

Abstract

A concise, modular, asymmetric synthesis of cis-2,6-disubstituted piperazines from readily available amino acid precursors is described. The key step in the synthesis is a Pd-catalyzed carboamination of a N1-aryl-N2-allyl-1,2-diamine with an aryl bromide. The products are obtained in 14-20:1 dr, with >97% ee, and the key cyclizations are the first examples of 6-membered ring formation via Pd-catalyzed carboamination reactions of unsaturated amines with aryl halides.
Substituted piperazines are of profound importance in medicinal chemistry and drug development. For example, a 2001 survey of biologically relevant templates in the MDDR revealed 2271 biologically active molecules that contained substituted N-aryl piperazines; 16 of these compounds were drugs on the market and an additional 23 were in phase II or III clinical trials.1 The presence of substituents on C2, C3, C5, and C6 of the ring has a significant influence on the biological activity of these molecules, and the ability to prepare substituted piperazines is of value for the optimization of biological properties.2
Although the asymmetric synthesis of monosubstituted or 2,5-disubstituted piperazines can be accomplished in a straightforward manner using readily available amino acid precursors,3 the stereoselective preparation of enantiomerically enriched 2,6-disubstituted piperazines remains challenging, particularly when one of the substituents is not a carboxylic acid or ester. There are few asymmetric routes to N,N-disubstituted-2,6-dialkylpiperazines, and the existing syntheses of these molecules typically require ≥6 steps.4,5 In this Letter we describe a concise, modular, asymmetric synthesis of 2,6-disubstituted piperazines that allows the stereoselective preparation of compounds bearing four different groups at N1, C2, N4, and C6.6
As shown in Scheme 1, our approach to the construction of 2,6-disubstituted piperazines employs a key Pd-catalyzed carboamination reaction,7,8 which generates the heterocyclic ring and forms two bonds simultaneously. Retrosynthetic analysis of the carboamination substrates (1) suggested these compounds could be prepared in a straightforward and modular fashion from simple precursors: amino acids (readily available in enantiopure form), allylic amines, and aryl halides. In addition to the synthetic utility of this strategy, the key cyclization reaction is a fundamentally interesting and challenging transformation. The generation of six-membered rings through Pd-catalyzed carboamination or carboetherification reactions between heteroatom-tethered alkenes and aryl/alkenyl halides has not been previously described, nor has the construction of heterocyclic rings bearing more than one nitrogen atom been achieved using this method. Thus, this transformation represents a significant advance in Pd-catalyzed alkene carboamination methodology.
Scheme 1. Retrosynthetic analysis.
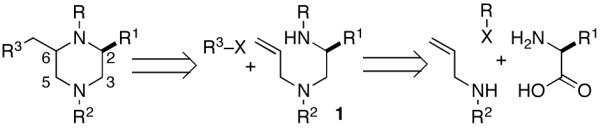
The starting materials for the carboamination reactions were prepared from commercially available amino acids using one of two 3-4 step sequences. As shown in Scheme 2, the first route employs unprotected amino acid starting materials, which were subjected to Cu-catalyzed N-phenylation using the anhydrous conditions developed by Ma, to generate 3 without significant degradation of enantiomeric purity.9 However, attempts to use biphasic conditions10 for the N-arylation of unhindered amino acids such as alanine or phenylalanine provided products with low enantiomeric purity (ca. 14-50% ee). Amide bond formation was achieved by coupling 3 with N- (benzyl)allylamine using Goodman’s DEPBT reagent.11 Reduction of amide 4 with LiAlH4 afforded 1a-b with ≥98% ee.
Scheme 2. Synthesis of substrates from unprotected amino acids.
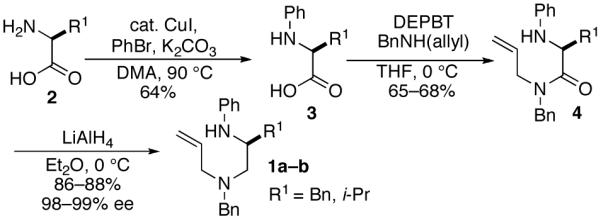
Alternatively, in some instances it was advantageous to employ N-Boc-protected amino acids as starting materials due to their commercial availability, or due to partial loss of enantiomeric purity during the Cu-catalyzed N-arylation reaction.12 These starting materials were converted to 7 in two steps via DCC-mediated coupling with an allylic amine followed by reduction with LiAlH4 at 0 °C (Scheme 3). Cleavage of the Boc group was accomplished by treatment of 7 with HCl/dioxane, and subsequent Pd-catalyzed N-arylation13 afforded the requisite substrates (1c-e) in high enantiomeric purity.
Scheme 3. Synthesis of substrates from protected amino acids.
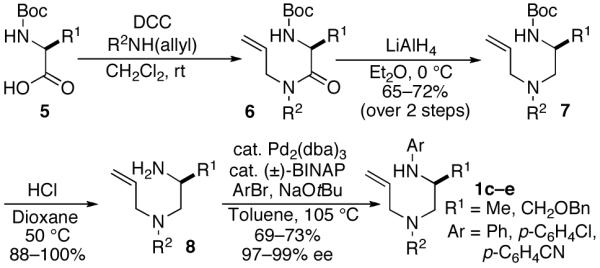
With suitable precursors in hand, we initially examined Pd-catalyzed reactions of phenylalanine-derived substrate 1a with 4-bromoanisole using a number of different phosphine ligands. In contrast to the analogous 2,5-disubstituted pyrrolidine-forming cyclizations, which were effectively catalyzed by mixtures of Pd2(dba)3 and dppe7a, use of catalysts supported by P(2-furyl)3 provided optimal results in the piperazine-forming reactions (Table 1). Use of a 4:1 ratio of phosphine to Pd led to complete consumption of the diamine starting material, whereas reactions that employed a 2:1 L:Pd ratio frequently halted at ca 85-90% conversion. The major side products generated in these transformations were unsaturated piperazines 10 and 11. Resubjection of 11 to the reaction conditions did not result in the formation of 9 or 10, and 9 was not converted to 10 under the reaction conditions.
Table 1.
Optimization Studies
 | ||||
|---|---|---|---|---|
| entry | ligandb | conversion (%)c | product ratiod | isolated yield |
| 1 | dppee | 50 | 68:0:32 | — |
| 2 | Dpe-Phos | 97 | 56:23:21 | — |
| 3 | Xantphos | 82 | 50:35:15 | — |
| 4 | P(o-tol)3 | 87 | 9:17:74 | — |
| 5 | P(2-furyl)3 | 97 | 71:7:21 | 62%e |
Conditions: 1.0 equiv 1a, 1.2 equiv Ar1Br, 1.2 equiv NaOtBu, 1 mol % Pd2(dba)3, 4 mol % chelating ligand or 8 mol % monodentate ligand, toluene (0.2 M), 105 °C, 8 h.
Dppe = 1,2-bis(diphenylphosphino)ethane, Dpe-Phos = 1,1-bis(diphenylphosphinophenyl)ether, Xantphos = 9,9-dimethyl-4,5-bis(diphenylphosphino)xanthene.
Conversion refers to % starting material consumed; 9-11 were the sole products detected in the crude reaction mixture.
Determined by 1H NMR analysis of crude reaction mixtures.
This yield was obtained after complete conversion.
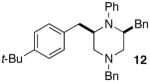
As shown in Table 2, the carboamination reactions are effective for transformations involving derivatives of several different amino acids including phenylalanine (1a), valine (1b), serine (1c), and alanine (1d-e). The reactions are tolerant of functionalized aryl groups on N-1 (e.g., p-chlorophenyl and p-cyanophenyl) and several different protecting groups on N-4 (e.g., benzyl, allyl, and p-methoxyphenyl).14 Electron-rich, -neutral, and -poor aryl bromides can be employed as coupling partners, although use of β-bromostyrene provided low yields (ca. 25%) of the desired allylpiperazine derivative. In most cases the cyclizations afforded cis-2,6-disubstituted piperazines with >20:1 dr, although cyclizations of 1c proceed with slightly lower (14:1) diastereoselectivity.15,16
Table 2.
Asymmetric Synthesis of 2,6-Disubstituted Piperazines
 | |||||
|---|---|---|---|---|---|
| entry | substrate | product | dr | ee | yieldc |
| 1 | 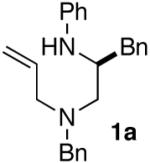 |
 |
>20:1 | 99% | 63% |
| 2 | 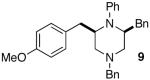 |
>20:1 | 99% | 62% | |
| 3 | 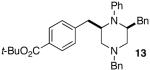 |
>20:1 | 98% | 59% | |
| 4 | 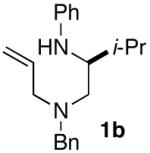 |
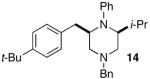 |
>20:1 | 99% | 51%c |
| 5 | 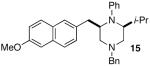 |
>20:1 | 99% | 50% | |
| 6 | 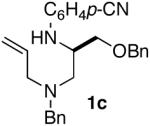 |
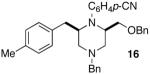 |
14:1 | 97% | 71% |
| 7 | 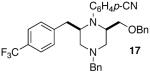 |
14:1 | 98% | 69% | |
| 8 | 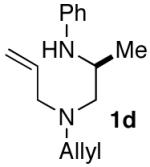 |
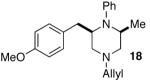 |
>20:1 | 99% | 53% |
| 9 | 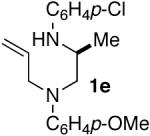 |
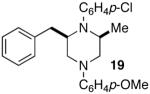 |
>20:1 | 99% | 51% |
Conditions: 1.0 equiv amine, 1.2 equiv ArBr, 1.2 equiv NaOtBu, 1 mol % Pd2(dba)3, 8 mol % P(2-furyl)3, toluene (0.2 M), 105 °C, 8-10 h.
Average isolated yields obtained from two or more experiments.
The reaction was conducted with 2 mol % Pd2(dba)3 and 16 mol % P(2-furyl)3.
A plausible mechanism for the Pd-catalyzed piperazine-forming reactions is shown below (Scheme 4). These transformations appear to be mechanistically analogous to previously described carboamination reactions of γ-aminoalkenes with aryl bromides,7 and are likely initiated by oxidative addition of the aryl bromide to Pd(0) to generate 20. This intermediate can react with the amine and NaOtBu to provide palladium (aryl)(amido) complex 21, which can undergo syn-aminopalladation7a,d to afford 22. Carbon-carbon bond-forming reductive elimination would then yield the observed piperazine product.
Scheme 4. Catalytic cycle.
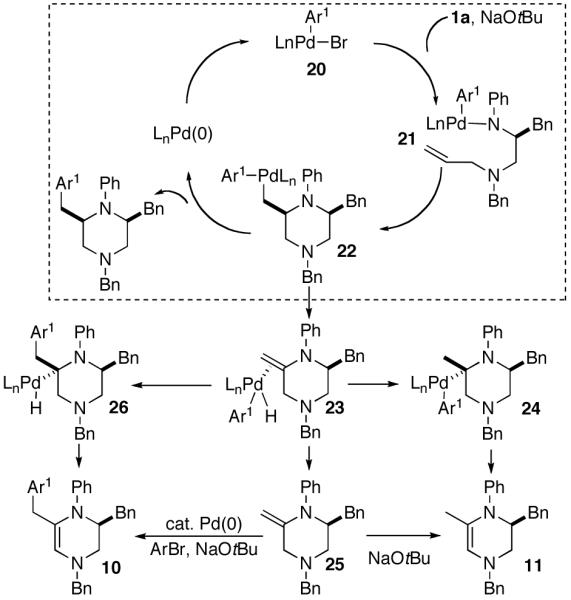
Side products 10 and 11 most likely derive from a common intermediate (23) that results from competing β-hydride elimination of 22. Compound 11 could be generated by insertion of the alkene into the Pd-H bond of 23 followed by β-hydride elimination from the C5 position of 24. Alternatively, displacement of Pd from 23 would afford 25, which could be converted to the more thermodynamically stable derivative 11 by reaction with NaOtBu. Side product 10 most likely results from Heck-type arylation of 25, although formation of 10 via carbopalladation of 23 followed by C5 β-hydride elimination from 26 cannot be ruled out.17 The generation of 10 and 11 provides further evidence to support the mechanism of 2,6-disubstituted piperazine formation shown above, rather than a mechanism involving alkene carbopalladation of 21.7a,d
Interestingly, the production of undesired compounds resulting from β-hydride elimination of 22 is much more problematic in the N-arylpiperazine-forming reactions than the analogous N-arylpyrrolidine-forming reactions.7a,18 This may be due to the fact that the transition state for β-hydride elimination from the five-membered pyrrolidine ring is more strained (due to re-hybridization of the C2-carbon from sp3 to sp2) than the analogous transition state for conversion of the six-membered ring 22 to 23.
The stereochemical outcome of the piperazine-forming reactions contrasts with our model for the analogous pyrrolidine-forming processes, which are believed to proceed via a transition state in which the C1-substutient is placed in a pseudoaxial orientation to minimize A(1,3) strain between the C1-group and the N-substituent (Ar, Boc, or Cbz).7a,g,j A similar transition state structure (27) should afford trans-2,6-disubstituted piperazines, which are not the major products in these reactions. Our current working hypothesis for piperazine-formation involves cyclization via transition state 28, in which the N1-Ar group is rotated such that N1 is pyramidalized. This eliminates the A(1,3) interaction and allows pseudoequatorial orientation of R1, which leads to the cis-2,6-disubstitued stereoisomers. This hypothesis is consistent with the observation that cyclizations of substrates bearing N1-(p-cyanophenyl) or N1-Boc groups, which should have a decreased degree of N1-pyramidalization, proceed with lower selectivity.16

In conclusion, we have developed a new, stereoselective, asymmetric synthesis of N-aryl-2,6-disubstituted piperazines. This strategy allows the modular construction of a number of derivatives containing different substituents at C2, C6, N1, and N4 from simple starting materials. The key cyclizations are the first examples of 6-membered ring formation in Pd-catalyzed carboamination reactions between alkene-tethered amines and aryl halides. Further studies on the scope, stereochemical outcome, and applications of these transformations are currently underway.
Supplementary Material
Acknowledgment
The authors thank the NIH-NIGMS (GM 071650) for financial support. Additional funding was provided by the Camille and Henry Dreyfus Foundation (New Faculty Award, Camille Dreyfus Teacher Scholar Award), Research Corporation (Innovation Award), Eli Lilly, Amgen, and 3M. We also thank the Vedejs research group at the University of Michigan for sharing chiral HPLC columns.
References
- 1.Nilsson JW, Thorstensson F, Kvarnström I, Oprea T, Samuelsson B, Nilsson I. J. Comb. Chem. 2001;3:546. doi: 10.1021/cc010013o. [DOI] [PubMed] [Google Scholar]
- 2.For examples of biologically active 2,6-disubstituted piperazines, see:Zheng G, Dwoskin LP, Deaciuc AG, Zhu J, Jones MD, Crooks PA. Bioorg. Med. Chem. 2005;13:3899. doi: 10.1016/j.bmc.2005.04.013.Chu-Moyer MY, Ballinger WE, Beebe DA, Berger R, Coutcher JB, Day WW, Li J, Mylari BL, Oates PJ, Weekly RM. J. Med. Chem. 2002;45:511. doi: 10.1021/jm010440g.
- 3.(a) Horton DA, Bourne GT, Smythe ML. Chem. Rev. 2003;103:893. doi: 10.1021/cr020033s. [DOI] [PubMed] [Google Scholar]; (b) Dinsmore CJ, Beshore DC. Tetrahedron. 2002;58:3297. [Google Scholar]; (c) Jung ME, Rohloff JC. J. Org. Chem. 1985;50:4909. [Google Scholar]
- 4.For syntheses of nonracemic 2,6-dialkylpiperazines, see:Schanen V, Cherrier M-P, de Melo SJ, Quirion J-C, Husson H-P. Synthesis. 1996:833.Mickelson JW, Belonga KL, Jacobsen EJ. J. Org. Chem. 1995;60:4177.Mickelson JW, Jacobsen EJ. Tetrahedron: Asymmetry. 1995;6:19.
- 5.For syntheses of racemic 2,6-dialkylpiperazines, see:Berkheij M, van der Sluis L, Sewing C, den Boer DJ, Terpstra JW, Hiemstra H, Bakker WII, van den Hoogenband A, van Maarseveen JH. Tetrahedron Lett. 2005;46:2369.Mouhtaram M, Jung L, Stambach JF. Tetrahedron. 1993;49:1391.
- 6.For selected recent approaches to the synthesis of substituted piperazine derivatives, see:Mercer GJ, Sigman MS. Org. Lett. 2003;5:1591. doi: 10.1021/ol034469l.Ferber B, Prestat G, Vogel S, Madec D, Poli G. Synlett. 2006:2133.Viso A, Fernandez de la Pradilla R, Flores A, Garcia A, Tortosa M, Lopez-Rodriguez ML. J. Org. Chem. 2006;71:1442. doi: 10.1021/jo052077h.and references cited therein.
- 7.(a) Ney JE, Wolfe JP. Angew. Chem., Int. Ed. 2004;43:3605. doi: 10.1002/anie.200460060. [DOI] [PubMed] [Google Scholar]; (b) Lira R, Wolfe JP. J. Am. Chem. Soc. 2004;126:13906. doi: 10.1021/ja0460920. [DOI] [PubMed] [Google Scholar]; (c) Bertrand MB, Wolfe JP. Tetrahedron. 2005;61:6447. [Google Scholar]; (d) Ney JE, Wolfe JP. J. Am. Chem. Soc. 2005;127:8644. doi: 10.1021/ja0430346. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Yang Q, Ney JE, Wolfe JP. Org. Lett. 2005;7:2575. doi: 10.1021/ol050647u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Ney JE, Hay MB, Yang Q, Wolfe JP. Adv. Synth. Catal. 2005;347:1614. doi: 10.1002/adsc.200505172. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Bertrand MB, Wolfe JP. Org. Lett. 2006;8:2353. doi: 10.1021/ol0606435. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Dongol KG, Tay BY. Tetrahedron Lett. 2006;47:927. [Google Scholar]; (i) Bertand MB, Leathen ML, Wolfe JP. Org. Lett. 2007;9:457. doi: 10.1021/ol062808f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Wolfe JP. Eur. J. Org. Chem. 2007:571. [PMC free article] [PubMed] [Google Scholar]; (k) Peng J, Lin W, Yuan S, Chen Y. J. Org. Chem. 2007;72:3145. doi: 10.1021/jo0625958. [DOI] [PubMed] [Google Scholar]
- 8.For examples of Cu-catalyzed intramolecular carboamination, see:Sherman ES, Fuller PH, Kasi D, Chemler SR. J. Org. Chem. 2007;72:3896. doi: 10.1021/jo070321u.Sherman ES, Chemler SR, Tan TB, Gerlits O. Org. Lett. 2004;6:1573. doi: 10.1021/ol049702+.For Pd(II)-catalyzed alkoxycarbonylation of alkenes bearing tethered nitrogen nucleophiles, see:Harayama H, Abe A, Sakado T, Kimura M, Fugami K, Tanaka S, Tamaru Y. J. Org. Chem. 1997;62:2113. doi: 10.1021/jo961988b.For carboamination reactions between alkenes and N-allylsulfonamides, see:Scarborough CC, Stahl SS. Org. Lett. 2006;8:3251. doi: 10.1021/ol061057e.For carboamination of vinylcyclopropanes, see:Larock RC, Yum EK. Synlett. 1990:529.For 1,1-carboamination of alkenes, see:Larock RC, Yang H, Weinreb SM, Herr RJ. J. Org. Chem. 1994;59:4172.
- 9.Ma D, Zhang Y, Yao J, Wu S, Tao F. J. Am. Chem. Soc. 1998;120:12459. [Google Scholar]
- 10.Lu Z, Twieg RJ. Tetrahedron Lett. 2005;46:2997. [Google Scholar]
- 11.DEPBT = 3-(diethoxyphosphoryloxy)-1,2,3-benzotriazin-4(3H)-one. Use of other reagents (e.g., DCC/HOBT or CDI) resulted in partial epimerization to afford products with ∼85-90% ee. See:Li H, Jiang X, Ye Y. -h., Fan C, Romoff T, Goodman M. Org. Lett. 1999;1:91. doi: 10.1021/ol990573k.
- 12.Cu-catalyzed N-arylation reactions of alanine afforded products with ≤ 93% ee. See reference 9.
- 13.(a) Muci AR, Buchwald SL. Top. Curr. Chem. 2002;219:131. [Google Scholar]; (b) Hartwig JF. In: Modern Arene Chemistry. Astruc D, editor. Wiley, VCH; Weinheim: 2002. p. 107. [Google Scholar]; (c) Schlummer B, Scholz U. Adv. Synth. Catal. 2004;346:1599. [Google Scholar]
- 14.Initial studies on carboaminations of amides such as 4 or 6 suggest these transformations may be feasible, but further optimization is required; low yields (ca. 10-20%) of the desired products were obtained.
- 15.Diastereomeric ratios observed in crude reaction mixtures were identical to those obtained upon isolation. In some cases the minor diastereomer could be separated by careful chromatography. See the Supporting Information for complete details.
- 16.Preliminary efforts to employ a N1 Boc-protected substrate resulted in the formation of a 1:1 mixture of diastereomers. Attempts to cyclize substrates bearing N1 Piv or Ac groups have thus far been unsuccessful.
- 17.Alkene insertion into late M-H bonds is usually much faster than alkene insertion into late M-C bonds. See:Brookhart M, Hauptman E, Lincoln DM. J. Am. Chem. Soc. 1992;114:10394.Siegbahn PEM, Stromberg S, Zetterberg K. Organometallics. 1996;15:5542.
- 18.The formation of unsaturated five-membered heterocycles analogous to10 and 11 is generally not observed in N-aryl-2-benzylpyrrolidine-forming reactions. Instead, the generation of regioisomeric N-aryl-2-methyl-3-arylpyrrolidine side products, which result from reversible β-hydride elimination/reinsertion processes, is observed. These side products are usually formed in ca. 10% yield, whereas the combined yields of10 and 11 is usually ca. 20-30%. For further discussion, see references 7a and 7j.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.