C6H4-CH2CH2CH2CH2), 4.00-4.13 (4 H, m, 2COOCH2CH3 ), 4.38-4.45 (1 H, m, Gluα-CH), 6.46 (2 H, s, 2-NH2 exch), 6.79 (1 H, s, 5-H), 7.28, 7.31-7.78, 7.80 (4H, dd, C6H4 ), 8.63-8.65 (1 H, d, CONH exch), 10.85 (1H, s, 3-NH exch).Anal. (C26H32N4O6S) C, H, N, S.
Diethyl N-{4-[5- (2-amino-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidin-6-yl)pentyl]benzoyl}-L-glutamate (47)
Using the General Procedure described above compound 47 (0.108 g, 68.7 %) was obtained as a white solid from 40 (0.10 g, 0.29 mmol); mp 144.2-144.8 °C; Rf 0.64 (MeOH/CHCl3, 1:6); 1H NMR (DMSO-d6): δ 1.14-1.21 (8 H, m, 2COOCH2CH3 and C6H4-CH2CH2CH2CH2CH2), 1.57-1.66 (4 H, p, C6H4-CH2CH2CH2CH2CH2), 1.93-2.17 (2 H, m, Gluβ-CH2), 2.41-2.46 (2 H, t, Gluγ-CH2), 2.61-2.72 (4 H, p, C6H4-CH2CH2CH2CH2CH2), 4.01-4.14 (4 H, m, 2COOCH2CH3 ), 4.38-4.46 (1 H, m, Gluα-CH), 6.46 (2 H, s, 2-NH2 exch), 6.79 (1 H, s, 5-H), 7.29, 7.31-7.78, 7.79 (4 H, dd, C6H4 ), 8.63-8.66 (1 H, d, CONH exch), 10.83 (1 H, s, 3-NH exch). HRMS (ESI, pos mode) m/z [M + Na]+ calcd for C27H34NO4S, 565.2097; found, 565.2134.
Diethyl N-{4-[6- (2-amino-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidin-6-yl)hexyl]benzoyl}-L-glutamate (48)
Using the General Procedure described above compound 48 (0.170 g, 80.57 %) was obtained as a white solid from 41 (0.14 g, 0.38 mmol); mp 153.7-154.4 °C; Rf 0.64 (MeOH/CHCl3, 1:6); 1H NMR (DMSO-d6): δ 1.12-1.19 (6 H, m, 2COOCH2CH3), 1.30-1.32 (4 H, m,C6H4-CH2CH2CH2CH2CH2CH2), 1.54-1.58 (4 H, m,C6H4-CH2CH2CH2 CH2CH2CH2), 1.94-2.14 (2 H, m, Gluβ-CH2), 2.39-2.44 (2 H, t, Gluγ-CH2), 2.58-2.69 (4 H, m, C6H4-CH2CH2CH2CH2CH2CH2), 3.99-4.12 (4 H, m, 2COOCH2CH3 ), 4.36-4.45 (1 H, m, Gluα-CH), 6.45 (2 H, s, 2-NH2 exch), 6.78 (1 H, s, 5-H), 7.27, 7.30-7.77, 7.80 (4 H, dd, C6H4 ), 8.63-8.65 (1 H, d, CONH exch), 10.86 (1 H, s, 3-NH exch). Anal. (C28H36N4O6S) C, H, N, S.
Diethyl N-{4-[7- (2-amino-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidin-6-yl)heptyl]benzoyl}-L-glutamate (49)
Using the General Procedure described above compound 49 (0.183 g, 66.8 %) was obtained as a semisolid from 42 (0.19 g, 0.48 mmol); Rf 0.65 (MeOH/CHCl3, 1:6); It was used directly in the next step without further characterization.
Diethyl N-{4-[8- (2-amino-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidin-6-yl)octyl]benzoyl}-L-glutamate (50)
Using the General Procedure described above compound 50 (0.170 g, 68.20 %) was obtained as a yellow solid from 43 (0.16 g, 0.40 mmol); Rf 0.64 (MeOH/CHCl3, 1:6); 1H NMR (DMSO-d6): δ 1.12-1.19 (6 H, m, 2COOCH2CH3), 1.32-1.56 (8 H, m,C6H4-CH2CH2CH2CH2CH2CH2CH2CH2), 1.58-1.62 (4 H, m,C6H4-CH2CH2CH2CH2CH2CH2CH2CH2), 1.98-2.24 (2 H, m, Gluβ-CH2), 2.41-2.45 (2 H, t, Gluγ-CH2), 2.59-2.69 (4 H, m, C6H4-CH2CH2CH2CH2CH2CH2CH2CH2), 4.01-4.12 (4 H, m, 2COOCH2CH3 ), 4.38-4.47 (1 H, m, Gluα-CH), 6.46 (2 H, s, 2-NH2 exch), 6.78 (1 H, s, 5-H), 7.27, 7.29-7.78, 7.80 (4 H, dd, C6H4 ), 8.63-8.65 (1 H, d, CONH exch), 10.83 (1 H, s, 3-NH exch). HRMS (ESI, pos mode) m/z [M + Na]+ calcd for C30H40N4O6S, 607.7212; found, 607.7233.
General Procedure for the Synthesis of Compounds 2-8
To a solution of 44-50 in ethanol (5-10 mL) was added aqueous 1 N NaOH and the reaction mixture stirred at room temperature for 3 h. The ethanol was evaporated under reduced pressure and the residue was dissolved in water (5-10 mL). The solution was cooled to 0 °C and carefully acidified to pH 3 with drop wise addition of 1 N HCl. The resulting suspension was left at 0 °C for 12 h and the residue was collected by filtration. Washed with water (5 mL) and dried over P2O5/vacuum at 50 °C to afford the free acids 2-8.
N-{4-[2- (2-amino-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidin-6-yl)ethyl]benzoyl}-L-glutamic acid (2)
Using the General Procedure described above compound 2 (0.064 g, 97.7 %) was obtained from 44 (0.075 g, 0.15 mmol) as a white solid; mp 167.7-168.4 °C; Rf 0.62 (MeOH/CHCl3, 1:6 + 1drop of gl. HOAc); 1H NMR (DMSO-d6): δ 1.89-2.12 (2 H, m, Gluβ-CH2), 2.32-2.37 (2 H, t, Gluγ-CH2), 2.97,2.99--3.02,3.04 (4 H, dd, C6H4-CH2CH2), 4.34-4.44 (1 H, m, Gluα-CH), 6.45 (2 H, s, 2-NH2 exch), 6.79 (1 H, s, 5-H), 7.32, 7.36-7.78, 7.80 (4 H, dd, C6H4 ), 8.51-8.53 (1 H, d, CONH exch), 10.81 (1 H, s, 3-NH exch), 12.40 (2 H, s, 2COOH exch). Anal. (C20H20N4O6S·0.5H2O) C, H, N, S.
N-{4-[3- (2-amino-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidin-6-yl)propyl]benzoyl}-L-glutamic acid (3)
Using the General Procedure described above compound 3 (0.071 g, 96.5 %) was obtained from 45 (0.083 g, 0.16 mmol) as a white solid; mp 155.5-156.1 °C; Rf 0.62 (MeOH/CHCl3, 1:6 + 1drop of gl. HOAc); 1H NMR (DMSO-d6): δ 1.91-2.10 (4 H, m, Gluγ-CH2 and C6H4-CH2CH2CH2 ), 2.32-2.36 (2 H, t, Gluβ-CH2), 2.66-2.73 (4 H, q, C6H4-CH2CH2CH2), 4.37-4.38 (1 H, m, Gluα-CH), 6.47 (2 H, s, 2-NH2 exch), 6.81 (1 H, s, 5-H), 7.29, 7.32-7.79, 7.82 (4 H, dd, C6H4 ), 8.49-8.51 (1 H, d, CONH exch), 10.83 (1 H, s, 3-NH exch), 12.51 (2 H, s, 2COOH exch). Anal. (C21H22N4O6S·0.5 CH3OH ) C, H, N, S.
N-{4-[4- (2-amino-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidin-6-yl)butyl]benzoyl}-L-glutamic acid (4)
Using the General Procedure described above compound 4 (0.086 g, 95.8 %) was obtained from 46 (0.10 g, 0.19 mmol) as a yellow solid; mp 154.5-154.9° C; Rf 0.64 (MeOH/CHCl3, 1:6 + 1drop of gl. HOAc); 1H NMR (DMSO-d6): δ 1.61-1.62 (4 H, m, C6H4-CH2CH2CH2CH2 ), 1.91-2.10 (2 H, m, Gluβ-CH2), 2.32-2.37 (2 H, t, Gluγ-CH2), 2.66-2.73 (4 H, q, C6H4-CH2CH2CH2CH2), 4.36-4.41 (1 H, m, Gluα-CH), 6.46 (2 H, s, 2-NH2 exch), 6.79 (1 H, s, 5-H), 7.28, 7.30-7.78, 7.81 (4 H, dd, C6H4 ), 8.51-8.54 (1 H, d, CONH exch), 10.83 (1 H, s, 3-NH exch), 12.41 (2 H, s, 2COOH exch). Anal. (C22H24N4O6S·0.5H2O) C, H, N, S.
N-{4-[5- (2-amino-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidin-6-yl)pentyl]benzoyl}-L-glutamic acid (5)
Using the General Procedure described above compound 5 (0.071 g, 97.9 %) was obtained from 47 (0.080 g, 0.15 mmol) as a white solid; mp 132.6-133 °C; Rf 0.64 (MeOH/CHCl3, 1:6 + 1drop of gl. HOAc); 1H NMR (DMSO-d6): δ 1.16-1.23 (2 H, m, C6H4-CH2CH2CH2CH2CH2 ), 1.59-1.63 (4 H, m, C6H4-CH2CH2CH2CH2CH2 ), 1.89-2.12 (2 H, m, Gluβ-CH2), 2.32-2.37 (2 H, t, Gluγ-CH2), 2.59-2.71 (4 H, m, C6H4-CH2CH2CH2CH2CH2), 4.37-4.39 (1 H, m, Gluα-CH), 6.47 (2 H, s, 2-NH2 exch), 6.79 (1 H, s, 5-H), 7.27, 7.30-7.77, 7.80 (4 H, dd, C6H4 ), 8.49-8.52 (1 H, d, CONH exch), 10.83 (1 H, s, 3-NH exch), 12.47 (2 H, s, 2COOH exch). Anal. (C23H26N4O6S·0.5C2H5OH) C, H, N, S.
N-{4-[6- (2-amino-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidin-6-yl)hexyl]benzoyl}-L-glutamic acid (6)
Using the General Procedure described above compound 6 (0.086 g, 98.6 %) was obtained from 48 (0.098 g, 0.17 mmol) as a solid; mp 139.7-141.1 °C; Rf 0.64 (MeOH/CHCl3, 1:6 + 1drop of gl. HOAc); 1H NMR (DMSO-d6): δ 1.32 (4 H, s, C6H4-CH2 CH2CH2CH2CH2CH2 ), 1.57 (4 H, s, C6H4-CH2CH2CH2CH2CH2CH2 ), 1.93-2.09 (2 H, m, Gluβ-CH2), 2.33-2.35 (2 H, m, Gluβ-CH2), 2.62-2.68 (4 H, m,C6H4-CH2CH2CH2CH2CH2CH2), 4.38-4.39 (1 H, m, Gluα-CH), 6.47 (2 H, s, 2-NH2 exch), 6.78 (1 H, s, 5-H), 7.27,7.29-7.78,7.80 (4 H, dd, C6H4 ), 8.50-8.52 (1 H, d, CONH exch), 10.85 (1 H, s, 3-NH exch), 12.40 (2 H, s, 2COOH exch). Anal. (C24H28N4O6S·0.4 H2O) C, H, N, S.
N-{4-[7- (2-amino-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidin-6-yl)heptyl]benzoyl}-L-glutamic acid (7)
Using the General Procedure described above compound 7 (0.287 g, 97.9 %) was obtained from 49 (0.326 g, 0.57 mmol) as a yellow solid; mp 134.9-135.5 °C; Rf 0.64 (MeOH/CHCl3, 1:6 + 1drop of gl. HOAc); 1H NMR (DMSO-d6): δ 1.35 (6 H, m, C6H4-CH2 CH2CH2CH2CH2CH2CH2), 1.59 (4 H, s, C6H4-CH2CH2CH2CH2CH2CH2CH2 ), 1.90-2.08 (2 H, m, Gluβ-CH2), 2.32-2.35 (2 H, m, Gluγ-CH2), 2.59-2.68 (4 H, m,C6H4-CH2CH2CH2CH2CH2CH2CH2), 4.36-4.40 (1 H, m, Gluα-CH), 6.44 (2 H, s, 2-NH2 exch), 6.77 (1 H, s, 5-H), 7.26-7.79 (4 H, dd, C6H4 ), 8.51 (1 H, d, CONH exch) 10.82 (1 H, s, 3-NH exch), 12.38 (2 H, s, 2COOH exch). Anal. (C25H30N4O6S·0.5H2O) C, H, N, S.
N-{4-[8- (2-amino-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidin-6-yl)octyl]benzoyl}-L-glutamic acid (8)
Using the General Procedure described above compound 8 (0.087 g, 97.6 %) was obtained from 50 (0.1 g, 0.17 mmol) as a brown solid; mp 122.2-123 °C; Rf 0.66 (MeOH/CHCl3, 1:6 + 1drop of gl. HOAc); 1H NMR (DMSO-d6): δ 1.38 (8 H, m, C6H4-CH2 CH2CH2CH2CH2CH2CH2CH2), 1.57 (4 H, s, C6H4-CH2CH2CH2CH2CH2CH2CH2CH2), 1.92-2.11 (2 H, m, Gluβ-CH2), 2.32-2.35 (2 H, m, Gluγ-CH2 ), 2.59-2.68 (4 H, m,C6H4-CH2CH2CH2 CH2CH2CH2CH2CH2), 4.35-4.39 (1 H, m, Gluα-CH), 6.44 (2 H, s, 2-NH2 exch), 6.77 (1 H, s, 5-H), 7.26-7.79 (4 H, dd, C6H4 ), 8.50-8.51 (1 H, d, CONH exch), 10.81 (1 H, s, 3-NH exch), 12.80 (2 H, s, 2COOH exch). Anal. (C26H32N4O6S·0.6 H2O) C, H, N, S.
Reagents for Biological Studies
[3′, 5′, 7-3H]MTX (20 Ci/mmol) and [3′, 5′, 7, 9-3H] folic acid (25 Ci/mmol), [14C(U)]-glycine (87mCi/mmol) were purchased from Moravek Biochemicals (Brea, CA). Unlabeled folic acid was purchased from the Sigma Chemical Co. (St. Louis, MO). Unlabeled MTX and LCV [(6R,S) 5-formyl tetrahydrofolate] were provided by the Drug Development Branch, National Cancer Institute, Bethesda, MD. Both labeled and unlabeled MTX were purified by HPLC prior to use.36 α,β-GAR and coenzyme 10-formyl-5,8-dideazafolic acid were provided by Dr. Richard Moran (Virginia Commonwealth University, Richmond, VA). Other chemicals were obtained from commercial sources in the highest available purity.
Cell Lines and Assays of Antitumor Drug Activities
RFC-, PCFT, and FRα-null MTXRIIOuaR2-4 (R2) CHO cells were a gift from Dr. Wayne Flintoff (University of Western Ontario)36 and were cultured in α-minimal essential medium (MEM) supplemented with 10% bovine calf serum (Invitrogen, Carlsbad, CA), penicillin-streptomycin solution and glutamine at 37°C with 5% CO2.
PC43-10 cells are R2 cells transfected with human RFC.37 RT16 cells are R2 cells transfected with human FRα, and D4 cells are R2 cells transfected with human FRβ. Both RT16 and D4 cells were described in a previous publication.18 The lack of endogenous hamster PCFT in the CHO R2, RT16, and PC43-10 sublines was confirmed by RT-PCR of PCFT transcripts (Supplement) and by transport assay at pH 5.5 and 7.2 with [3H]MTX (see Supplement). R2/PCFT4 cells and R2/VC cells were prepared by transfection with human PCFT cDNA in pCDNA3.1 vector and with empty pCDNA3.1, respectively (below). All the R2 transfected cells (PC43-10, RT16, D4, and R2/PCFT4) were routinely cultured in α-MEM plus 1.5 mg/ml G418. Prior to the cytotoxicity assays (see below), RT16 and D4 cells were cultured in complete folate-free RPMI 1640 (without added folate) for three days.
KB human cervical cancer were purchased from the American Type Culture Collection (Manassas, VA) whereas IGROV1 ovarian carcinoma cells were a gift of Dr. Manohar Ratnam (Medical University of Ohio). Cells were routinely cultured in folate-free RPMI 1640 medium, supplemented with 10% fetal bovine serum, penicillin-streptomycin solution, 2 mM glutamine at 37°C with 5% CO2. FR and RFC levels in IGROV1 and KB cells were previously reported18. Human PCFT levels were measured by real-time PCR with gene specific primers and the results will be reported elsewhere (S. Kugel Desmoulin, Y. Wang, A. Gangjee, and L.H. Matherly manuscript in preparation).
For growth inhibition assays, cells (CHO, KB, or IGROV1) were plated in 96 well plates (∼5000 cells/well, total volume of 200 μl medium) with a broad range of antifolate concentrations. The medium was RPMI 1640 (contains 2.3 μM folic acid) with 10% dialyzed serum and antibiotics for experiments with R2 and PC43-10 cells. For RT16, D4, KB, and IGROV1 cells, cells were cultured in folate-free RPMI media with 10% dialyzed fetal bovine serum (Invitrogen) and antibiotics supplemented with 2 nM LCV. The requirement for FR-mediated drug uptake in these assays was established in parallel incubation including 200 nM folic acid. For R2/PCFT4 cells, the medium used routine growth inhibition assays was folate-free RPMI 1640 (pH 7.2) containing 25 nM LCV, supplemented with 10% dialyzed fetal bovine serum and antibiotics. For all cell lines, incubations were routinely up to 96 h; metabolically active cells (a measure of cell viability) were assayed with CellTiter-blue Cell Viability Assay (Promega, Madison, WI) and fluorescence was measured (590 nm emission, 560 nm excitation) with a Molecular Dynamics fluorescence plate reader. Data were exported from Softmax Pro software to an Excel spreadsheet for analysis and determinations of IC50s, corresponding to the drug concentrations that result in 50% loss of cell growth.
For some of the in vitro growth inhibition studies, the inhibitory effects of the antifolate inhibitors on de novo thymidylate biosynthesis (i.e., thymidylate synthase) and de novo purine biosynthesis (GARFTase and AICARFTase) were tested by co-incubations with thymidine (10 μM) and adenosine (60 μM), respectively. For de novo purine biosynthesis, additional protection experiments used AICA (320 μM) as a means of distinguishing inhibitory effects at GARFTase from those at AICARFTase.18,34
For assays of colony formation in the presence of the antifolate drugs, KB cells were harvested in log phase, diluted and 100 cells were plated into 60 mm dishes in folate-free RPMI1640 medium supplemented with 2 nM LCV, 10% dialyzed fetal bovine serum, penicillin-streptomycin, and 2 mM glutamine, in the presence or absence of the thieno[2,3-d]pyrimidine compounds and MTX (from 1 to 1000 nM). The highest concentrations (1000 nM) of antifolate drugs were also tested in the presence of folic acid (100 μM) to examine the protection effect. The dishes were incubated at 37°C with 5% CO2 for 10 days. At the end of the incubations, the dishes were rinsed with Dulbecco’s phosphate-buffered saline (DPBS), 5% trichloroacetic acid, borate buffer (10 mM, pH 8.8), followed by 30 min incubation in 1% methylene blue in the borate buffer. The dishes were rinsed with borate buffer and colonies were enumerated for calculating percent colony-forming efficiency normalized to control.
FR Binding Assay
[3H]Folic acid binding was used to assess levels of surface FRs.38 Briefly, cells (e.g., RT16 or D4) (∼1.6×106) were rinsed twice with DPBS followed by a quick rinse with an acetate buffer (10 mM sodium acetate, 150 mM NaCl, pH 3.5) to remove FR-bound folates. Cells were washed twice with ice-cold 2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)-buffered saline (20 mM HEPES, 140 mM NaCl, 5 mM KCl, 2 mM MgCl2, 5 mM glucose, pH7.4) (HBS). Cells were incubated in HBS with [3H]folic acid (50 nM, specific activity 0.5 Ci/mmol) in the presence and absence of unlabeled folic acid or antifolate for 15 min at 0°C. The dishes were rinsed three times with ice-cold HBS, after which the cells were solubilized with 0.5 N sodium hydroxide and aliquots measured for radioactivity and protein contents. Protein concentrations were measured with Folin phenol reagent.39 Bound [3H]folic acid was calculated as pmol/mg protein. Relative binding affinities for assorted folate/antifolate substrates were determined as previously described18 and were calculated as the inverse molar ratio of unlabeled ligands required to inhibit [3H]folic acid binding by 50%. By definition, the relative affinity of folic acid is 1.
PCFT-mediated transport transport assay
To assess PCFT transport activity in R2/PCFT4 and R2/VC cells, transport was assayed at pH 7.2 in HEPES-buffered saline (20 mM HEPES, 140 mM NaCl, 5mM KCl, 2 mM MgCl2, 5 mM glucose) and at pH 5.5 in 4-morphilinopropane sulfonic acid (MES)-buffered saline (20 mM MES, 140mM NaCl, 5mM KCl, 2mM MgCl2, and 5 mM glucose) in 2 ml suspension (1.5 × 107 cells total). Uptake of [3H]MTX (0.5 μM) was measured at 37° C for 2 min, then quenched with 10 ml of ice-cold DPBS. Cells were washed with ice-cold DPBS (3x) and solubilized with 0.5 N NaOH. Levels of intracellular radioactivity were expressed as pmol/mg protein, calculated from direct measurements of radioactivity and protein contents of cell homogenates. Protein concentrations were measured with Folin phenol reagent.39
In vitro GARFT Enzyme Inhibition Assay
Purified recombinant mouse GARFTase enzyme was a gift from Dr. Richard Moran (Virginia Commonwealth University, Richmond, VA).40 Enzyme activity was assayed spectrophotometrically at 37°C using GARFTase (0.75 nM), α,β-GAR (11 μM) and coenzyme 10-formyl-5,8-dideazafolic acid (10 μM) in HEPES buffer (75 mM, pH 7.5) with or without antifolate inhibitor (10 to 30,000 nM). The absorbance of the reaction product, 5,8-dideazafolic acid, was monitored at 295 nm over the first minute as a measure of the initial rate of enzyme activity. IC50s were calculated as the concentrations of inhibitors that resulted in a 50% decrease in the initial velocity of the GARFTase reaction.
In situ GARFT Enzyme Inhibition Assay
Incorporation of [14C]glycine into [14C]FGAR as an in situ measure of endogenous GARFTase activity, was described by Beardsley et al.34 as modified by Deng et al.18 for studies with the GARFTase inhibitor LMX. For these experiments, KB cells were seeded in 4 ml complete folate-free RPMI 1640 plus 2 nM LCV in 60 mm dishes at a density of 2×106 cells per dish. On the next day, the medium was replaced with 2 ml fresh complete folate-free RPMI 1640 plus 2 nM LCV (without supplementing glutamine). Azaserine (4 μM final concentration) was added in the presence and absence of the antifolate inhibitors (0.1, 1, 10, 100, 1000 nM). After 30 min, glutamine (final concentration, 2 mM) and [14C]glycine (final specific activity 0.1mCi/L) were added. Incubations were at 37°C for 15 h, at which time cells were washed (one time) with ice-cold folate-free RPMI 1640 plus serum. Cell pellets were dissolved in 2 ml 5% trichloroacetic acid at 0°C. Cell debris was removed by centrifugation (the cell protein contents in the pellets were measured), and the supernatants were extracted twice with 2 ml of ice-cold ether. The aqueous layer was passed through a 1 cm column of AG1x8 (chloride form), 100-200 mesh (BioRad, Carlsbad, CA), washed with 10 ml of 0.5N formic acid and then 10 ml of 4N formic acid, and finally eluted with eight ml 1N HCl solution. The elutants were collected and determined for radioactivity. The accumulation of radioactive FGAR was calculated as pmol per mg protein over a range of inhibitor concentrations. IC50s were calculated as the concentrations of inhibitors that resulted in a 50% decrease in FGAR synthesis.
Statistical Analyses
Statistical analyses were performed with GraphPad Prism 4.0.
Supplementary Material
Acknowledgement
This work was supported in part by grants from the National Institutes of Health, National Cancer Institute, CA125153 (AG) and CA53535 (LHM). We thank Dr. Wayne Flintoff for the gift of the MTXRIIOuaR2-4 Chinese hamser ovary cells, Dr. Manohar Ratnam for providing a culture of IGROV1 cells, and Dr. Richard Moran for providing recombinant mouse GARFTase and assays reagents.
Abbreviations
- AICA
5-amino-4-imidazolecarboxamide
- AICARFTase
5-amino-4-imidazole-carboxamide ribonucleotide formyltransferase
- CHO
Chinese hamster ovary
- DPBS
Dulbecco’s phosphate-buffered saline
- EI
electron impact
- GARFTase
glycinamide ribonucleotide formyltransferase
- FGAR
formyl glycinamide ribonucleotide
- GAR
glycinamide ribonucleotide
- HBSS
Hank’s balanced salts solution
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HBS
HEPES-buffered saline
- HRMS
high resolution mass spectra
- IC50
50% inhibitory concentration
- LCV
leucovorin
- LMX
Lometrexol
- MTX
methotrexate
- MES
2-(N-morpholino)ethanesulfonic acid
- PMX
Pemetrexed
- RTX
Raltitrexed
- RFC
reduced folate carrier
- TLC
thin layer chromatography
References
- 1.Stokstad ELR. Historical Perspective On Key Advances in the Biochemistry and Physiology of Folates. In: Piccian MF, Stokstad ELR, Gregory JF, editors. Folic Acid Metabolism in Health and Disease. Wiley-Liss; New York: 1990. pp. 1–21. [Google Scholar]
- 2.Matherly LH, Goldman ID. Membrane Transport of Folates. Vitam. Horm. 2003;66:403–456. doi: 10.1016/s0083-6729(03)01012-4. [DOI] [PubMed] [Google Scholar]
- 3.Matherly LH, Hou Z, Deng Y. Human Reduced Folate Carrier: Translation of Basic Biology to Cancer Etiology and Therapy. Cancer Metastasis Rev. 2007;26:111–128. doi: 10.1007/s10555-007-9046-2. [DOI] [PubMed] [Google Scholar]
- 4.Salazar MD, Ratnam M. The Folate Receptor: What Does It Promise in Tissue-Targeted Therapeutics? Cancer Metastasis Rev. 2007;26:141–152. doi: 10.1007/s10555-007-9048-0. [DOI] [PubMed] [Google Scholar]
- 5.Zhao R, Goldman ID. The Molecular Identity and Characterization of a Proton-Coupled Folate Transporter-PCFT; Biological Ramifications and Impact on the Activity of Pemetrexed. Cancer Metastasis Rev. 2007;26:129–139. doi: 10.1007/s10555-007-9047-1. [DOI] [PubMed] [Google Scholar]
- 6.Zhao R, Min SH, Wang Y, Campanella E, Low PS, Goldman ID. A Role for the Proton-Coupled Folate Transporter (PCFT -SLC46A1) in Folate Receptor-Mediated Endocytosis. J. Biol. Chem. 2009 doi: 10.1074/jbc.M807665200. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Monahan BP, Allegra CJ. Antifolates. In: Chabner BA, Longo DL, editors. Cancer Chemotherapy and Biotherapy. 4th Edison Lippincott-Raven; Philadelphia: pp. 109–148. [Google Scholar]
- 8.Chu E, Callender MA, Farrell MP, Schmitz JC. Thymidylate Synthase Inhibitors as Anticancer Agents: From Bench to Bedside. Cancer Chemother. Pharmacol. 2003;52(Suppl 1):S80–89. doi: 10.1007/s00280-003-0625-9. [DOI] [PubMed] [Google Scholar]
- 9.Hazarika M, White RM, Johnson JR, Pazdur R. FDA Drug Approval Summaries: Pemetrexed (Alimta) Oncologist. 2004;9:482–488. doi: 10.1634/theoncologist.9-5-482. [DOI] [PubMed] [Google Scholar]
- 10.Cohen MH, Johnson JR, Wang YC, Sridhara R, Pazdur R. FDA Drug Approval Summary: Pemetrexed for Injection (Alimta) for the Treatment of Non-Small Cell Lung Cancer. Oncologist. 2005;10:363–368. doi: 10.1634/theoncologist.10-6-363. [DOI] [PubMed] [Google Scholar]
- 11.Taylor EC, Harrington PJ, Fletcher SR, Beardsley GP, Moran RG. Synthesis of the Antileukemic Agents 5,10-Dideazaaminopterin and 5,10-dideaza-5,6,7,8-tetrahydroaminopterin. J. Med. Chem. 1985;28:914–921. doi: 10.1021/jm00145a012. [DOI] [PubMed] [Google Scholar]
- 12.Stock CC, Reilly HC, Buckley SM, Clarke DA, Rhoads CP. Azaserine, a New Tumour-Inhibitory Substance; Studies with Crocker Mouse Sarcoma 180. Nature. 1954;173:71–72. doi: 10.1038/173071a0. [DOI] [PubMed] [Google Scholar]
- 13.Lewis LR, Robins RK, Cheng CC. The Preparation and Antitumor Properties of Acylated Derivatives of 6-Thiopurine Ribosides. J. Med. Chem. 1964;7:200–204. doi: 10.1021/jm00332a015. [DOI] [PubMed] [Google Scholar]
- 14.Hertz R, Tullner WW. Inhibition of Estrogen-induced Growth in the Genital Tract of the Female Chick by a Purine Antagonist; Reversal by Adenine. Science. 1949;109:539. doi: 10.1126/science.109.2839.539. [DOI] [PubMed] [Google Scholar]
- 15.Jackson RC, Harkrader RJ. The Contributions of de novo and Salvage Pathways of Nuceotide Biosynthesis in Normal and Malignant Cells. In: Tattersall MHN, Fox RM, editors. Nucleosides and Cancer Treatment. Academic Press; Sydney: 1981. pp. 18–31. [Google Scholar]
- 16.Tonkinson JL, Marder P, Andis SL, Schultz RM, Gossett LS, Shih C, Mendelsohn LG. Cell Cycle Effects of Antifolate Antimetabolites: Implications for Cytotoxicity and Cytostasis. Cancer Chemother. Pharmacol. 1997;39:521–531. doi: 10.1007/s002800050608. [DOI] [PubMed] [Google Scholar]
- 17.Smith SG, Lehman NL, Moran RG. Cytotoxicity of Antifolate Inhibitors of Thymidylate and Purine Synthesis to WiDr Colonic Carcinoma Cells. Cancer Res. 1993;53:5697–5706. [PubMed] [Google Scholar]
- 18.Deng Y, Wang Y, Cherian C, Hou Z, Buck SA, Matherly LH, Gangjee A. Synthesis and Discovery of High Affinity Folate Receptor-Specific Glycinamide Ribonucleotide Formyltransferase Inhibitors with Antitumor Activity. J. Med. Chem. 2008;51:5052–5063. doi: 10.1021/jm8003366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mauritz R, Peters GJ, Kathmann I, Teshale H, Noordhuis P, Comijn EM, Pinedo HM, Jansen G. Dynamics of Antifolate Transport via the Reduced Folate Carrier and the Membrane Folate Receptor in Murine Leukaemia Cells in vitro and in vivo. Cancer Chemother. Pharmacol. 2008 doi: 10.1007/s00280-008-0683-0. published online, Feb. 19, 2008. [DOI] [PubMed] [Google Scholar]
- 20.Elnakat H, Ratnam M. Distribution, Functionality and Gene Regulation of Folate Receptor Isoforms: Implications in Targeted Therapy. Adv. Drug Deliv. 2004;56:1067–1084. doi: 10.1016/j.addr.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 21.Leamon CP, Reddy JA, Vlahov IR, Westrick E, Dawson A, Dorton R, Vetzel M, Santhapuram HK, Wang Y. Preclinical Antitumor Activity of a Novel Folate-Targeted Dual Drug Conjugate. Mol. Pharm. 2007;4:659–667. doi: 10.1021/mp070049c. [DOI] [PubMed] [Google Scholar]
- 22.Lu Y, Wu J, Gonit M, Yang X, Lee A, Xiang G, Li H, Liu S, Marcucci G, Ratnam M, Lee RJ. Role of Formulation Composition in Folate Receptor-Targeted Liposomal Doxorubicin Delivery to Acute Myelogenous Leukemia Cells. Mol. Pharm. 2007;4:707–712. doi: 10.1021/mp070058l. [DOI] [PubMed] [Google Scholar]
- 23.Muller C, Forrer F, Schibli R, Krenning EP, de Jong M. SPECT Study of Folate Receptor-Positive Malignant and Normal Tissues in Mice Using a Novel 99mTc-Radiofolate. J. Nucl. Med. 2008;49:310–317. doi: 10.2967/jnumed.107.045856. [DOI] [PubMed] [Google Scholar]
- 24.Jansen G. Receptor-and Carrier-Mediated Transport Systems for Folates and Antifolates: Exploitation for Folate Chemotherapy and Immunotherapy. In: Jackman AL, editor. Anticancer Development Guide: Antifolate Drugs in Cancer Therapy. Humana Press Inc.; Totowa, NJ: 1999. pp. 293–321. [Google Scholar]
- 25.Ray MS, Muggia FM, Leichman CG, Grunberg SM, Nelson RL, Dyke RW, Moran RG. Phase I Study of (6R)-5,10-Dideazatetrahydrofolate: A Folate Antimetabolite Inhibitory to de novo Purine Synthesis. J. Natl. Cancer Inst. 1993;85:1154–1159. doi: 10.1093/jnci/85.14.1154. [DOI] [PubMed] [Google Scholar]
- 26.Jones TR, Calvert AH, Jackman AL, Brown SJ, Jones M, Harrap KR. A Potent Antitumour Quinazoline Inhibitor of Thymidylate Synthetase: Synthesis, Biological Properties and Therapeutic Results in Mice. Eur. J. Cancer. 1981;17:11–19. doi: 10.1016/0014-2964(81)90206-1. [DOI] [PubMed] [Google Scholar]
- 27.Jones TR, Calvert AH, Jackman AL, Eakin MA, Smithers MJ, Betteridge RF, Newell DR, Hayter AJ, Stocker A, Harland SJ, et al. Quinazoline Antifolates Inhibiting Thymidylate Synthase: Variation of the N10 Substituent. J. Med. Chem. 1985;28:1468–1476. doi: 10.1021/jm00148a016. [DOI] [PubMed] [Google Scholar]
- 28.Jackman AL, Taylor GA, Gibson W, Kimbell R, Brown M, Calvert AH, Judson IR, Hughes LR. ICI D1694, A Quinazoline Antifolate Thymidylate Synthase Inhibitor That is a Potent Inhibitor of L1210 Tumor Cell Growth in vitro and in vivo: A New Agent for Clinical Study. Cancer Res. 1991;51:5579–5586. [PubMed] [Google Scholar]
- 29.Jackman AL, Farrugia DC, Gibson W, Kimbell R, Harrap KR, Stephens TC, Azab M, Boyle FT. ZD1694 (Tomudex): A New Thymidylate Synthase Inhibitor with Activity in Colorectal Cancer. Eur. J. Cancer. 1995;31A:1277–1282. doi: 10.1016/0959-8049(95)00166-g. [DOI] [PubMed] [Google Scholar]
- 30.Bavetsias V, Marriott JH, Melin C, Kimbell R, Matusiak ZS, Boyle FT, Jackman AL. Design and Synthesis of Cyclopenta[g]quinazoline-based Antifolates as Inhibitors of Thymidylate Synthase and Potential Antitumor Agents. J. Med. Chem. 2000;43:1910–1926. doi: 10.1021/jm991119p. [DOI] [PubMed] [Google Scholar]
- 31.Bavetsias V, Marriott JH, Theti DS, Melin JC, Wilson SC, Jackman AL. Cyclopenta[g]quinazoline-based Antifolates: The Effect of the Chirality at the 6-position on the Inhibition of Thymidylate Synthase (TS) Bioorg Med. Chem. Lett. 2001;11:3015–3017. doi: 10.1016/s0960-894x(01)00612-6. [DOI] [PubMed] [Google Scholar]
- 32.Larock CR, Leung W, Stolz-Dunn S. Synthesis of Aryl-substituted Aldehydes and Ketones via Palladium-catalyzed Coupling of Aryl Halides and Non-allylic Unstaturated Alcohols. Teteahedron Lett. 1989;30:6629–6632. [Google Scholar]
- 33.Rosowsky A, Chen KK, Lin M. 2,4-Diaminothieno [2,3-d]pyrimidines as Antifolates and Antimalarials. Synthesis of 5,6-Disubstituted Derivatives and Related Tetracyclic Analogs. J. Med. Chem. 1973;16:191–194. doi: 10.1021/jm00261a004. [DOI] [PubMed] [Google Scholar]
- 34.Beardsley GP, Moroson BA, Taylor EC, Moran RG. A New Folate Antimetabolite, 5,10-Dideaza-5,6,7,8-tetrahydrofolate is a Potent Inhibitor of de novo purine Synthesis. J. Biol. Chem. 1989;264:328–333. [PubMed] [Google Scholar]
- 35.Fry DW, Yalowich JC, Goldman ID. Rapid Formation of Poly-Gamma-glutamyl Derivatives of Methotrexate and Their Association with Dihydrofolate Reductase as Assessed by High Pressure Liquid Chromatography in the Ehrlich Ascites Tumor Cell in vitro. J. Biol. Chem. 1982;257:1890–1896. [PubMed] [Google Scholar]
- 36.Flintoff WF, Davidson SV, Siminovitch L. Isolation and Partial Characterization of Three Methotrexate-Resistant Phenotypes from Chinese Hamster Ovary Cells. Somatic Cell Genet. 1976;2:245–261. doi: 10.1007/BF01538963. [DOI] [PubMed] [Google Scholar]
- 37.Wong SC, Proefke SA, Bhushan A, Matherly LH. Isolation of Human cDNAs that Restore Methotrexate Sensitivity and Reduced Folate Carrier Activity in Methotrexate Transport-Defective Chinese Hamster Ovary Cells. J. Biol. Chem. 1995;270:17468–17475. doi: 10.1074/jbc.270.29.17468. [DOI] [PubMed] [Google Scholar]
- 38.Chattopadhyay S, Wang Y, Zhao R, Goldman ID. Lack of Impact of the Loss of Constitutive Folate Receptor Alpha Expression, Achieved by RNA Interference, on the Activity of the New Generation Antifolate Pemetrexed in HeLa Cells. Clin. Cancer Res. 2004;10:7986–7993. doi: 10.1158/1078-0432.CCR-04-1225. [DOI] [PubMed] [Google Scholar]
- 39.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein Measurement with the Folin Phenol Reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 40.Sanghani SP, Moran RG. Tight Binding of Folate Substrates and Inhibitors to Recombinant Mouse Glycinamide Ribonucleotide Formyltransferase. Biochemistry. 1997;36:10506–10516. doi: 10.1021/bi970825u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.