

Abstract
Biliary atresia (BA) is a disease of the newborn which results in obstruction of the biliary tree. The cause of BA remains unknown; however, recent studies using the murine model of biliary atresia have found that rotavirus infection of the biliary epithelial cell (cholangiocyte) triggers an inflammatory response. We hypothesized that rotavirus infection of cholangiocytes results in the release of chemokines, important mediators of the host immune response.
Methods
In vivo, Balb/c pups were injected with rhesus rotavirus (RRV) or saline, and, their extrahepatic bile ducts were microdissected 2,5, 7, and 14 days after injection. Next, an immortalized cholangiocyte cell line (mCl) was incubated with RRV or serum free media. Qualitative and quantitative chemokine assement was performed using ELISA, PCR, and immunohistchemistry.
Results
In vivo, increased levels of the chemokines MIP-2, MCP-1, KC and RANTES were found in RRV-infected murine bile ducts. In vitro, infected mCl cells produced increasing amounts of these same chemokines in relation to dose and time.
Conclusion
These novel results suggest that chemokine expression by RRV-infected cholangiocytes may trigger a host inflammatory process that causes bile duct obstruction. Understanding how viral infection initiates this response may shed light on the pathogenesis of biliary atresia.
Introduction
Biliary atresia is a disease of the newborn that results in progressive fibrosis and ultimately obliteration of the bile ducts. Without treatment afflicted children progress to end-stage liver failure and death. Although the etiology of biliary atresia is unknown, potential causes include a genetic abnormality, abnormal prenatal circulation and exposure to an environmental toxin or virus(1). A viral etiology enjoys the greatest amount of support as viruses such as rotavirus, reovirus, Cytomegalovirus, Epstein-Barr Virus and Human Papilloma Virus(2–5) have been isolated from the livers of children afflicted with biliary atresia. In addition, a murine model of biliary atresia exists in which newborn mice infected with rhesus rotavirus (RRV) develop a disease process that shares striking similarities to human disease (6, 7). These similarities include biliary obstruction and inflammation within the extra-hepatic bile duct(8–10).
Recent studies have shown that in the murine model of biliary atresia RRV targets the biliary epithelium for infection(9, 10). Following infection, mice develop an obstructive cholangiopathy in which there is marked infiltration of the biliary tract with mononuclear inflammatory cells. The mononuclear cells contribute to the pathogenesis of the disease process as immune deficient mice infected with RRV do not develop biliary obstruction(8, 11). The mechanism by which this inflammatory process is initiated has not been established.
Rotavirus infection of intestinal epithelial cells results in increased levels of chemokine release and mononuclear cell migration(12). Chemokines are small proteins that attract mononuclear cells. They are classified as CXC or CC chemokines according to the highly conserved position of four cysteine residues. Mouse Macrophage Inflammatory Protein 2 (MIP-2) and KC are prototypical CXC chemokines and functional analogs to human IL-8 with potent chemotactic activity for neutrophils. Regulated upon Activation, Normal T Expressed and Secrete (RANTES), Monocyte Chemotactic Protein 1 (MCP-1) and Thymus and Activation Regulated Chemokine (TARC) are prototypical CC chemokines which attract monocytes, memory T lymphocytes, basophils, and NK cells(13).
In the current study, we hypothesized that RRV infection of cholangiocytes causes chemokine release initiating the inflammatory response that causes biliary obstruction. To test this hypothesis, we determined in vivo and in vitro the effect of RRV infection on cholangiocyte chemokine production. By understanding how viral infection initiates the inflammatory process that result in biliary tract obstruction we may better understand the pathogenesis of biliary atresia.
Methods
Mice
Breeding pairs of BALB/c mice were kept in micro-isolator cages in a virus-free environment. They were fed and watered daily with sterilized chow and water. Upon arrival, the mice were held separately in micro-isolators for 1 week prior to breeding. Females were separated approximately 1 week prior to their expected delivery.
Cells, culture media, and viruses
A mouse cholangiocyte cell line (mCl) derived from primary cholangiocytes, harvested from BALB/c mice and immortalized with SV-40 large T cell antigen was provided by the laboratory of Dr. James Boyer (Yale Liver Care Center, Hartford, CT). These cells express GGT and cytokeratin-7 consistent with their biliary epithelium origin (data not shown). The cell line was maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Cellgro, Herndon, VA) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA), penicillin (10,000 U/ml)-streptomycin (10,000 μg/ml) (Invitrogen), 1% L-glutamine (Invitrogen) and amphotericin B (250 μg/ml) (Cellgro). The rotavirus strain RRV originally obtained from Dr. Harry Greenberg (Stanford University, Palo Alto, CA) was maintained in MA104 cells.
Experimental biliary atresia
In vivo
All animal studies were performed in accordance with institutional animal welfare guidelines using an approved animal protocol. The murine model of experimental biliary atresia was generated using previously described techniques. In brief, newborn pups were injected into the intraperitoneal cavity (i.p.) with 1.25 × 106 fluorescent focus forming units (FFU) of RRV/gram body weight diluted in saline within 24 hours of birth. Pups injected with saline served as controls. Pups were sacrificed 2, 5, 7 and 14 days post-infection and their extrahepatic biliary tract were micro dissected, homogenized in Earle’s Balanced Salt Solution (EBSS) and frozen at −80°C until analyzed by ELISA. A separate subset of mice had their hepatobiliary tract dissected and snap frozen in liquid nitrogen for extraction of total RNA and analysis by RT-PCR for the presence of chemokine mRNA. Another subset of mice had their livers harvested, embedded in Histo Prep embedding media (Fisher Diagnostic, Fairlawn, New Jersey) and stored at −80°C until analyzed by immunohistochemical techniques.
In vitro
mCl cells were seeded in 14ml culture tubes (Corning Inc., Corning, NY) at a density of 5 × 105 cells per tube and grown to 80–90% confluence in a monolayer in DMEM + 10% FBS for 48 hours at 37°C and 5% CO2. For viral infection, cells were washed in serum-free medium and overlaid with 200ul of serum-free DMEM and 4ul/ml trypsin containing live virus at a multiplicities of infection (MOI) ranging from 0 to 100. An MOI of 1 indicates a concentration of one virus per cell, and an MOI of 100 indicates 100 viruses per cell. The cells were incubated with virus for 1 hour at 37°C, washed and overlaid with serum-free DMEM and 4ul/ml trypsin. In dose response experiments, tubes were incubated for 24 hours with the described MOIs. In time course experiments, tubes were incubated for 0, 6, 12, 24 and 48 hours with virus. In both experiments, supernatants were then aspirated and stored at −20°C until analyzed by ELISA. Total RNA was immediately extracted from the cells for further analysis by reverse transcription polymerase chain reaction (RT-PCR).
Chemokine analysis
Quantitative assessment of MIP-2, KC, RANTES, MCP-1 and TARC chemokines was done using R&D Quantikine sandwich enzyme immunoassay kits (Minneapolis, MN). Fifty microliters of standard, control or sample (supernatant fluid from mCl cell line or biliary homogenates) were used according to manufacturer’s instructions. The optical density was determined using a microplate reader (Spectramax Plus, Molecular Devices) set to 450nm and chemokine concentrations were calculated by a computer program (Softmax Pro Version 2.2.1, Molecular Devices Corp.) and recorded as pg/bile duct.
PCR amplification of chemokine mRNA
Following the aspiration of the supernatant, the cell monolayer was treated with 1ml of Trizol (Invitrogen, Carlsbad, CA). The hepatobiliary trees of infected mice and controls from the various time points were homogenized in 3ml of Trizol. Total RNA was extracted using standard protocols and fractionated on a 1.5% agarose formaldehyde gel and stained with 1mg/ml of ethidium bromide to confirm integrity of the RNA. cDNA was made from each sample using Invitrogen reagents in a total reaction volume of 50 μl (Invitrogen Life Technologies, U.S.A.). The cDNAs were amplified in a PTC-200 Peltier thermal cycler (MJ Research, Watertown, MA). Each sample underwent 30 cycles of 30 sec. at 94°C, 30 sec. at 55°C and 1 min. at 72°C. The final cycle consisted of 1 min. at 94°C, 30 sec. at 55°C and 1 min. at 72°C. Each PCR product was fractionated on a 1.7% agarose gel containing 0.5mg/ml of ethidium bromide, viewed with a Kodak digital science Image station-440 and Kodak 1D-imaging software (Eastman Kodak, New Haven, CT). Mouse β-actin served as the internal reverse transcription control.
The murine primers used for PCR were:
| MIP-2 (300-bp product): | sense, 5′-CCCAGACAGAAGTCATAGCCACTC-3′ |
| anti-sense, 5′-CTTGCCTTTGTTCAGTATCCTTTTGG-3 | |
| β-actin (233-bp product): | sense, 5′TCCATCATGAAGTGTGACG-3′ |
| anti-sense, 5′-ACATCTGCTGGAAGGTGG-3′ |
Immunohistochemistry
At the time of sacrifice (2, 7, or 14 days post-infection), the livers of pups infected with RRV or saline were excised, rinsed in PBS and immediately mounted in Histo Prep mounting medium and stored at −80°C. Sections were cut at 7um, immediately fixed in cold acetone and allowed to air dry. Sections were rehydrated in PBS for 5 min.
Endogenous peroxidase activity was inhibited by incubating the slides in a solution of 2.7% H2O2 (30% solution) and 97.3% methanol for 15 min. Sections were surrounded using a PAP pen, then rabbit serum diluted per manufacturer’s recommendations (Vector Laboratories PK-4005, Burlingame, CA) in PBS was incubated for 1 hour on rotation. Avidin and biotin blockers were serially added for 15 min each. Slides were washed and goat anti-mouse MIP-2 (R&D Systems, Catalog #AF-452-NA) diluted 1:100 in PBS was applied and incubated for 1 hour. Slides were washed and horse-radish peroxidase conjugated rabbit anti-goat antibody diluted 1:500 in PBS was applied for 30 min. Slides were washed and ABC reagent was applied to the tissue sections and incubated for 30 min. Slides were washed and DAB substrate applied and incubated for 3 min., the reaction was stopped by soaking the slides in diH2O. Slides were counterstained in Harris Hematoxylin. Serial washes were performed in increasing concentrations of alcohol and xylene and coverslips applied with Permount (Fisher, Fairlawn, NJ).
The slides were photographed on a Nikon Eclipse E600 fluorescent microscope equipped with a Nikon DXM 1200F camera (Fryer Co. Inc., Cincinnati, OH) and captured using Nikon ACT-1 version 2.62.
Statistics
Analysis of non-continuous variables was done using Chi-square and Fisher Exact tests. Results of continuous variables were expressed as mean +/− SEM and analyzed using ANOVA with post-hoc testing where appropriate. A p< 0.05 was considered significant.
Results
The effect of RRV infection on chemokine expression in vivo
In the murine model of biliary atresia, mice become symptomatic between five and seven days post infection, a time when virus can be found within the biliary epithelium(10). Chemokine expression following infection was determined in extrahepatic biliary samples procured 2, 5, 7 and 14 days post-infection (Figure 1). At all time points studied, there was a marked increase in levels of detectable MIP-2. The amount of RANTES, MCP-1 and KC increased over controls beginning at 5 days post infection and remain elevated through day 14 post-infection (infected mice perish 14–18 days post infection). The peak level of RANTES, MCP-1, and MIP-2 occurred at 7 days post-infection. KC levels peaked five days after infection. While there was basal levels of KC and RANTES detected in uninfected mice, no MCP-1 or MIP-2 were found in control mice at any time point tested. In contrast to these findings, the chemokine TARC was not detected in control or infected mice until 14 days post infection. At this time point, similar levels were noted in both control and infected mice indicating that RRV infection caused no effect on TARC expression.
Figure 1. Chemokine levels (panels A–E) in extra-hepatic bile ducts following infection with RRV.
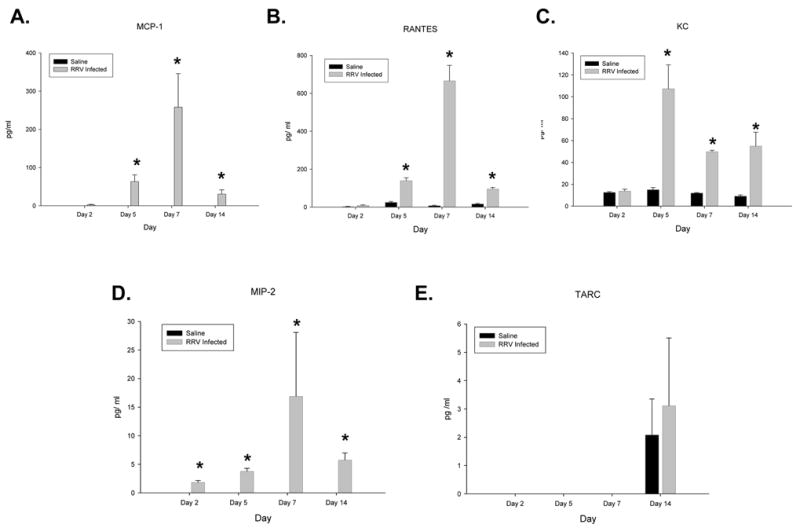
Bile ducts were harvested from new born mice 2, 5, 7 and 14 days post infection with RRV and homogenized (n=3–7). Chemokine levels were determined by ELISA and expressed as means with standard error. Significantly higher levels of RANTES, KC, and MCP-1 were observed starting at day 5 and continuing through day 14 (p<0.05). RANTES and MCP-1 peaked at day 7 while KC peaked at day 5. MIP-2 was significantly higher at all time points tested (p<0.05), peaking at day 7.
The effect of RRV infection on chemokine secretion in mCl cells
The studies performed in vivo revealed increased chemokine levels in the extra-hepatic biliary tract following RRV infection. The extra-hepatic biliary tract is comprised of a variety of cell types. To precisely determine if cholangiocytes infected with RRV release chemokines, we measured chemokine production in a recently established in vitro model where RRV is used to infect a cholangiocyte cell line. In this in vitro model, RRV is better able to replicate in cholangiocytes than in hepatocytes (data not shown). Infection of mCl cells resulted in increased chemokine production of MIP-2, MCP-1, KC and RANTES but not the chemokine TARC.
Dose response experiments were performed to evaluate the extent of chemokine expression following RRV infection of cholangiocytes (Figure 2). There was an increase in the levels of MIP-2, KC, RANTES and MCP-1 in a dose dependent fashion. The most dramatic increase was in MIP-2 levels which increased eight fold over controls at the highest concentration of virus tested (MOI 100). At this high MOI, RANTES levels increased four fold whereas KC and MCP-1 levels increased by 2-fold. In contrast, there was a basal level of TARC chemokine release that did not vary following RRV infection.
Figure 2. Chemokine levels in the supernatant (panels A–E) of in vitro cholangiocytes infected with increasing amounts of RRV.

In vitro, cholangiocyte cells were infected with increase amounts of RRV (n=3–5 tubes). Chemokine expression in the supernatants was determined by ELISA and expressed as means with standard error. Some chemokine expression was seen in the absence of infection in controls, though virus at MOIs of 50 and 100 resulted in significant increases in the quantity of MIP-2, RANTES and KC found in supernatant fluid from cholangiocytes (p<0.05) 24 hours after infection. MCP-1 was found to be significantly elevated over background expression at an MOI of 100 (p<0.05). Experiment conditions repeated 3 times.
The temporal response of chemokine expression by mCl cells after exposure to RRV was assessed at an MOI of 10 (Figure 3). At 6 and 12 hours post-infection, levels of all chemokines remained similar between infected and control treated cholangiocytes. At twenty-four and forty-eight hours post-infection, the levels of all chemokines increased significantly over control (p<0.05). Once again, the largest increase was in MIP-2.
Figure 3. Chemokine levels over time in the supernatant (panels A–D) of in vitro cholangiocytes infected with RRV.

Cholangiocytes were exposed to RRV at an MOI of 100 for 1 hour, washed and let incubated for 6, 12, 24 and 48 hour periods (n=3–7). Chemokine expression in the supernatants was determined by ELISA and expressed as means with standard error. There was no difference in chemokine levels expressed by the cholangiocytes at 6 and 12 hours post infection. Conversely at 24 and 48 hours post infection significantly more of all four chemokines were observed (p<0.05).
MIP-2 mRNA expression and protein localization
The change in expression of MIP-2 was the most dramatic among chemokines assayed both in vivo and in vitro. MIP-2 is the murine analog of IL-8, the primary chemoattractant to neutrophils which are found through-out the extrahepatic biliary tract following RRV infection. Because of this, further studies focused on this chemokine. RT-PCR for MIP-2 mRNA was performed on the mCL cell line. MIP-2 mRNA was detectable in hepatobiliary tract samples procured from mice at day 5 and 7 days post-infection (Figure 4a) corresponding to the detectable increases in MIP-2 protein levels found by ELISA. No MIP-2 mRNA was found in saline treated control tissue. It was found that MIP-2 mRNA increased in cholangiocytes exposed to RRV when compared to control cells after 24 hours. β-actin served as a loading control (Figure 4b).
Figure 4. A. MIP-2 mRNA in the extra-hepatic biliary tract harvested from mice infected with RRV.

RT-PCR on mRNA extracted from samples harvested at day 2, 5 and 7 days post RRV infection of new born mice for MIP-2 and Beta-actin. mRNA for MIP-2 was only detectable on the infected samples of day 5 and 7 post infection. B. In vitro expression of MIP-2 mRNA in cholangiocytes after RRV infection. mRNA from cholangiocytes infected with RRV at an MOI of 100 for 1 hour. After 24 hours of incubation MIP-2 was unregulated in the infected cells.
Immunohistochemistry for MIP-2 on hepatobiliary tissue demonstrated presence of protein 7 days post infection (Figure 5). The intensity of signal appeared greatest surrounding the bile ducts within the portal tracts in a pattern similar to what is seen when staining for RRV. At Day 14 MIP-2 signal was not evident.
Figure 5. Immunohistochemistry for MIP-2 in the hepatobiliary system following RRV infection.

Hepatobiliary tissue was harvested from mice on day 2, 7 and 14 post infection with RRV and stained with an anti-mouse MIP-2 anti-body. Positive staining, small arrows ( ) were observed in liver samples harvested from mice on post infection day 2 and day 7. Large arrow (
) were observed in liver samples harvested from mice on post infection day 2 and day 7. Large arrow ( ) indicates portal vein.
) indicates portal vein.
Discussion
The etiology of biliary atresia has not been defined; however, recent studies suggest a viral infection triggering a host immune response could be the basis for the pathogenesis of this devastating disease process (14). In the murine model of biliary atresia, it has been shown that RRV targets the biliary epithelial cell for infection(10). Following infection, there is infiltration of the extrahepatic biliary tract by mononuclear cells(9). This host immune response plays a critical role in the resulting obstruction as the depletion of specific T-cell subsets resulted in alterations in biliary obstruction(9). For the host immune response to contribute to the pathogenesis of bile duct injury, signals must be sent by the infected biliary epithelium attracting mononuclear effector cells to the extra-hepatic biliary tract. In this study, it was found that the infected cholangiocyte produced chemokines which may be that initiating signal.
In vivo, the increase in detectable chemokines paralleled the progression of disease. The localization of chemokines to tissue surrounding the portal triad and the similarity of the pattern of chemokines released to what was found in vitro support a potential role of the infected cholangiocyte as the initiating source in the progression of disease. In vitro, infection of cholangiocytes with RRV led to an increased release of chemokines in a dose and time dependent fashion. The finding that TARC did not increase was important indicating that RRV infection resulted in the release of specific chemokines, not a generalized release of many pro-inflammatory chemokines. Further investigation into MIP-2 demonstrated that the increase in protein correlates with increased transcriptional activity demonstrated by elevated mRNA rather than the release of constitutive protein. Though this work is descriptive in nature, the results provide the framework for future studies designed to test whether inhibition of chemokine secretion directly contributes to biliary obstruction. Previously published data indicates that modulation of the inflammatory signaling cascade, specifically cytokine secretion, may attenuate the severity of biliary atresia in the murine model. Shivakumar demonstrated that RRV-infected, IFN-γ deficient Balb/c mice had a much lower mortality rate than the wild type animals(9). Administration of recombinant IFN-γ subsequently recapitulated the obstructive phenotype in knockout mice. Interestingly though, studies involving two other mouse strains deficient in Th1 inflammatory cytokines, specifically TNF-α and IL-12, did not show any modulation of biliary epithelial cell injury or duct obstruction(15, 16). Given the redundancy of signaling molecules as well as their receptors, modulation of cytokine/chemokine secretion may be exceedingly complex.
Previously, we have demonstrated that mitogen-activated protein kinase (MAPK) signaling was increased in in vivo and in vitro models of experimental biliary atresia. (17). Additionally, it has been shown that MAPK activation occurs in other epithelial cells lines infected by viruses (18, 19). In a study by Pazdrak et al, respiratory syncytial virus replication resulted in RANTES production that increased in a temporal dependent relationship with ERK phosphorylation (19). Future experiments will be performed to determine if RRV induced MAPK signaling leads to the chemokine production we report in the current study.
Chemokines have been implicated in the pathogenesis of a variety of hepatobiliary diseases including biliary atresia. Elevated levels of IL-8 have been found in the serum of jaundiced children with biliary atresia(20). Within liver samples of children afflicted with biliary atresia, mRNA expression of IL-8 was also increased(21). Increased amounts of RANTES have been found in patients with primary biliary cirrhosis, and the therapeutic benefits of fibrates in this biliary disease may be through the inhibition of this chemokine with its resultant effects on inflammatory cell migration(22). It is also thought to be a mediator of dendritic cell expansion in a murine model of obstructive jaundice(23). MCP-1 was found to be elevated in serum samples of patients with biliary atresia with liver dysfunction or portal hypertension(24). Both KC and its receptor CXCR2 may have a direct hepatotoxic role through activation of proinflammatory and profibrotic genes(25). CXCR3+ (the receptor for CXCL9, 10, and 11) mononuclear cells have been detected in the excised biliary remnants of children with biliary atresia(26). In all of these studies, the source for increased chemokine production was not determined. While it has been shown that viral infection of mononuclear cells can induced expression of chemokines(27, 28) and that rotavirus infected human peripheral blood mononuclear cells will release an array of cytokines(29), the results of our current study suggest that the source could also be the injured cholangiocyte initiating the hepatobiliary disease.
Infection of newborn mice with RRV leads to an obstructive cholangiopathy dominated by the presence of inflammatory cells. Cholangiocytes infected with RRV expressed chemokines which may lead to chemotaxis of mononuclear cells to the area of rotavirus infection. Further studies are necessary to more clearly understand this process and extend these results to afflicted children.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Balistreri WF, Grand R, Hoofnagle JH, Suchy FJ, Ryckman FC, Perlmutter DH, et al. Biliary atresia: current concepts and research directions. Summary of a symposium. Hepatology. 1996;23(6):1682–92. doi: 10.1002/hep.510230652. [DOI] [PubMed] [Google Scholar]
- 2.Riepenhoff-Talty M, Gouvea V, Evans MJ, Svensson L, Hoffenberg E, Sokol RJ, et al. Detection of group C rotavirus in infants with extrahepatic biliary atresia. J Infect Dis. 1996;174(1):8–15. doi: 10.1093/infdis/174.1.8. [DOI] [PubMed] [Google Scholar]
- 3.Domiati-Saad R, Dawson DB, Margraf LR, Finegold MJ, Weinberg AG, Rogers BB. Cytomegalovirus and human herpesvirus 6, but not human papillomavirus, are present in neonatal giant cell hepatitis and extrahepatic biliary atresia. Pediatr Dev Pathol. 2000;3(4):367–73. doi: 10.1007/s100240010045. [DOI] [PubMed] [Google Scholar]
- 4.Drut R, Drut RM, Gomez MA, Cueto Rua E, Lojo MM. Presence of human papillomavirus in extrahepatic biliary atresia. J Pediatr Gastroenterol Nutr. 1998;27(5):530–5. doi: 10.1097/00005176-199811000-00007. [DOI] [PubMed] [Google Scholar]
- 5.Fjaer RB, Bruu AL, Nordbo SA. Extrahepatic bile duct atresia and viral involvement. Pediatr Transplant. 2005;9(1):68–73. doi: 10.1111/j.1399-3046.2005.00257.x. [DOI] [PubMed] [Google Scholar]
- 6.Petersen C, Grasshoff S, Luciano L. Diverse morphology of biliary atresia in an animal model. Journal of Hepatology. 1998;28:603–7. doi: 10.1016/s0168-8278(98)80283-3. [DOI] [PubMed] [Google Scholar]
- 7.Riepenhoff-Talty M, Schaekel K, Clark HF, Mueller W, Uhnoo I, Rossi T, et al. Group A rotaviruses produce extrahepatic biliary obstruction in orally inoculated newborn mice. Pediatr Res. 1993;33(4 Pt 1):394–9. doi: 10.1203/00006450-199304000-00016. [DOI] [PubMed] [Google Scholar]
- 8.Mack CL, Tucker RM, Sokol RJ, Karrer FM, Kotzin BL, Whitington PF, et al. Biliary atresia is associated with CD4+ Th1 cell-mediated portal tract inflammation. Pediatr Res. 2004;56(1):79–87. doi: 10.1203/01.PDR.0000130480.51066.FB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shivakumar P, Campbell KM, Sabla GE, Miethke A, Tiao G, McNeal MM, et al. Obstruction of extrahepatic bile ducts by lymphocytes is regulated by IFN-gamma in experimental biliary atresia. J Clin Invest. 2004;114(3):322–9. doi: 10.1172/JCI21153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Allen SR, Jafri M, Donnelly B, McNeal M, Witte D, Bezerra J, et al. Effect of rotavirus strain on the murine model of biliary atresia. J Virol. 2007;81(4):1671–9. doi: 10.1128/JVI.02094-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shivakumar P, Bezerra JA. Biliary atresia and Th1 function: linking lymphocytes and bile ducts: commentary on the article by Mack et al. on page 79. Pediatr Res. 2004;56(1):9–10. doi: 10.1203/01.PDR.0000129655.02381.F0. [DOI] [PubMed] [Google Scholar]
- 12.Casola A, Estes MK, Crawford SE, Ogra PL, Ernst PB, Garofalo RP, et al. Rotavirus infection of cultured intestinal epithelial cells induces secretion of CXC and CC chemokines. Gastroenterology. 1998;114(5):947–55. doi: 10.1016/s0016-5085(98)70314-2. [DOI] [PubMed] [Google Scholar]
- 13.Rollins BJ. Chemokines. Blood. 1997;90(3):909–28. [PubMed] [Google Scholar]
- 14.Mack CL. The pathogenesis of biliary atresia: evidence for a virus-induced autoimmune disease. Semin Liver Dis. 2007;27(3):233–42. doi: 10.1055/s-2007-985068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mohanty SK, Shivakumar P, Sabla G, Bezerra JA. Loss of interleukin-12 modifies the pro-inflammatory response but does not prevent duct obstruction in experimental biliary atresia. BMC Gastroenterol. 2006;6:14. doi: 10.1186/1471-230X-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tucker RM, Hendrickson RJ, Mukaida N, Gill RG, Mack CL. Progressive biliary destruction is independent of a functional tumor necrosis factor-alpha pathway in a rhesus rotavirus-induced murine model of biliary atresia. Viral Immunol. 2007;20(1):34–43. doi: 10.1089/vim.2006.0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jafri M, Donnelly B, McNeal M, Ward R, Tiao G. MAPK signaling contributes to rotaviral-induced cholangiocyte injury and viral replication. Surgery. 2007 Aug;142(2):192–201. doi: 10.1016/j.surg.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 18.Kujime K, Hashimoto S, Gon Y, Shimizu K, Horie T. p38 mitogen-activated protein kinase and c-jun-NH2-terminal kinase regulate RANTES production by influenza virus-infected human bronchial epithelial cells. J Immunol. 2000 Mar 15;164(6):3222–8. doi: 10.4049/jimmunol.164.6.3222. [DOI] [PubMed] [Google Scholar]
- 19.Pazdrak K, Olszewska-Pazdrak B, Liu T, Takizawa R, Brasier AR, Garofalo RP, et al. MAPK activation is involved in posttranscriptional regulation of RSV-induced RANTES gene expression. Am J Physiol Lung Cell Mol Physiol. 2002 Aug;283(2):L364–L372. doi: 10.1152/ajplung.00331.2001. [DOI] [PubMed] [Google Scholar]
- 20.Honsawek S, Chongsrisawat V, Vejchapipat P, Thawornsuk N, Tangkijvanich P, Poovorawan Y. Serum interleukin-8 in children with biliary atresia: relationship with disease stage and biochemical parameters. Pediatr Surg Int. 2005;21(2):73–7. doi: 10.1007/s00383-004-1329-x. [DOI] [PubMed] [Google Scholar]
- 21.Huang YH, Chou MH, Du YY, Huang CC, Wu CL, Chen CL, et al. Expression of toll-like receptors and type 1 interferon specific protein MxA in biliary atresia. Lab Invest. 2007;87(1):66–74. doi: 10.1038/labinvest.3700490. [DOI] [PubMed] [Google Scholar]
- 22.Hirano Y, Hirano F, Fujii H, Makino I. Fibrates suppress chenodeoxycholic acid-induced RANTES expression through inhibition of NF-kappaB activation. Eur J Pharmacol. 2002;448(1):19–26. doi: 10.1016/s0014-2999(02)01902-7. [DOI] [PubMed] [Google Scholar]
- 23.Bleier JI, Katz SC, Chaudhry UI, Pillarisetty VG, Kingham TP, 3rd, Shah AB, et al. Biliary obstruction selectively expands and activates liver myeloid dendritic cells. J Immunol. 2006;176(12):7189–95. doi: 10.4049/jimmunol.176.12.7189. [DOI] [PubMed] [Google Scholar]
- 24.Kobayashi H, Tamatani T, Tamura T, Kusafuka J, Koga H, Yamataka A, et al. The role of monocyte chemoattractant protein-1 in biliary atresia. J Pediatr Surg. 2006;41(12):1967–72. doi: 10.1016/j.jpedsurg.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 25.Stefanovic L, Stefanovic B. Mechanism of direct hepatotoxic effect of KC chemokine: sequential activation of gene expression and progression from inflammation to necrosis. J Interferon Cytokine Res. 2006;26(10):760–70. doi: 10.1089/jir.2006.26.760. [DOI] [PubMed] [Google Scholar]
- 26.Shinkai M, Shinkai T, Puri P, Stringer MD. Increased CXCR3 expression associated with CD3-positive lymphocytes in the liver and biliary remnant in biliary atresia. J Pediatr Surg. 2006;41(5):950–4. doi: 10.1016/j.jpedsurg.2006.01.060. [DOI] [PubMed] [Google Scholar]
- 27.Hall DJ, Bates ME, Guar L, Cronan M, Korpi N, Bertics PJ. The role of p38 MAPK in rhinovirus-induced monocyte chemoattractant protein-1 production by monocytic-lineage cells. J Immunol. 2005 Jun 15;174(12):8056–63. doi: 10.4049/jimmunol.174.12.8056. [DOI] [PubMed] [Google Scholar]
- 28.Malmgaard L, Melchjorsen J, Bowie AG, Mogensen SC, Paludan SR. Viral activation of macrophages through TLR-dependent and -independent pathways. J Immunol. 2004 Dec 1;173(11):6890–8. doi: 10.4049/jimmunol.173.11.6890. [DOI] [PubMed] [Google Scholar]
- 29.Mesa MC, Rodriguez LS, Franco MA, Angel J. Interaction of rotavirus with human peripheral blood mononuclear cells: plasmacytoid dendritic cells play a role in stimulating memory rotavirus specific T cells in vitro. Virology. 2007 Sep 15;366(1):174–84. doi: 10.1016/j.virol.2007.04.007. [DOI] [PubMed] [Google Scholar]