

Abstract

Dirhodium(II) caprolactamate exhibits optimal efficiency for the production of the tert-butylperoxy radical, which is a selective reagent for hydrogen atom abstraction. These oxidation reactions occur with aqueous tert-butyl hydroperoxide (TBHP) without rapid hydrolysis of the caprolactamate ligands on dirhodium. Allylic oxidations of enones yield the corresponding enedione in moderate to high yields, and applications include allylic oxidations of steroidal enones. Although methylene oxidation to a ketone is more effective, methyl oxidation to a carboxylic acid can also be achieved. The superior efficiency of dirhodium(II) caprolactamate as a catalyst for allylic oxidations by TBHP (mol % catalyst, % conversion) is described in comparative studies with other metal catalysts that are also reported to be effective for allylic oxidations. That different catalysts produce essentially the same mixture of products with the same relative yields suggests that the catalyst is not involved in product forming steps. Mechanistic implications arising from studies of allylic oxidation with enones provide new insights into factors that control product formation. A previously undisclosed disproportionation pathway, catalyzed by the tert-butoxy radical, of mixed peroxides for the formation of ketone products via allylic oxidation has been uncovered.
Keywords: Selective hydrogen atom abstraction, enediones, steroidal enones, disproportionation, mixed peroxides, cholesterol oxidation
Introduction
Allylic oxidations are of fundamental importance in synthetic organic chemistry, and a variety of reagents have been used for this transformation.1,2 Although stoichiometric selenium dioxide3 and chromium(VI)4 reagents have long been used to convert the methylene adjacent to a carbon-carbon double bond to a carbonyl group, the most promising oxidant, applicable to catalytic methods, is tert-butyl hydroperoxide (TBHP).5,6,7 Selectivity for allylic oxidation by TBHP is due to the ability of the tert-butylperoxy radical to remove a hydrogen atom from the activated site having the lowest carbonhydrogen bond dissociation energy.8 However, application of this methodology for allylic oxidation with compounds of increasing complexity has been demonstrated in very few cases.
We have reported that dirhodium caprolactamate, Rh2(cap)4, is a highly effective catalyst for allylic oxidations by TBHP (eq 1),9 and we have recently shown that this methodology can be used for highly selective allylic oxidations with Δ5-steroidal alcohols.10 During our initial investigation on the oxidation of cycloalkenes, we observed that those substrates bearing electron-withdrawing substituents were more difficult to oxidize than those with electron-donating substituents. A similar inhibition was reported in groundbreaking studies of TBHP oxidations of α,β-enones to 1,4-enediones facilitated by Pearlman’s catalyst [20% Pd(OH)2 on carbon].11 Substrates having electron-withdrawing substituents generally require greater amounts of TBHP to reach 100% conversion and, with Rh2(cap)4,9 require higher catalyst loading.
 |
(1) |
Another feature of allylic oxidations using TBHP is the frequent use of the oxidant in decane or benzene6,7,9,11,12 as an anhydrous solution, instead of the much safer and less expensive 70% TBHP in water (T-HYDRO®). We began our investigations with TBHP in decane based on our belief that hydrolysis of carboxamidate ligands on the Rh2(cap)4 catalyst would be avoided under these conditions.9 However, we subsequently reported effective allylic oxidations by T-HYDRO® catalyzed by Rh2(cap)4,10 and we recently described propargylic oxidations that employed water as the reaction solvent.13 The catalytically active rhodium species remains intact for sufficient periods under these conditions.
To fully understand the scope, limitations, and mechanism of this oxidative methodology we chose to focus our attention toward relatively unreactive α,β-unsaturated carbonyl compounds as substrates, and we were particularly attentive to those that have not been commonly explored. Curiously, and with few exceptions, the majority of allylic oxidations have been limited to cyclic systems. Moreover, there have not been any reported examples of the oxidation of a methyl group adjacent to a carbon-carbon double bond.
In addition to the use of palladium catalysis for allylic oxidation by TBHP,11 copper(II) and iron(III) salts have also been employed,14,15 as have chromium(VI),16 manganese(III),17 copper iodide,18 cobalt acetate,19 and ruthenium(III).20 Although the tert-butylperoxy radical is the most likely hydrogen abstraction agent,8–10 the possibility of oxo-metal involvement, especially with ruthenium catalysts,21 persists. For that reason, having a model system able to distinguish between these mechanistic pathways would provide a better understanding of this oxidative process. We now report the results of this investigation
Results and Discussion
 |
(2) |
Oxidation of trans-3-nonene-2-one, whose fire bee toxin22 product 1 has been previously prepared by allylic oxidation,9 was selected for optimization (eq 2). Using T-HYDRO® as the oxidant and Rh2(cap)4 as the catalyst at 40 °C, various solvents were examined for suitability. 1,2-Dichloroethane (DCE), nitromethane, and water all led to comparable conversions to 1 after 16 hours when 8 equivalents of TBHP were employed. In contrast to previous reports of catalytic TBHP oxidations under anhydrous conditions,6,8 the addition of weak bases such as sodium bicarbonate, pyridine, or triethylamine lowered percent conversions. Two factors limited the efficiency of this allylic oxidation: (1) the slow rate of oxidation of the substrate relative to radical chain decomposition of TBHP (kP/kD, Scheme 1)23 and (2) the decrease in the concentration of the catalytically active dirhodium species (Figure 1). For the former, increasing the number of molar equivalents of TBHP increased percent conversion, and increasing the reaction temperature from room temperature to 40 °C increased the rate of oxidation. For the latter, adding Rh2(cap)4 in two portions, 0.5 mol % to initiate the reaction and the second 0.5 mol % portion after 16 h, ensured complete conversion and high product yield. Subsequent studies revealed that complete conversion could be achieved using as little as 0.1 mol % Rh2(cap)4 if applied twice, each with 5.0 molar equiv of TBHP, after the initial addition (22 and 44 h), but these conditions were not further pursued.
Scheme 1.

Figure 1.

Dirhodium (II,III) caprolactamate visible spectrum as a function of time (1, 3, 5, and 7 h): TBHP (0.27 M) in DCE with 1.0 mol % Rh2(cap)4 without olefinic substrate. Note that dirhodium (II,III) caprolactamate (λmax 507 and 974 nm) is clearly visible even after 5 h and that dirhodium (II,II) caprolactamate (λmax 607 nm) becomes visible as the reaction proceeds.
The role of dirhodium caprolactamate in these oxidation reactions is suggested from the spectral observations revealed in Figure 1. The conversion of Rh2(cap)4 to its oxidized Rh2(cap)4(OH) form by TBHP has been previously reported,10 and we speculated that Rh2(cap)4(OH) would be converted back to Rh2(cap)4 via TBHP. The observation of Rh2(cap)4 in Figure 1, combined with confirmation of its initial formation and presence throughout the course of oxidation through HPLC analysis, confirms the catalytic role for Rh2(cap)4 that is described in Scheme 2. Thus, dirhodium caprolactamate is effective and efficient for the production of the tert-butylperoxy radical.
Scheme 2.

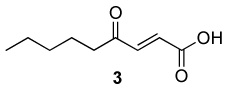
Optimum conditions were selected for oxidation of α,β-unsaturated substrates (0.54 M in DCE) that included initial addition of 0.5 mol % Rh2(cap)4 and 4.0 or 5.0 equiv T-HYDRO® followed by the same portion of catalyst and oxidant after 16 hours. Reactions were performed at 40 °C and terminated 40 hours after the initial catalyst/oxidant addition. Percent conversion at 16 hours was only half that at 40 hours, and using acetonitrile or nitromethane as solvent resulted in lower conversions than that achieved in DCE. Selected α,β-unsaturated carbonyl compounds were oxidized according to this methodology, and the results are reported in Table 1. Noteworthy are the relatively high yields of oxidized product obtained with acyclic compounds. For example, trans-4-oxo-2-nonenoic acid (3), reported to have antibiotic activity,24 is efficiently prepared in one transformation (entry 3), improving upon the previously described multistep syntheses.25 Oxidation of methyl crotonate under the same conditions yielded monomethyl fumaric acid (8) in modest isolated yield.
Table 1.
Oxidation of α,β-Unsaturated Carbonyl Compounds by T-HYDRO® catalyzed by Rh2(cap)4.a
| Entry | Substrate | Product | Equiv. TBHP |
%Yield b |
|---|---|---|---|---|
| 1 | 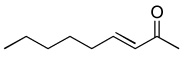 |
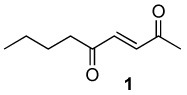 |
10.0 8.0 |
84 76 |
| 2 | 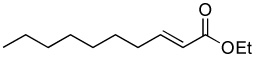 |
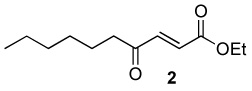 |
10.0 | 76 |
| 3 | 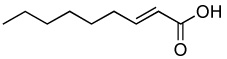 |
 |
10.0 | 87 |
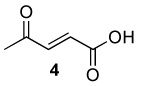
| 4 | 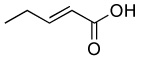 |
 |
10.0 8.0 |
85 82 |
| 5 | 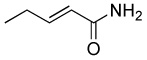 |
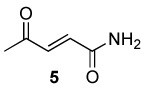 |
8.0 | 65 |
| 6 | 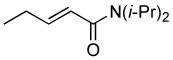 |
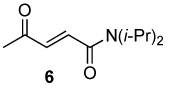 |
8.0 | 70 |
| 7 | 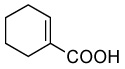 |
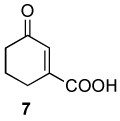 |
8.0 | 64 |
| 8 | 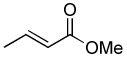 |
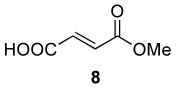 |
8.0 | 32 |
Reactions were performed with 1.36 mmol of substrate in 2.5 mL of DCE to which was added 4.0 or 5.0 equiv of TBHP (70% in water) and 0.50 mol % Rh2(cap)4 and the solution was heated at 40 °C. After 16 hours an additional 4.0 or 5.0 equiv of TBHP (70% in water) and 0.50 mol % Rh2(cap)4 was added, and reaction was continued for an additional 24 hours.
Isolated yield after column chromatography; carboxylic acids were purified by recrystallization from ethyl acetate and the mass yield after recrystallation is given.
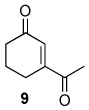
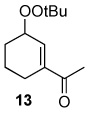
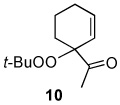
The oxidation reactions reported in Table 1 are remarkably free of by-products. However, while investigating the T-HYDRO® oxidation of 1-acetylcyclohexene (a substrate that was reported to be cleanly oxidized by TBHP in decane using Rh2(cap)4 aided by potassium carbonate8) we uncovered three unreported mixed peroxides (10, 11, and 12) in addition to the previously reported 1310 and desired enedione 9. Two of these mixed peroxides (10 and 11) were revealed during a study of the influence of TBHP molar amount on product formation (Figure 2); mixed peroxide 12 was isolated from a reaction that employed 0.5 mol % Rh2(cap)4 and 4.0 molar equivalents TBHP. That enone 11 results from allylic oxidation of 10 is clearly suggested from the data in Figure 2.
Figure 2.

Oxidation of 1-acetylcyclohexene (0.27 M in DCE, 40 °C) by T-HYDRO®, catalyzed by Rh2(cap)4. The relative yield of oxidation products as a function of molar equivalents of TBHP was determined after 16 hours.
Mixed peroxide 10 is formed in competition with mixed peroxide 13 and enedione 9. Formation of mixed peroxides 10, 11, 12, and 13 is consistent with the generation of intermediate allyl radicals 14 and 15 (Scheme 3) that subsequently react with the tert-butylperoxy radical. As can be seen from overall product formation, intermediate 15 is more prevalent than 14. Conversely, oxidations of cyclohexene derivatives bearing electron-donating substituents at the 1-position (e.g., AcO, tert-butyl) under identical conditions did not form detectable amounts of mixed peroxide products.9
Scheme 3.

Further support that the pathway described in Scheme 3 is operative in Rh2(cap)4 catalyzed oxidations by TBHP was demonstrated through oxidation of enantiomerically pure (R)-(+)-limonene with T-HYDRO® in water (eq 3). Unlike previous TBHP oxidations of 1-substituted cyclohexenes, from which the dominant product resulted from oxidation at the 3-position,5–9 the dominant product of the oxidation of (R)-(+)-limonene was racemic carvone (16, 44 % isolated yield). The observed carvone 16 arose from hydrogen atom abstraction from the 6-position of limonene to produce a racemic free radical that underwent subsequent transformation. The expected oxidation at the 3-position of limonene, affording isopiperitinone 17, was observed as the minor reaction product (7 % isolated yield). Other oxidation products were also formed, but none in yields of 5 % or greater. These minor products were part of an inseparable residue; however, resonances in the 13C NMR spectrum of the residue provided suggested TBHP capture at the multiple allylic positions of limonene, but this was not further pursued. The preference for hydrogen atom abstraction from the 6-position, rather than from the 3-position, suggests the operation of steric factors. Although allylic oxidations of limonene, other than those by selenium dioxide,26 have been investigated, those using TBHP have been reported to form carvone and related compounds derived from hydrogen abstraction at the 6-position, but there has not been prior mention of their optical purity.27
 |
(3) |
To determine the eventual fate of reaction products, mixed peroxides 10, 12, and 13 were isolated, purified, and then subjected to reaction conditions similar to those reported in Table 1. The mixed peroxide and dirhodium catalyst were diluted with CD2Cl2 (to 0.27 M), then treated with 70 % TBHP in D2O. Biphenyl was added as an internal standard and the reactions were monitored by 1H NMR. The results of these experiments are described in equation 4–equation 6 which show that further oxidation occurs, resulting in either ketone formation (9, 11, and 18/eq 5) or in the production of a second mixed peroxide (18/eq 6, 19, and 20). Perhaps the most remarkable result is that of equation 6 whereby mixed peroxide 13 is converted, in relatively high yield, to diketone 9, supporting a previously undisclosed catalytic disproportionation pathway for the formation of ketone products via allylic oxidation (Scheme 4). Indeed, this pathway may explain the common inability to detect mixed peroxide products in oxidations by TBHP.
 |
(4) |
 |
(5) |
 |
(6) |
Scheme 4.

The relative stability of allylic oxidation product 9 is seen in the minor extent of its conversion to mixed peroxide 16 (9 → 16: 13% yield at 13% conversion after 20 h using the same reaction conditions as in equation 4–equation 6). This result is consistent with the low reactivity of α,β-unsaturated carbonyl compounds towards allylic oxidation (see Scheme 1) and to the importance of steric influences in hydrogen atom abstraction reactions of the tert-butylperoxy radical. Interestingly, oxidation of 9 is an order of magnitude less pronounced than oxidation of 1-acetylcyclohexene.
TBHP oxidation of 1-acetylcyclohexene, in association with intermediate allyl radicals 14 and 15 (Scheme 3), presents an opportunity to easily compare the Rh2(cap)4 catalyzed oxidations with those from other metal catalysts. Oxidation products from reactions of TBHP with 1-acetylcylohexene using several common catalysts using a limited amount of TBHP (4.0 equiv), under exactly the same conditions, are reported in Table 2. Comparison with the prior Rh2(cap)4 catalytic methodology from our laboratory9 using TBHP in decane is also provided. Enedione 9, as well as mixed peroxides 10 and 13, are easily identified by their distinctive 1H NMR chemical shifts. Particularly noticeable is the variability in percent conversion, which is a reflection of the turnover rate for production of the active oxidant. Even at 0.1 mol % Rh2(cap)4 percent conversion is exceptional among the catalysts examined. Of special significance is the low variability in the ratio of 9 + 13 to 10, which is approximately 1.9 ± 0.3 for all catalysts examined. This value, applicable to all of the catalysts and changing slightly with the amount of catalyst used, suggests that these allylic oxidations occur through the same reaction pathway - via the allyl free radical from hydrogen atom abstraction from the 3-position of 1-acetylcyclohexene - and that the trapping of this intermediate occurs in the same fashion by the same radical species. That the catalyst is not involved in the product forming step is also drawn from the low variability in the ratio of 9 + 13 to 10, and this conclusion is possibly applicable to prior claims7a,9,11,18 of metal catalyst involvement in the product determining step in allylic oxidation reactions of TBHP. In addition, intermediate oxo-ruthenium(IV) complexes are often implicated in alkane oxidation reactions catalyzed by ruthenium(III) chloride,28 but the present results suggest that such species are not involved in the allylic oxidation process involving TBHP in water. Also expressed in Table 2, percent conversion of 1-acetylcyclohexene is highest with Rh2(cap)4, and CuI appears to be only slightly less efficient, but at much higher catalyst loading.
Table 2.
Oxidation of 1-acetylcyclohexene by TBHP in CD2Cl2 at 40 °C with various catalysts.a
| Catalyst | Oxidant | % Conversion b | % Yieldc | Relative %yieldd | Ratio (9 + 13)/10 | ||
|---|---|---|---|---|---|---|---|
 |
 |
 |
|||||
| 0.5 mol % Rh2(cap)4 | 70 % TBHP in water | 80 | 71 | 27 | 34 | 39 | 1.6 |
| 0.1 mol % Rh2(cap) | 70 % TBHP in water | 80 | 63 | 40 | 30 | 30 | 2.3 |
| 0.5 mol % Rh2(cap)4 0.5 equiv. K2C03 | 6.7 M TBHP in decane | 82 | 49 | 45 | 22 | 33 | 2.0 |
| 2.0 mol % RuCl3•nH2O | 70 % TBHP in water | 59 | 42 | 21 | 38 | 40 | 1.5 |
| 2.0 mol % CuI | 70 % TBHP in water | 71 | 51 | 24 | 37 | 39 | 1.6 |
| 5.0 mol % Pd(OH)2 | 70 % TBHP in water | 38 | 36 | 17 | 50 | 33 | 2.0 |
| 5.0 mol % Pd(OH)2 0.25 equiv. K2CO3 | 70 % TBHP in water | 62 | 42 | 40 | 31 | 29 | 2.4 |
Reactions were performed with 1-acetylcyclohexene (0.27 M in CD2C12), using 4.0 equiv 70 % TBHP in D20 and the specified amount of catalyst with 1.0 equiv of biphenyl as internal standard. The reactions were performed in a standard NMR tube, heated to 40 °C, and were monitored by 1H NMR.
Percent conversion for each reaction was measured by the amount of 1-acetylcyclohexene remaining after 20 hours, relative to the internal standard.
Percent yield was determined by the sum of products formed after 20 hours, relative to the internal standard.
The results from duplicate runs were reproducible within 5 % of the reported values.
The mechanistic pathway for the Rh2(cap)4 mediated allylic oxidation that is consistent with the reported data is outlined in Scheme 5 using cyclohexene as the model olefinic substrate. Initial oxidation occurs between TBHP and Rh2(cap)4 to form the oxidized dirhodium(II,III) intermediate and the tert-butoxy radical that, in turn, abstracts a hydrogen atom from TBHP at a rate that is much faster than hydrogen atom abstraction from the allylic position of the alkene.29,30 The tert-butylperoxy radical undergoes selective hydrogen atom abstraction from the hydrocarbon substrate: a process that is well documented and universally accepted.9, 10,31,32 The existence of an allylic radical in these reactions is also consistent with the conversion of optically pure limonene into racemic carvone (eq 3). Capture of the allyl radical by the tert-butylperoxy radical33 forms the mixed peroxide that is susceptible to tert-butoxy radical catalyzed disproportionation.
Scheme 5.

However, mixed peroxides are not observed as reaction intermediates in results from the allylic oxidation of cholesterol 21 to form 7-ketocholesterol 22 and, instead, 7-hydroxycholesterol 23 and the 7-hydroperoxycholesterol 24 in both α and β-stereoisomeric forms are obtained.10,34,35 Mixed peroxides analogous to 24 from TBHP oxidation of cholesteryl acetate have been reported as minor products (< 5%),36 and they may be present in lower amounts in related reactions with cholesterol.34 Both 23 and 24 are understood to be products of dioxygen capture of an allyl radical in one pathway for the formation of the enone 22.34 Upon close analysis of reaction products from the oxidation of 21, we were able to identify and isolate α– and β-23 and 24 (eq 7), but we were not able to confirm the presence of mixed peroxides. In addition, based on the seminal work of Schenck37 and similar subsequent reports,38 the intermediate allyl radical from hydrogen atom abstraction at the 7-position of cholesterol may form tertiary 5-hydroperoxycholesterol, but this product is not observed due to rapid [2,3]-sigmatropic rearrangement to 7-hydroperoxycholesterol 24.
 |
(7) |
An explanation for the hydroperoxide, diol, and enone oxidation products from cholesterol is that in this case the allyl radical reacts with dioxygen to form an intermediate alkylperoxy radical (Scheme 6, shown with cyclohexene as substrate). The alkylperoxy radical either abstracts a hydrogen atom from the allylic position of another hydrocarbon to form hydroperoxide or undergoes oxidation of Rh24+ to form the enone product directly. This direct conversion explains why alcohols are either not observed9 or are generally very minor products in TBHP oxidations that are catalyzed by Rh2(cap)4.10 This pathway provides a viable alternative to bimolecular disproportionation of two peroxy radicals that would form an alcohol and a ketone in equivalent amounts along with dioxygen (the Russell mechanism).39 However, the question remains regarding the relative ability of an allyl radical to capture either the tert-butyl peroxy radical or dioxygen, and this question is not answered in this study.
Scheme 6.

The oxidations of testosterone (eq 8), 17-acetyltestosterone (eq 9), 4-androstene-3,17-dione (eq 10), and 4-cholesten-3-one (eq 11) were performed under the same conditions as those reported in Table 1. Although using 1,2-dichloroethane as solvent was initially believed to be necessary to dissolve the water-insoluble steroidal compounds, examination of the oxidation process using water as solvent actually led to an improvement in product yield (e.g., 68 % yield for 26 after 48 h with 12 equiv of T-HYDRO®). In contrast to oxidations of cholesterol in which the secondary alcohol at the 3-position was stable to oxidative conversion to a ketone, oxidation of the secondary alcohol at the 17-position of testosterone was competitive with allylic oxidation of the enone, and both androst-4-ene-3,17-dione and androst-4-ene-3,6,17-trione accompanied formation of 25. This oxidative process appears to be general for strained secondary alcohols (cyclopentanol, endo-norbornanol, borneol, for example), but optimization of these reactions has not been pursued. In an attempt to effect sequential oxidation on both sides of the carbon-carbon double bond, 3β-acetoxyandrost-5-en-17-one was subjected to sequential treatments with TBHP and Rh2(cap)4 (eq 12); the desired 3β-acetoxyandrost-5-en-4,7,17-trione 27 was formed in 54% yield.
 |
(8) |
 |
(9) |
 |
(10) |
 |
(11) |
 |
(12) |
In summary, dirhodium(II) caprolactamate is an efficient engine for the production of the tert-butylperoxy radical, which is a selective reagent for hydrogen atom abstraction. This efficiency is exemplified in comparative studies with other metal catalysts that are reported to be operative for allylic oxidations with TBHP. As a result, allylic oxidations, even of normally recalcitrant substrates (such as enones), can effectively produce the corresponding keto-products including steroidal enones. Mechanistic implications arising from studies of allylic oxidation with less reactive enones have provided new insights into factors that control product formation.
Experimental Section
General Methods
HPLC grade solvents were used for extraction and purification. Unless otherwise indicated, all reactions were run under atmospheric conditions. All reagents were obtained from commercial sources. Dirhodium(II) caprolactamate [Rh2(cap)4·2CH3CN] was prepared as previously described.40 Silica gel plates (0.25 mm, 60 F254) were used for analytical thin layer chromatography and spots were visualized using 254 nm ultraviolet light before using potassium permanganate, vanillin, or anisaldehyde stains as visualizing agents. Chromatographic purifications were performed using silica gel (60 microns, 40–63 mesh) according to the method of Still.41 Yields reported are for isolated compounds according to their mass unless otherwise noted. All products were characterized and in agreement with those that were previously described. 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were obtained on a 400 MHz spectrometer as solutions in CDCl3 unless otherwise noted. Chemical shifts are reported in parts per million (ppm, δ) relative to internal Me4Si (δ 0.00) for 1H and relative to internal chloroform (δ 77.0) for 13C; coupling constants are reported in Hertz (Hz).
General Procedure for the Oxidation of α,β-Unsaturated Carbonyl Compounds
A 10 mL vial equipped with a stirbar was charged with substrate (1.36 mmol) and Rh2(cap)4 (5 mg, 0.007 mmol). Solvent (2.5 mL) was added followed by the addition of TBHP (0.75 mL, 5.5 mmol, 4 equiv). The vial was loosely capped and stirred for 16 hrs, then the second portion of Rh2(cap)4 (5 mg, 0.007 mmol) and TBHP (0.75 mL, 5.5 mmol, 4 equiv) was added. After an additional 24 hrs, the solution was concentrated and purified by column chromatography to obtain analytically pure compounds whose spectral characteristics were identical to those previously reported: trans-3-nonen-2,5-dione (1),11 ethyl (E)-4-oxo-2-decenoate (2).42 (E)-4-oxo-2-pentenamide (5),43 4-androsten-17β-3,6-dione (25),44 17β-acetoxyandrost-4-en-3,6-dione (26),45 4-androstene-3,6,17-trione (27),46 4-cholesten-3,6-dione (28)47 and acids were purified by recrystallization in ethyl acetate and matched with those in the literature: (E)-4-oxo-2-nonenoic acid (3),25a (E)-4-oxo-2-pentenoic acid (4),48 2-cyclohexenone-1-carboxylic acid (7).49 and fumaric acid monomethyl ester (8).50
Oxidation of 1-Acetylcyclohexene - Isolation and Characterization of 10, 11, 12, 13 and 18
1-Acetylcyclohexene (0.750 g; 6.04 mmol) and Rh2(cap)4 (22 mg; 0.030 mmol) were dissolved in 22 mL of 1,2-dichloroethane in a 100 mL round-bottom flask. T-HYDRO® (3.31 mL; 24.1 mmol) was slowly added (1 mL/minute), and the color of the reaction solution turned from purple to brown-red in color. The flask was capped with a rubber septum, vented with a 18-gauge needle, and heated at 40 °C in an oil bath. After 17.5 hours, the reaction was concentrated under reduced pressure to yield 2.11 g of a red-brown oil. The oil was purified via silica gel column chromatography (100 % hexanes to 2:1 hexanes: ethyl acetate, gradient). Only the purest fractions were used for full characterization: 55 mg of 10, 20 mg of 11, 18 mg of 12, 32 mg of 13, 20 mg of 16, and 312 mg of 3-acetyl-2-cyclohexenone (9).
1-(1-tert-Butylperoxy)cyclohex-2-enyl)ethanoate (10)
Rf 0.69 (20% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) δ 6.12 (ddd, J = 3.2, 4.4, 10.0 Hz, 1 H), 5.66 (ddd, J = 2.0, 2.0, 10.0 Hz, 1 H), 2.31 (s, 3 H), 2.07–2.02 (comp, 2 H), 1.97–1.91 (comp, 2 H), 1.73–1.65 (comp, 2 H), 1.23 (s, 9 H); 13C NMR (100 MHz, CDCl3) δ 210.0, 135.6, 123.0, 85.2, 79.7, 26.9, 26.6, 25.3, 24.4, 18.2; FTIR (thin film) 2979, 2935, 2871, 1720, 1363, 1197, 735 cm−1. Exact mass calculated for C12H20O3 + H (ES) 213.1490; found 213.1115.
4-(tert-Butylperoxy)-4-ethanoylcyclohex-2-enone(11)
Rf 0.37 (20% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) δ 6.99 (d, J = 10.4 Hz, 1 H), 6.17 (d, J = 10.4 Hz, 1 H), 2.57 (m, 1 H) 2.46–2.44 (comp, 2 H), 2.38 (s, 3 H), 2.27–2.22 (comp, 3 H), 1.25 (s, 9 H); 13C NMR (100 MHz, CDCl3) δ 207.6, 198.0, 145.2, 131.8, 84.4, 81.06, 33.3, 28.7, 26.4, 24.6; FTIR (thin film) 3056, 2980, 2937, 1720, 1688, 1365, 1192, 874, 737 cm−1. Exact mass calculated for C12H18O4 + H (ES) 227.1283; found 227.1288.
1-(6-tert-Butylperoxy)cyclohex-1-enyl)ethanoate (12)
Rf 0.58 (20% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) δ 7.08 (dd, J = 4.8, 2.8 Hz, 1 H), 5.00 (bs, 1 H), 2.40–2.38 (comp, 2 H), 2.35 (m, 1 H), 2.32 (s, 3 H), 2.20–2.17 (m, 1 H), 1.75–1.78 (m, 1 H), 1.63–1.66 (m, 1 H), 1.26 (s, 9 H); 13C NMR (100 MHz, CDCl3) δ 198.0, 146.1, 136.2, 80.1, 72.1, 26.5, 26.3, 26.0, 25.4, 15.7; FTIR (thin film) 3060, 2928, 2977, 1674, 1362, 1258, 1240, 1196, 736 cm−1. Exact mass calculated for C12H20O3 + H (ES) 213.1490; found 213.1481.
1-(3-tert-Butylperoxy)cyclohex-1-enyl)ethanoate (13)
Rf 0.62 (20% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) δ 6.86 (t, J = 1.5 Hz, 1 H), 4.63 (bs, 1 H), 2.34 (s, 3 H), 2.25–2.19 (comp, 2 H), 1.87–1.70 (comp, 3 H), 1.63–1.61 (m, 1 H), 1.28 (s, 9 H); 13C NMR (100 MHz, CDCl3) d 199.7, 142.3, 136.3, 80.4, 77.4, 26.4, 26.3, 25.6, 23.3, 18.9; FTIR (thin film) 3055, 2980, 2930, 1672, 1265, 1235, 734 cm−1. Exact mass calculated for C12H20O3 (CI) 214.1412; found 214.1481.
4-(tert-Butylperoxy)-3-ethanoylcyclohex-2-enone (18)
Rf 0.33 (20% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) δ 6.53 (s, 1 H), 5.16 (t, J = 3.2 Hz, 1 H), 2.81 (ddd, J = 5.2, 12.8, 17.6 Hz, 1 H), 2.62–2.60 (m, 1 H), 2.47–2.45 (m, 1 H), 2.44 (s, 3 H), 2.00 (m, 1 H), 1.23 (s, 9 H); 13C NMR (100 MHz, CDCl3) δ 200.7, 198.9, 149.4, 133.6, 81.0, 71.41, 32.9, 26.8, 26.3, 25.7; FTIR (thin film) 2982, 2937, 2253, 1685, 1365, 1222, 1191, 907, 736 cm−1 . Exact mass calculated for C12H18O4 + H (ES) 227.1283; found 227.1259.
Oxidation of Cholesterol (19) - Isolation of 22, 23, and 24
Cholesterol (0.525 g; 1.36 mmol) and Rh2(cap)4 (10 mg; 0.013 mmol) were dissolved in 5 mL of 1,2-dichloroethane in a 6 dram vial equipped with a stirbar. T-HYDRO® (0.74 mL; 5.43 mmol) was added, and the color of the reaction solution turned from purple to brown-red in color. The vial was loosely capped and stirred at room temperature. After 15 hours, the reaction was concentrated under reduced pressure to 1.01 g of a red-brown semisolid. The crude material was purified via silica gel column chromatography (100 % hexanes to 1:1 hexanes: ethyl acetate, gradient) to afford 164 mg of 22, 62 mg of α-23, 13 mg of β-23, 57 mg of α-24, 25 mg of β-24, and 106 mg of starting material (21). The full characterization of 22, 23, and 24 has been reported.10, 38a
Oxidation of 17β-Acetoxyandrost-4-en-3-one in Water
17β-Acetoxyandrost-4-en-3-one51 (1.32 g, 4.0 mmol) was stirred vigorously in 12 mL of water at room temperature in a 50 mL flask. Then Rh2(cap)4 (29.5 mg, 0.040 mmol) was added, followed by addition of T-HYDRO® (4.6 mL; 32 mmol). The reaction became dark purple-red in color. The flask was closed with a rubber septum with a balloon and stirred for 24 h in a 40 °C oil bath. At that time additional portions of Rh2(cap)4 (15 mg, 0.02 mmol) and T-HYDRO® (2.3 mL; 16 mmol) were added, and the reaction solution was stirred for another 24 h. The reaction was extracted into diethyl ether (3 × 20 mL). The organic extracts were combined, dried over anhydrous MgSO4, filtered, then concentrated under reduced pressure to yield a crude yellow oil that was purified by silica gel chromatography (25 % ethyl acetate in hexanes) to yield 938 mg (2.72 mmol, 68 %) of a white solid (26) m.p. 193–195 °C; 192–194 °C (lit.).45
3β-Acetoxyandrost-5-en-4,7,17-trione (27)
3β-Acetoxyandrost-5-en-17-one (330 mg, 1.0 mmol) and Rh2(cap)4 (7.4 mg, 10 µmol) were added into a vial together with 3 mL of water was added, followed by T-HYDRO® (1.14 mL, 8.0 mmol), and the stirred reaction mixture was heated in an oil bath at 40 °C. After 24 h and 48 h, additional portions of Rh2(cap)4 (7.4 mg, 10 µmol) and T-HYDRO® (1.14 mL, 8.0 mmol) were added. The reaction was extracted into diethyl ether (3 × 20 mL), the organic extracts were combined, dried over anhydrous MgSO4, filtered, then concentrated, under reduced pressure, to yield a crude yellow oil that was purified by silica gel chromatography (33 % ethyl acetate in hexanes) to give 195 mg (0.54 mmol, 54 %) of a white solid: m.p. 216–218 °C; 218–220 °C (lit.).45 1H NMR (CDCl3, 400 MHz): defining absorptions are at δ 6.15 (s, 1H), 5.28 (dd, J = 12.0, 8.0 Hz, 1H), 2.77 (ddd, J = 12.0, 8.0, 4.0 Hz, 1H), 2.50 (d, J = 8.0 Hz, 1H), 2.45 (d, J = 8.0 Hz, 1H), 2.24–2.31 (m, 1H), 2.18 (s, 3H), 1.22 (s, 3H), 0.90 (s, 3H).
Supplementary Material
Procedures, product characterization, and analysis of compounds, especially steroidal products. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgement
We are grateful for financial support for this research from the National Institutes of Health (GM 46503) and from the National Science Foundation (CHE-0456911).
References
- 1.(a) Bulman-Page PC, McCarthy TJ. In: Comprehensive Organic Synthesis. Trost BM, editor. Vol. 7 Oxford, UK: Pergamon; 1991. [Google Scholar]; (b) Olah GA, Molnar A. Hydrocarbon Chemistry. 2nd. Hoboken: Wiley; 2003. [Google Scholar]
- 2.This discussion is unrelated to that for the elegant oxidative methods that are emerging with with palladium and ruthenium catalysts: Chen MS, White MC. J. Am. Chem. Soc. 2004;126:1346. doi: 10.1021/ja039107n. Delcamp JH, White MC. J. Am. Chem. Soc. 2006;128:15076. doi: 10.1021/ja066563d. Arends IWCE, Kodama T, Sheldon RA. In: Ruthenium Catalysts and Fine Chemistry. Bruneau C, Dixneuf PH, editors. Heidelberg, Germany: Springer-Verlag; 2004.
- 3.(a) Umbreit MA, Sharpless KB. J. Am. Chem. Soc. 1977;99:5526. [Google Scholar]; (b) Rabjohn N. Org. React. 1976;24:261. [Google Scholar]; (c) Crich D, Zou Y. Org. Lett. 2004;6:775. doi: 10.1021/ol036501h. [DOI] [PubMed] [Google Scholar]; (c) Zeni G, Stracke MP, Lissner E, Braga AL. Synlett. 2003;12:1880. [Google Scholar]
- 4.(a) Shing TMK, Yeung YY. Angew. Chem. Int. Ed. 2005;44:7981–7984. doi: 10.1002/anie.200502763. [DOI] [PubMed] [Google Scholar]; b Hua Z, Carcache DA, Tian Y, Li Y-M, Danishefsky SJ. J. Org. Chem. 2005;70:9849. doi: 10.1021/jo051556d. [DOI] [PubMed] [Google Scholar]; c Salmond WG, Barta MA, Havens JL. J. Org. Chem. 1978;43:2057. [Google Scholar]
- 5.(a) Rothenberg G, Weiner H, Sasson Y. J. Mol. Catal. A: Chemical. 1998;136:253–262. [Google Scholar]; (b) Jurado-Gonzalez M, Sullivan AC, Wilson JRH. Tet. Lett. 2003;44:4283–4286. [Google Scholar]; (c) Arsenou ES, Koutsourea AI, Fousteris MA, Nikolaropoulos SS. Steroids. 2003;68:407. doi: 10.1016/s0039-128x(03)00042-4. [DOI] [PubMed] [Google Scholar]
- 6.a) Yu J-Q, Corey EJ. Org. Lett. 2002;4:2727. doi: 10.1021/ol0262340. [DOI] [PubMed] [Google Scholar]; (b) Yu J-Q, Wu HC, Corey EJ. Org. Lett. 2005;7:1415. doi: 10.1021/ol050284y. [DOI] [PubMed] [Google Scholar]
- 7.(a) Shing TKM, Yeung YY, Su PL. Org. Lett. 2006;8:3149. doi: 10.1021/ol0612298. [DOI] [PubMed] [Google Scholar]; (b) Blay G, Fernández I, Giménez T, Pedro JR, Ruiz R, Pardo E, Lioret F, Muñoz MC. Chem. Comm. 2001:2102–2104. doi: 10.1039/b105132f. [DOI] [PubMed] [Google Scholar]; c Salvador JAR, Clark JH. Chem. Comm. 2001:33. [Google Scholar]
- 8.Blanksby SJ, Ellison GB. Acc. Chem. Res. 2003;36:255. doi: 10.1021/ar020230d. [DOI] [PubMed] [Google Scholar]
- 9.Catino J, Forslund RE, Doyle MP. J. Am. Chem. Soc. 2004;126:13622. doi: 10.1021/ja045330o. [DOI] [PubMed] [Google Scholar]
- 10.Choi H, Doyle MP. Org. Lett. 2007;9:5349. doi: 10.1021/ol7025284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu J-Q, Corey EJ. J. Am. Chem. Soc. 2003;125:3232. doi: 10.1021/ja0340735. [DOI] [PubMed] [Google Scholar]
- 12.Salvador JAR, Silvestre SM. Tetrahedron Lett. 2005;46:2581. [Google Scholar]
- 13.McLaughlin EC, Doyle MP. J. Org. Chem. 2008;73:4317. doi: 10.1021/jo800382p. [DOI] [PubMed] [Google Scholar]
- 14.Bravo A, Bjørsvik H-R, Fontana F, Liguori L, Minisci F. J. Org. Chem. 1997;62:3849. doi: 10.1021/jo970302s. [DOI] [PubMed] [Google Scholar]
- 15.Caudle MT, Riggs-Gelasco P, Gelasco AK, Penner-Hahn JE, Pecoraro VL. Inorg. Chem. 1996;35:3577. [Google Scholar]
- 16.Fousteris MA, Koutsourea AI, Nikolaropoulos SS, Riahi A, Muzart J. J. Mol. Catal. A: Chem. 2006;250:70. [Google Scholar]
- 17.Shing TKM, Yeung YY, Su PL. Org. Lett. 2006;8:3149. doi: 10.1021/ol0612298. [DOI] [PubMed] [Google Scholar]
- 18.Arsenou ES, Koutsourea AI, Fousteris MA, Nikolaropoulos SS. Steroids. 2003;68:407. doi: 10.1016/s0039-128x(03)00042-4. [DOI] [PubMed] [Google Scholar]
- 19.Salvador JAR, Clark JH. Chem. Commun. 2001:33. [Google Scholar]
- 20.Miller RA, Li W. Humphrey. Tet. Lett. 1996;37:3429. [Google Scholar]
- 21.Balini R, Astolfi P. Liebigs Ann. 1996:1879. [Google Scholar]
- 22.(a) Russell GA. J. Am. Chem. Soc. 1957;79:3871. [Google Scholar]; b Caudle MT, Riggs-Gelasco P, Gelasco AK, Penner-Hahn JE, Pecoraro VL. Inorg. Chem. 1996;35:3577. [Google Scholar]
- 23.Timmons D, Doyle MP. In: Metal Bonds Between Metal Atoms. 3rd ed. Cotton FA, Murillo CA, Walton RA, editors. New York: Springer Science and Business Media; 2005. Chapter 13. [Google Scholar]
- 24.Pfefferle C, Kempter C, Metzger JW, Fiedler H–P. J. Antibiotics. 1996;49:826. doi: 10.7164/antibiotics.49.826. [DOI] [PubMed] [Google Scholar]
- 25.(a) Ballini R, Bosica G. J. Nat. Prod. 1998;61:673. doi: 10.1021/np970504j. [DOI] [PubMed] [Google Scholar]; (b) Shet J, Tilve S. Synthesis. 2004;11:1859. [Google Scholar]; c Obrect D, Weiss B. Helv. Chim. Acta. 1989;72:117. [Google Scholar]
- 26.Selective oxidation occurs at the 4-position: Wilson CA, III, Shaw PE. J. Org. Chem. 1973;38:1684. Jensen HP, Sharpless KB. J. Org. Chem. 1975;40:264.
- 27.(a) Lempers HEB, Sheldon RA. Appl. Catal. A: General. 1996;143:137. [Google Scholar]; (b) Silva AD, Patitucci ML, Bizzo HR, D’Elia E, Antunes OAC. Catal. Commun. 2002;3:435. [Google Scholar]
- 28.(a) Murahashi S-I, Komiya N, Oda Y, Kuwabara T, Naota T. J. Org. Chem. 2000;65:9186. doi: 10.1021/jo001348f. [DOI] [PubMed] [Google Scholar]; (b) Lempers HEB, Garcia AR, Sheldon RA. J. Org. Chem. 1998;63:1408. [Google Scholar]
- 29.Avila DV, Ingold KU, Lusztyk J, Green WA, Procopio DR. J. Am. Chem. Soc. 1995;117:2929. [Google Scholar]
- 30.Bravo A, Bjørsvik H-R, Fontana F, Liguori L, Miniski F. J. Org. Chem. 1997;62:3849. doi: 10.1021/jo970302s. [DOI] [PubMed] [Google Scholar]
- 31.(a) Snelgrove DW, Lusztyk J, Banks JT, Mulder P, Ingold KU. J. Am. Chem. Soc. 2001;123:469. [Google Scholar]; (b) MacFaul PA, Arends IWCE, Ingold KU, Wayner DDM. J. Chem. Soc. Perkin Trans. 1997;2:135–145. [Google Scholar]
- 32.Chavez FA, Mascharak PK. Acc. Chem. Res. 2000;33:539–545. doi: 10.1021/ar990089h. [DOI] [PubMed] [Google Scholar]
- 33.(a) Koola JD, Kochi JK. J. Org. Chem. 1987;52:4545. [Google Scholar]; (b) Srinavasan K, Perrier S, Kochi JK. J. Mol. Catal. 1986;38:297. [Google Scholar]
- 34.Miller RA, Li W, Humphrey GR. Tetrahedron Lett. 1996;37:3429. [Google Scholar]
- 35.Nielsen JH, Olsen CE, Skibsted LH. Food Chem. 1996;56:33. [Google Scholar]
- 36.Arsenou ES, Koutsourea AI, Fousteris MA, Nikolaropoulos SS. Steroids. 2003;68:407. doi: 10.1016/s0039-128x(03)00042-4. [DOI] [PubMed] [Google Scholar]
- 37.Schenck GO, Neumüller OA, Eisfeld W. Justus Liebigs Ann. Chem. 1958;618:202. [Google Scholar]
- 38.(a) Beckwith ALJ, Davies AG, Davison IGE, Maccoll A, Mruzek MH. J. Chem. Soc. Perkin Trans. 1989;2:815. [Google Scholar]; (b) Dang H, Davies AG, Davison IGE, Schiesser CH. J. Org. Chem. 1990;55:1432. [Google Scholar]; (c) Ponce MA, Ramirez JA, Galagovsky LR, Gros EG, Erra-Balsells R. J. Chem. Soc. Perkin Trans. 2000;2:2351. [Google Scholar]
- 39.Miyamoto S, Martinez GR, Medeiros MHG, Di Mascio P. J. Am. Chem. Soc. 2003;125:6172. doi: 10.1021/ja029115o. [DOI] [PubMed] [Google Scholar]
- 40.Doyle MP, Westrum LJ, Wolthuis WNE, See MM, Boone WP, Bagheri V, Pearson MM. J. Am. Chem. Soc. 1993;115:958. [Google Scholar]
- 41.Still WC, Kahn M, Mitra AJ. J. Org. Chem. 1978;43:2923. [Google Scholar]
- 42.Manfredini S, Simoni D, Zanirato V, Casolari A. Tetrahedron Lett. 1988;29:3997. [Google Scholar]
- 43.Scheffold R, Dulos P. Helv. Chim. Acta. 1967;50:798–807. [Google Scholar]
- 44.Jasiczak J. J. Chem. Soc. Perkin Trans. 1. 1988;10:2687. [Google Scholar]
- 45.Marwah P, Marwah A, Lardy HA. Green Chem. 2004:570. [Google Scholar]
- 46.Kiran I. J. Chem. Res. 2004;3:208. [Google Scholar]
- 47.Hunter CA, Priest S–M. Steroids. 2006;71:30. doi: 10.1016/j.steroids.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 48.Lüönd RM, Walker J, Neier RW. J. Org. Chem. 1992;57:5005. [Google Scholar]
- 49.Webster FX, Silverstein RM. Synthesis. 1987;10:922. [Google Scholar]
- 50.Davis RA. J. Nat. Prod. 2005;68:769. doi: 10.1021/np050025h. [DOI] [PubMed] [Google Scholar]
- 51.17β-Acetoxyandrost-4-en-3-one was prepared according to: Krauser JA, Guengerich FP. J. Biol. Chem. 2005;280:19496. doi: 10.1074/jbc.M501854200.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Procedures, product characterization, and analysis of compounds, especially steroidal products. This material is available free of charge via the Internet at http://pubs.acs.org.