

Abstract
Retinoic acid (RA) is a critical signaling molecule that regulates gene transcription and the cell cycle. Understanding of RA signaling has increased dramatically over the past decades, but the connection between whole body RA homeostasis and gene regulation in individual cells is still unclear. It has been proposed that cytochrome P450 family 26 (CYP26) enzymes have a role in determining the cellular exposure to RA by inactivating RA in cells that do not need RA. The CYP26 enzymes have been shown to metabolize RA efficiently and they are also inducible by RA in selected systems. However, their expression patterns in different cell types and a mechanistic understanding of their function is still lacking. Based on preliminary kinetic data and protein expression levels, one may predict that if CYP26A1 is expressed in the liver at even very low levels, it will be the major RA hydroxylase in this tissue. As such, it is an attractive pharmacological target for drug development when one aims to increase circulating or cellular RA concentrations. To further the understanding of how CYP26 enzymes contribute to the regulation of RA homeostasis, structural information of the CYP26’s, commercially available recombinant enzymes and good specific and sensitive antibodies are needed.
Keywords: CYP26, retinoic acid, homeostasis, clearance
1. Introduction
Retinoic acid (RA, Figure 1) is the biologically active metabolite of vitamin A (retinol). Naturally occurring derivatives or synthetic compounds that have structures or activities similar to vitamin A are referred to as retinoids [1]. RA can chemically exist as several different geometric isomers including: all-trans RA (at-RA), 9-cis-RA, 11-cis-RA, 13-cis-RA, and 9,13-dicis-RA [2–5].
Figure 1.
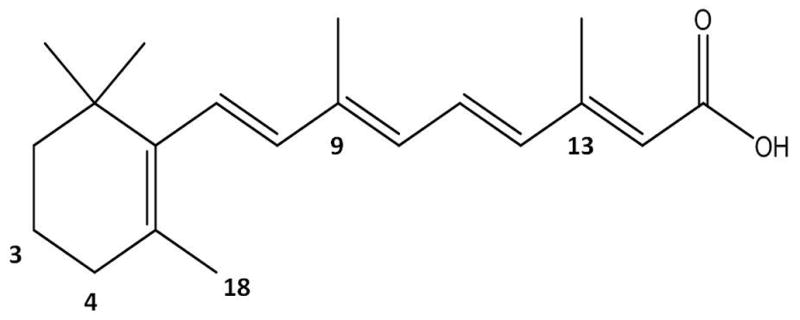
Structure of all-trans retinoic acid with carbon positions labeled.
There is general consensus that the main source of RA in humans, excluding therapeutic dosing, is via synthesis from dietary precursors such as β-carotene and retinyl palmitate [2, 6] but some RA can also be obtained from the diet, primarily from ingestion of liver. Vitamin A is stored primarily in liver stellate cells as retinyl esters, which are hydrolyzed in hepatocytes by retinyl ester hydrolases (REH) to retinol [7]. Retinol, the precursor of RA, is the main circulating retinoid and typically circulates at concentrations of 1–3 μM [8, 9]. The circulating concentrations of both endogenous at-RA and 13-cis-RA in human volunteers were 3–13 nM [4, 10]. In mice, serum concentrations of at-RA and 13-cis-RA were 1–3 nM [3, 11]. Tissue concentrations for RA isomers were greater, and ranged from 7–40 nM for at-RA and 1–6 nM for 13-cis-RA. 9,13-dicis-RA concentrations in tissues ranged from 3–23 nM and 9-cis-RA was not detected in serum or the tissues analyzed [3]. RA is obtained from retinol via a two step synthesis in which the conversion of retinol to retinal is the rate limiting step [6]. Retinol is oxidized intracellularly by retinol dehydrogenases (ADH or RDH) to retinal, and retinal is then metabolized by NAD/NADP dependant retinal dehydrogenases (RALDH) and by retinal oxidase enzymes to RA[12]. There is also in-vitro evidence that P450 enzymes can perform the biosynthesis of retinal from retinol, and RA from retinal, but their contribution under physiological conditions is predicted to be insignificant [13, 14]. The major route of RA elimination is via metabolism but the exact enzymes that regulate RA clearance are not well characterized. Cytochrome P450 enzymes, namely CYP26, CYP3A, and CYP2C, are most frequently proposed to metabolize RA.
Epidemiological data as well as animal studies have shown that RA is essential for a wide variety of biological processes including maintenance of skin and epithelial cells, regulation of apoptosis, maintenance and regulation of immunity, placental development and maintenance, and embryogenesis. [1, 15]. Both too much and too little RA has been shown to be detrimental for health in both developing embryos and adults. During development, marginal excess RA has been linked to embryonic malformations such as cleft palate, neural tube defects and limb malformations [1, 15, 16]. In adults, excess vitamin A is correlated with an increased risk of hip fracture; for every 1 mg increase in vitamin A intake, the risk for hip fracture increased by 68% [17].
Vitamin A deficiency leads to poor immunity and deaths due to infections [18]. The World Health Organization (WHO) estimates that approximately 250 million pre-school age children are deficient in vitamin A and that WHO’s intervention program, consisting of promotion of breastfeeding and vitamin A supplementation, has reduced mortality by 23% overall, and up to 50% for acute measles sufferers[19]. Sufficient amounts of RA are important in adults as well. RA is needed to maintain proper brain function and a decrease in RA exposure is associated with decreases in memory and increased risk of Alzheimer’s disease [20–22]. Additionally, skin conditions, such as phrynoderma, are associated with vitamin A deficiency[23].
The precise mechanism of how RA regulates biological processes is not yet fully understood. It has been shown that RA acts as a signaling molecule and regulates gene expression by binding to the nuclear retinoic acid receptors (RARα, RARβ, and RARγ) [24], which then bind DNA as a heterodimer with the retinoid X receptors (RXRα, RXRβ, and RXRγ). At-RA also activates peroxisome proliferator-activated receptors (PPARβ/δ) [25, 26]. It is believed that at-RA binds to RAR and 9-cis RA binds to either RAR or RXR [15, 25], but the biological role of 9-cis RA in humans is controversial as 9-cis RA has not been detected in human plasma or tissues but has been detected in locust embryos [27].
Based on the biological effects of deficiency and excess exposure of RA, there is a clear need to regulate the concentrations of RA at both the cellular and total body levels. How cellular, organ and total body exposure to RA is regulated is not fully understood. It is generally believed that retinol, circulating in plasma, is actively taken up into cells and oxidized to RA. Once RA is formed, two general models, one paracrine and one autocrine, describing RA action and metabolism have been proposed. In the paracrine model, RA is generated in one cell and then is taken up by either a target tissue where it binds to its receptor, causing pharmacological action, or a non-target tissue where RA is metabolized [28]. In the autocrine model, RA is synthesized, binds to its receptor and is metabolized in the same cell [29]. Based on existing evidence it is likely that both models apply under specific circumstances during fetal development or adult life. A better understanding of the applicability of these models is critical to our understanding of how the expression and activity of CYP26 enzymes contribute to overall RA signaling.
In the cellular environment, RA is believed to be bound to cellular retinoic acid binding proteins (CRABPs). The roles that the CRABPs play in regulating RA actions and in-vivo metabolism are, however, unclear. CRABPII has been shown to have a role in delivering RA from the cytosol to RAR in the nucleus [30] and CRABPI may have a role in facilitating RA metabolism although the details are not yet fully understood [31, 32]. Either CRABPI or CRABPII could potentially contribute to sequestering of RA in cells and to directing RA’s pharmacological effects.
The models of RA homeostasis and action proposed raise many questions: Does metabolism occur in the tissue where RA exerts its effects, or in non-target tissues? Is the liver a target tissue or a metabolic tissue, or both? And, which P450s are responsible for the metabolism of RA, especially if metabolism occurs outside of the liver? Better understanding of the processes that control RA clearance is important, as selective metabolism of RA in specific cells or tissues could function as a mechanism to control the biological activity of RA. This review focuses on the current knowledge of CYP26 enzymes and their role in elimination of RA.
2. CYP26 family of enzymes
2.1. Biochemistry of CYP26 enzymes
The CYP26 family of enzymes was discovered in studies screening for proteins that contribute to RA dependant regeneration of damaged zebrafish fins [33, 34]. Although the sequence of the cloned cDNA from the zebrafish fins was less than 30% homologous with other P450 enzymes, the sequence contained a heme binding domain establishing the clone, P450RAI, as a CYP enzyme [34]. This CYP was believed to be a RA hydroxylase as it was inducible by RA and metabolized RA. P450RAI was later assigned as CYP26A1. After its initial discovery in zebrafish, the CYP26A1 gene was cloned and characterized in humans [35, 36], as well as other species, including mouse [37], rat [38], chicken [39], and cow [40]. Two other members of the family, CYP26B1 and CYP26C1 were later identified in humans [41, 42] and other species [43–46]. A fourth member of the CYP26 family has been published in the literature, CYP26D1 [47, 48], however, it has only been identified in zebrafish and it appears identical with zebrafish CYP26C1 [43]. From our analysis of the available amino acid sequences, zebrafish CYP26C1 and CYP26D1 have 99% sequence identity and are therefore the same enzyme.
Figure 2A and B show an analysis of inter-CYP26 identity between CYP26A1, CYP26B1, and CYP26C1 and the interspecies identity between human, mouse, and zebrafish based on previously published alignments of the amino acid sequences [36, 41, 42, 44–46]. Of the three enzymes, CYP26B1 appears to be the most highly conserved between human, mouse, and zebrafish. The human to mouse sequence identity is >80% for each of the three isomers (CYP26A1, CYP26B1 and CYP26C1). As shown in Figure 2A the sequence identity between species for CYP26A1, CYP26B1 and CYP26C1 is less than 98% and hence, these enzymes are technically not the same in each species. For example, the common drug metabolizing CYPs are assigned as different isoforms if sequence identity is <98% [49]. In contrast to the drug metabolizing CYPs, which typically have variable numbers of isoforms in each subfamily in different species, each species studied has only three CYP26 enzymes. Hence, it appears plausible to assign these as orthologs with the same names, despite the distinct sequence differences and the fact that the sequence identity does not meet the 80% threshold for orthologs assigned by the P450 naming guidelines [49].
Figure 2.
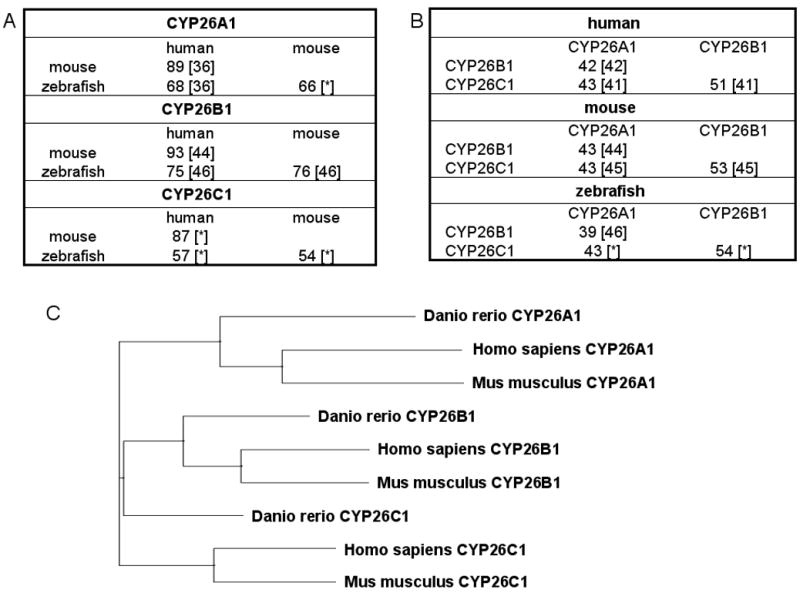
Analysis of CYP26 sequences in human, mouse and zebrafish. Comparison of amino acid sequence identity (percent identity) across species for CYP26A1, CYP26B1, and CYP26C1 (A) and across CYP26 subfamilies for human, mouse, and zebrafish (B). Numbers in brackets indicate the reference for gene alignment and % identity. [*] indicates alignments done for this manuscript using ClustalW. Panel (C) shows a phylogenetic three of human, mouse and zebrafish CYP26A1, CYP26B1, and CYP26C1. The CYP26A1 (Hs CAH72804.1, Dr NP_571221.2 and Mm NP_031837.2), CYP26B1 (Hs NP_063938.1, Dr AAQ82596.1, Mm AAH59246.1) and CYP26C1 (Hs NP_899230.2, Dr AAI29132.1, Mm AAI51107.1) genes were aligned using Vector NTI (Invitrogen) and phylogenetic tree was constructed based on the multiple alignment. Neither rigorous calculation of evolutionary distances nor phylogenetic relationship can be inferred with confidence from this tree.
Within species, CYP26B1 and CYP26C1 have higher sequence homology with each other than they do with CYP26A1 (Figure 2B). In humans, CYP26A1 and CYP26C1 are located in chromosome 10 and CYP26B1 in chromosome 2 [15]. Based on their chromosomal location, it has been proposed that CYP26C1 is a result of gene duplication of CYP26A1 [50]. Figure 2C depicts a phylogenetic tree of the CYP26 genes in human, mouse, and zebrafish, similar to those previously published [43, 46, 48]. The phylogenetic tree demonstrates a divergence of CYP26A1 from CYP26B1 and CYP26C1, and also a divergence of zebrafish enzymes from mouse and human. A more detailed analysis of chromosomal locations and sequence alignments would be necessary to fully propose an evolutionary path of CYP26A1, CYP26B1, and CYP26C1.
We are unaware of X-ray crystal structures of any of the CYP26 enzymes. In the absence of crystal structures for CYP26, the existing crystal structures of CYP2C8, CYP2C9 and CYP3A4 were used to construct a homology model for CYP26A1[51]. The amino acid environment of the resulting model was further tested and ligands of CYP26A1, such as at-RA and the inhibitor (S)-R115866 were docked in the homology model allowing for further optimization of the structure of the active site. Homology models for the other CYP26 enzymes have not been published.
2.2. Biology of CYP26 in vivo expression and function
Studies using knock-out animals have shown that both CYP26A1 and CYP26B1 are essential proteins. Cyp26a1−/−, Cyp26b1−/−, and Cyp26c1−/− mice have all been produced using homologous recombination [50, 52–54]. Cyp26a1−/− and Cyp26b1−/− were lethal, whereas Cyp26c1−/− mice were viable and did not show any alterations in embryonic development or phenotype at birth [50]. Cyp26a1 and Cyp26c1 appear to have some functional redundancy as double knock-out of Cyp26c1 and Cyp26a1 had a more severe phenotype than Cyp26a1 −/−single knock-outs.
While both Cyp26a1−/− and Cyp26b1−/− mice died, there were distinct differences between the phenotype of the knock-outs suggesting these two enzymes are not functionally redundant. Cyp26a1−/− mice died during mid-gestation [52], whereas Cyp26b1−/− mice died shortly after birth, which was attributed to respiratory failure [54]. Embryos of both knock-outs had limb and facial deformities. Cyp26a1−/− mice had abnormalities similar to those observed in teratogenesis caused by RA excess [52]. Many of the Cyp26a1−/− mice had truncated or otherwise deformed tails, abnormalities of the cervical vertebrae, or spina bifida. Additionally, some Cyp26a1−/−fetuses had malpositioned hindlimbs or showed exencephaly. Interestingly, the lethal and phenotypic malformations observed in Cyp26a1−/− mice could be rescued by heterozygous disruption of Aldh1a2 [53]. Cyp26b1−/− mice showed greater defects in limb bud rather than tail bud development as was observed in Cyp26a1−/− mice. As a result, Cyp26b1−/− mice showed severe malformations of the forelimbs and hindlimbs [54].
Based on the knock-out data, it has been of interest why Cyp26a1−/− and Cyp26b1−/− are lethal and what role these enzymes play during development. The differences in the deformities observed in Cyp26a1−/− and Cyp26b1−/− mice can possibly be explained by differences in CYP26A1 and CYP26B1 expression during development. Using in-situ hybridization, CYP26 mRNA has been identified in murine embryos in a developmental stage and tissue specific manner [37, 44, 52, 54–56]. CYP26A1 gene expression has been detected in murine embryos as early as 6 days post-coitum (d.p.c.) [37] with the posterior-anterior gradient changing dramatically in a short period between 7.25 and 8.5 d.p.c. [37, 44, 56]. This expression pattern of CYP26A1 has been shown to create an uneven distribution of RA concentrations in the embryo, which then directs the development and patterning of the hindbrain, vertebrae, and tail bud [56]. During 8 d.p.c., CYP26A1 is expressed specifically in rhombomere 2 and the tail bud [52], whereas CYP26C1 is expressed in rhombomeres 2 and 4 [45]. Between 9.5 and 10.5 d.p.c. CYP26A1 is expressed mainly in the hindgut, neural crest, the neural plate and tail bud [37] and CYP26C1 is expressed in the branchial arch and remains in rhombomere 2 but not in rhombomere 4 [45]. In contrast to CYP26A1 and CYP26C1, CYP26B1 is first detected at embryonic 8 d.p.c. and is detectable in rhombomeres 3 and 5 [44]. By 9.5 d.p.c. CYP26B1 is detected in rhombomeres 5 and 6. Between 9.5–11.5 d.p.c., CYP26B1 is expressed in the developing limb buds [44, 54]. During 12–18.5 d.p.c. CYP26A1 mRNA was present in rib and vertebral cartilage, neural retina, oral epithelium, limbs, and the stomach [55]. In comparison, CYP26B1 mRNA was more highly expressed in the craniofacial region, hindbrain, forebrain, spinal cord, lung, kidney, spleen, thymus, testis, and skin [55].
In contrast to expression in mice, CYP26B1 mRNA was relatively low and CYP26A1 mRNA was highest in the human fetal brain, compared to other tissues [57]. The highest expression of CYP26B1 in the human fetus was in the kidney and muscle. CYP26C1 was not detected in human fetal liver or brain tissue [57]. The fact that the developmental stage of the fetuses was not reported could possibly explain discrepancies between the expression data in humans and mice due to the dramatic changes in CYP26 expression with gestational age. For example, in a study of CYP26 (isoform not specified) mRNA expression in human prenatal cephalic tissues, there was a gradual 11-fold increase in CYP26 mRNA between days 51 and 110 of gestation [58].
In the adult human, CYP26A1 mRNA expression was highest in the liver and CYP26C1 was present in the brain and liver [57]. CYP26B1 mRNA was not detected at all in adult human liver and was instead most abundantly expressed in the placenta, ovary, testes, and intestine [57]. Overall, CYP26B1 expression was more ubiquitous in expression than CYP26A1 or CYP26C1 in human tissues screened. The expression pattern is in agreement with earlier work that reported most tissues having detectable levels of CYP26B1 mRNA, with highest levels detected in the brain, specifically the pons and cerebellum, whereas CYP26A1 showed low levels of expression in most tissues and was absent from the brain [42]. It should be noted that due to the lack of proven specific antibodies against CYP26A1, CYP26B1 or CYP26C1, presence and expression levels of the CYP26 proteins have not been demonstrated and the published localization of the enzymes is based on mRNA data from single individuals.
The differences in the expression of CYP26 isoforms is likely the result of different regulation of the three enzymes. It has been documented that there is up-regulation of RA metabolism when cells or animals are treated with RA [33, 34, 36, 42, 59, 60]. This is likely due to RA’s auto-induction of its own metabolism. In early studies, CYP26 induction was observed in RA treated mouse liver, but not mouse brain [35]. Using Northern blot and RT-PCR techniques, CYP26A1 mRNA in rat liver samples was shown to have a positive correlation with RA and dietary vitamin A status [60]. A correlation between the age of the rat and CYP26A1 mRNA was also observed. It is generally believed that RAR is responsible for the induction of CYP26 [61, 62], but in some cell lines RA has no effect on CYP26 mRNA expression [34, 36, 42], suggesting a more complex regulatory mechanism.
3. Metabolism of RA
At-RA is metabolized by several different enzymes to form multiple products such as ketones, epoxides, and hydroxylated products [1, 63]. The most common metabolite mentioned in the literature is 4-OH-RA, which can then be further oxidized to 4-oxo-RA and diOH-RA. Other identified metabolites include 18-OH-RA, 3-OH-RA and 5,6-epoxy-RA. There are reports that some RA metabolites, for example, 4-oxo-RA and 5,6-epoxy-RA have biological activity [64–67], but the general consensus is that RA, not the metabolites, regulate development [53]. While the hydroxylated and subsequently oxidized metabolites are primarily formed through cytochrome P450 catalyzed mechanisms, the 5,6 epoxide appears to be generated through peroxyl radical-dependent mechanisms [68]. RA, its isomers, and its metabolites can also undergo phase II metabolism by UGT2B7 to form RA-glucuronides. While at-RA, 5,6-epoxy-RA, and 4-oxo-RA are exclusively carboxyl-linked to the glucuronide, 4-OH-RA can be glucuronidated at either the carboxyl or the hydroxyl function [68].
3.1 RA metabolism by P450s
Several other P450s, such as CYP3A4, CYP3A5, CYP3A7, CYP2C8, CYP2C9, CYP2C18 and CYP4A11, have all been shown to metabolize at-RA to 4-OH-RA [69–72]. The 4-OH-RA metabolite appeared to be the primary metabolite formed by these CYPs with either lymphoblastoid cells overexpressing P450s or in cDNA-derived CYPs expressed in baculovirus-infected insect cells (supersomes™). CYP2C19 was reported to metabolize at-RA to 4-OH-RA in only one of the three papers which evaluated this enzyme [72]. CYP2D6, CYP2E1, CYP1A1, CYP2B6, and CYP2A6 did not metabolize at-RA in any of the studies [70–72]. Subsequent metabolism of 4-OH-RA to 4-oxo-RA was observed in at least one, but not all studies when RA was incubated with CYP3A4, CYP3A7, CYP2C8, CYP2C9, CYP1A1, and CYP4A11. The 18-OH-RA metabolite was only reported in one of the screening panels[70] and was described to be formed by CYP3A5, CYP3A7, CYP2C8, CYP2C9, CYP2C18, CYP1A1, and CYP4A11. In the same study, the 5,6-epoxy-RA metabolite was reported to be formed by the same enzymes that formed 4-OH-RA, however based on other publications, this oxidation could be non-P450 mediated.
Of the P450 enzymes routinely tested, CYP2C8 was most efficient in metabolizing at-RA to 4-OH-RA [70–72]. The obtained kinetic parameters for CYP2C8 in the three reported studies are shown in Table 1. Vmax, Km, and Clint values ranged twelve-, thirty-six-, and fifty-eight-fold, respectively, between studies, but when efficiency of different isoforms was compared, CYP2C8 was consistently most efficient. CYP3A4 and CYP2C9 were the next most efficient enzymes at metabolizing at-RA (Clint 2.5–20 μL/min/nmolP450). The large inter-study variability observed for CYP2C8 was observed with CYP3A4 and CYP2C9, as well.
Table 1.
Kinetic parameters for formation of 4-OH-RA from all trans-RA by CYP2C8 in various in vitro systems. Clint is defined as Vmax/Km.
The amount of protein used, length of incubation, concentration of at-RA, and cell system used could all account for the differences in kinetics observed between studies. For example, it is reported that at-RA is highly protein bound [73] and therefore if excess protein is used during incubations, extensive non-specific binding could lead to a depletion of free at-RA in solution, artificially increasing the Km value. Additionally, human lymphoblastoid cells may express other native P450s or other catalytic enzymes that contribute to at-RA metabolism.
In addition to forming oxidized products, at-RA can isomerize to form 13-cis-RA, 9-cis-RA, and 9,13-dicis-RA [3, 74]. This isomerization is classically induced by white light, but can also be conducted by glutathione-S-transferase. In lymphoblast microsomes expressing single CYP enzymes, CYP3A7, CYP2C8, CYP4A11, and others metabolized 13-cis-RA to 4-OH-13-cis-RA, whereas CYP2C9, CYP2C8, and CYP3A7 were shown to metabolize 9-cis-RA to 4-OH-9-cis-RA[75]. Metabolites of 9-cis-RA and 13-cis-RA have also been detected in plasma after administration of corresponding isomers [8].
3.2 Catalytic activity of CYP26
All CYP26 enzymes have been shown to metabolize RA, despite having different mRNA expression patterns and regulation mechanisms [34, 36, 41, 42, 47]. Based on the phenotype of knockout animals, inducibility by RA, and apparent efficacy in metabolizing RA, CYP26 enzymes are all believed to be responsible for clearing RA from various tissues and the body as a whole. Based on the earlier studies, RA is the primary retinoid metabolized by CYP26A1 and CYP26B1, however at μM concentrations, 9-cis-RA, 13-cis-RA, retinal, and retinol do also appear to be ligands of CYP26s [42]. Interestingly, 9-cis-RA is a much better substrate for CYP26C1 than for CYP26A1 or CYP26B1 [41].
Similar to what has been observed for other P450 enzymes, 4-OH-RA is the primary metabolite of RA formed by CYP26A1 and CYP26B1. The formation of the 4-oxo-RA and 18-OH-RA, as well as a series of more polar, presumably secondary metabolites was observed [42]. Based on HPLC retention times, CYP26A1 and CYP26B1 expressed in COS-1 cells formed 4-OH-RA, 18-OH-RA and 4-oxo-RA when incubated with RA. The identity of the metabolites formed by CYP26A1 have been confirmed by LC-MS-MS [63]. Mass spectra of 4-OH-RA, 4-oxo-RA and 18-OH-RA, in addition to other oxidized products, were obtained from extracts of CYP26A1 transfected mammalian cell lines incubated with RA. Additional oxidized products were detected and proposed to contain multiple hydroxylations and/or oxo-products. Whether these products are formed sequentially from 4-OH-RA and other oxidized metabolites by CYP26, or by other metabolic enzymes in the cell lines, is currently unknown. LC-MS data from incubations performed with CYP26C1 transfected cells have shown that RA is metabolized by CYP26C1 to products similar to those seen in CYP26A1 and CYP26B1 transfected cells [41]. Early work in transiently transfected cells showed that all CYP26 isoforms metabolize at-RA to several oxidized products. Many of the studies performed to identify at-RA metabolites and determine preliminary kinetics have been done in COS, HEK, or other mammalian cell systems that either over-express or transiently express CYP26. While these immortalized cell lines are readily available and easy to culture and transfect, only limited data can be obtained due to variable expression levels and the presence of other native proteins. Although expression of CYP26 in immortalized mammalian cell lines can give a qualitative analysis of the role of CYP26 in RA depletion, these systems do not allow for detailed metabolite identification or kinetic characterization. The low expression levels make it difficult to prepare microsomal fractions or purify the protein in sufficient quantities. Expression at quantities sufficient for isolation of the P450 of interest is necessary to perform detailed biochemical studies of CYP26 function and activity. Furthermore, to compare the relative contribution of CYP26 isoforms to other P450s, the CYP26s need to be expressed in a system that allows quantification of the P450 concentration.
Attempts to express CYP26A1 in E. coli have been unsuccessful [76]. Recently, CYP26A1 was expressed in baculovirus-infected insect cells and purified. The kinetics of at-RA turnover was characterized [77]. Using this recombinant system, at-RA was confirmed to be a tight binding ligand of CYP26A1 resulting in a typical type I binding spectrum upon binding to CYP26A1. The depletion Km of at-RA was 9.4 nM and the Vmax 11.3 pmoles/min/pmole P450. As expected for a microsomal P450 enzyme, catalysis by CYP26A1 was oxidoreductase dependant. However, cytochrome b5 had no effect on CYP26A1 activity. A key point of metabolism of at-RA by CYP26A1 is that at-RA displays higher binding affinity to CYP26A1 in comparison to other P450s, and the intrinsic clearance (1.2 mL/min/pmol P450) for RA depletion by CYP26A1 is more than 800-fold higher than previously reported for the other P450s. Similar kinetic and biochemical characterization of CYP26B1 and CYP26C1 is yet to be conducted.
3.3 Relative importance of CYP26 in RA clearance
Since the discovery of CYP26 enzymes, it has been widely accepted that clearance of endogenous (non-therapeutic) RA is mediated primarily by CYP26 enzymes. This claim has not, however, been supported by detailed kinetic analysis or protein expression data in various tissues. A classic experiment using specific inhibitors of CYP enzymes to identify the contribution of various P450s involved provided equivocal results [71, 72]. Quinidine, a CYP2D6 inhibitor, showed some of the highest % inhibition compared to control activity, but CYP2D6 has been shown not to metabolize RA. Conversely, sulfaphenazole, an inhibitor of CYP2C9, showed little to no inhibition of RA metabolism in the two studies although CYP2C9 does metabolize RA.
In the absence of CYP26 specific inhibitors and evidence of specificity of known P450 inhibitors, we predicted the relative contribution of the individual liver P450 enzymes shown to metabolize RA using published Clint (Vmax/Km) values [70–72] and amounts of various P450s present in human liver [78]. Figure 3 shows the predicted contributions of CYP26A1, CYP2C8, CYP2C9 and CYP3A4 to RA clearance at physiological concentrations of RA. In the absence of data on CYP26A1 expression in the human liver we used either an expression level of 5 pmol/mg of liver of CYP26A1 or no expression of CYP26A1 in our predictions. This expression level is extrapolated from the expression of other P450 enzymes in the liver (10–130 pmol/mg liver). It is likely that CYP26A1 expression in the human liver will be highly variable as CYP26A1 in rat liver appears to be regulated by RA exposure. A characterization of CYP26A1 protein expression in the human liver, as well as inter-individual variability, is necessary to obtain a more accurate prediction of CYP26 contribution to RA clearance. From our predictions it is, however, evident that if expressed to a detectable level in the liver, CYP26A1 will be the major P450 enzyme contributing to RA clearance in humans. In the absence of CYP26A1 expression, CYP2C8 and possibly CYP3A4 will be the major RA hydroxylases in human liver.
Figure 3.
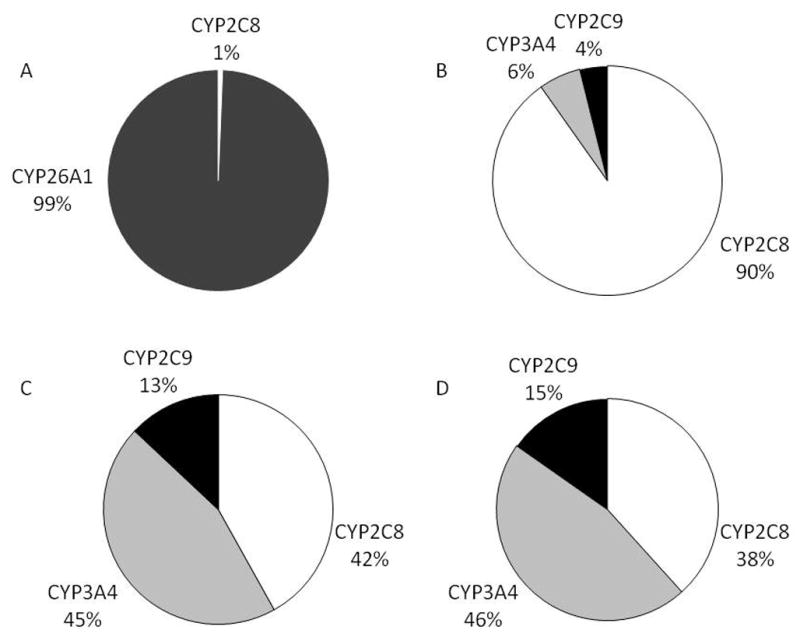
Prediction of the relative contribution of CYP26A1, CYP2C8, CYP2C9, and CYP3A4 to liver clearance of at-RA using published Clint values and published expression levels of CYP2C8, CYP2C9 and CYP3A4. CYP26A1 concentration was set at 5 pmoles/mg liver (A) or 0 pmoles/mg liver (B–D) and 24 pmoles CYP2C8, 111 pmoles CYP3A4, and 73 pmoles of CYP2C9 per mg of liver was assumed in all predictions [78]. Intrinsic clearance values for RA metabolism by CYP3A4, CYP2C8 and CYP2C9 were obtained from [71] (A–B), [70] (C), and [72] (D). The different relative % contributions of CYP2C8, CYP3A4 and CYP2C9 are due to variable enzyme kinetic data for these proteins between publications.
4 Therapeutic applications of at-RA and 13-cis-RA
While RA is synthesized in the body from dietary precursors, it may also be administered therapeutically. Both at-RA or tretinoin (Vesanoid®) and 13-cis-RA or isotretinoin (Accutane®) are administered clinically. Tretinoin is approved to induce cytodifferentiation and decrease proliferation of acute promyelocytic leukemia [79] whereas isotretinoin is approved for the treatment of severe nodular acne that has been unresponsive to other forms of treatment [80]. These and other retinoids, primarily RAR ligands, are currently being evaluated for additional therapeutic applications and a thorough review on this topic is available [81]. Both at-RA and 13-cis-RA are believed to circulate, highly bound to albumin (tretinoin >95%, isotretinoin 99%). A summary of their pharmacokinetic parameters is presented in Table 2 [73, 82, 83]. The terminal half life of tretinoin (0.5 – 2 hours) is much shorter than that of isotretinoin (21 hours) and is most likely due to its higher clearance. As the concentrations of tretinoin and isotretinoin detected in the plasma of patients receiving therapy are reported to be in the low or sub μM range, the CYP26 enzymes are likely to be saturated at these concentrations and we predict the role of other P450s to be more significant during therapy than at lower concentrations of RA.
Table 2.
Kinetic parameters for the metabolism of therapeutically administered RA. Numbers in brackets refer to the reference.
| Compound | all trans-retinoic acid | 13-cis-retinoic acid |
|---|---|---|
| Generic name | tretinoin | isotretinoin |
| Trade name | Vesanoid ® | Accutane ® |
| Cl (mL/min) | 998*[83] | 133*[82] |
| fu (%) | <5 [73] | <0.1 [82] |
| t ½ (hr) | 0.5 – 2 [73] | 21 [82] |
| AUC(ng*hr/mL) | 1313 [83] | 10,000 [82] |
| Cmax (ng/mL) | 347 [73], 508 [83] | 862 [82] |
| Tmax (hr) | 1–2 [73], 3.9 [83] | 5.3 [82] |
Cl/F calculated from dose/AUC
[83] Subjects received single oral at-RA dose of 45 mg/m2 (average body surface area was 1.93 m2), values were obtained on day 1 of study
[73] APL patients dosed with 45 mg/m2 (~80 mg) daily
[82] Healthy volunteers receiving a single 80 mg dose with standardized high fat meal. Some kinetic parameters were different under fasted conditions.
Auto-induction of RA clearance during RA therapy is a significant clinical challenge in cancer patients, and likely to involve induction of CYP26A1 mediated clearance. It is generally believed that the decreased peak plasma concentrations and AUCs observed after repeated dosing of RA are due to induction of metabolic enzymes [84, 85]. To overcome this metabolic resistance, instead of administering more RA, P450 inhibitors have been co-administered with RA to decrease its clearance [86]. In two female acute promyelotic leukemia patients, treatment with fluconazole caused a two to four-fold increase in at-RA AUC [87]. In a second case study, at-RA toxicity was observed after co-administration with fluconazole requiring 70% dose reduction of at-RA [88]. Similarly, the AUC of at-RA was increased approximately two-fold higher after ketoconazole administration [89]. At present, the identity of the specific P450 inhibited is not known. The fact that RA metabolism could be inhibited in-vivo, has lead to the development of specific structural analogues of RA modified with functional groups designed to inhibit P450 metabolism. These compounds are referred to as RA metabolism blocking agents (RAMBAs) [90–92]. While they represent a wide class of structures, they all have a common purpose: to inhibit the metabolism of RA so that effective RA concentrations are maintained even after auto-induction of RA metabolism. IC50s for the RAMBAs towards various RA metabolizing systems range from low nM to μM.
The most studied RAMBA is liarozole, which has been shown to inhibit at-RA hydroxylation in-vitro and to cause a two-fold increase in at-RA AUC in-vivo [93]. However, use of liarozole in humans is associated with side effects and significant efforts are made to obtain more specific RAMBA’s (For review see [91]). Several groups have reported the development of compounds that are potent, nanomolar, inhibitors of at-RA hydroxylation in-vitro [91, 94–98]. Some of the reported compounds, such as R116010, R115866 (rambazole) and some 2,6-disubstituted naphtalenes were demonstrated to have some degree of selectivity towards at-RA metabolizing enzymes although the data were from rat tissues or transfected cell lines [94, 95, 98].
Ideally, RAMBAs should inhibit selectively the CYP26 enzymes. However, the promiscuity of P450 enzymes and the efficiency of CYP3A and CYP2C8 to clear at-RA complicates the design of new selective compounds, as multiple P450’s are likely to contribute to at-RA clearance when at-RA is administered at therapeutic concentrations. CYP3A4 is most likely the enzyme that is responsible for the increased clearance and decreased circulating at-RA concentrations in adults undergoing chronic treatment with the enzyme inducing antiepileptic drugs phenytoin and carbamazepine [99]. In infants and children treated with phenytoin, phenobarbital, carbamazepine and ethosuximide, the plasma concentrations of at-RA were more than 70% lower than in untreated children, suggesting that these drugs significantly induce at-RA metabolism [100].
5 Conclusions
Over the last ten years, significant progress has been made in identifying CYP26 genes in various species, and improving the understanding of the function these enzymes play during development. An overall knowledge of the biochemistry of these enzymes has also been obtained. Current data allows approximate predictions of the relative contributions of various P450 enzymes in comparison to CYP26A1 in RA clearance, although significant variability between studies does exist. It is apparent that when CYP26A1 is expressed in the human liver it will be the major contributor to endogenous RA clearance. However, at present it is not known at what levels CYP26A1 protein is expressed in the liver and how much inter- and intra-individual variability there is in the population. During therapeutic administration, the contribution of CYP26A1 in clearing RA isomers has not been quantitatively assessed. Finally, the lack of structural information and commercially available recombinant enzyme hinder the development of novel therapeutic approaches that would take advantage of the increasing understanding of CYP26 biology.
6. Expert opinion
The greatest gaps in the understanding of the function of the CYP26 enzymes are perhaps in determining their expression and activity after birth and during adult life. The processes that regulate the tissue and spatio-temporal expression of the CYP26 enzymes in adults are very poorly characterized as well as the function of these enzymes during adult life. Despite the fact that increasing information related to alternative mechanisms of CYP26 regulation has become available, the most unequivocal data on regulation of CYP26 expression remains to be the induction of CYP26A1 and to lesser extent CYP26B1 by RA in some tissue types. Why RA does not affect the expression of these enzymes in all cell types is yet to be explained. It is important to note that all currently available expression data refers to mRNA levels, which do not necessarily correlate with protein expression levels of activity. In addition, a significant caveat of the mRNA expression data in humans is that it usually refers to a single donor. As the regulation of these enzymes is expected to be dynamic, these single donor expression levels may be very misleading. It is clear that more thorough studies that determine the expression of CYP26 protein in various adult tissues are needed to further the understanding of overall RA homeostasis. While difficult to conduct, studies which test whether CYP26 enzymes function in cells to prevent RA action or to deplete RA after release from nuclear RAR are needed.
Another important unknown for these enzymes is their subcellular localization. P450 enzymes exist in both mitochondria and endoplasmic reticulum (ER). For CYP26A1, indirect evidence using reconstituted systems with ER P450 reductase has provided support for the notion that this enzyme is an ER P450. However, no such data exists for CYP26B1 and CYP26C1. As microsomal incubations from human liver, by definition, have only the ER membrane fraction, results from such incubations will fail to recognize CYP26 contribution if it is localized in the mitochondria.
The progress of research in the area of CYP26 biochemistry is greatly hindered by two factors. First, all but one (4-oxo-RA) of the metabolites identified as products of RA oxidation by CYP26 are not commercially available for researchers and second, these enzymes have proven to be very difficult to express and purify from heterologous systems. These difficulties have prevented detailed enzyme kinetic and pharmacokinetic analyses as well as validation of potential antibodies. The facts that RA isomerizes under white light and chemical synthesis of the proposed metabolites are not well established, make this area exceptionally difficult to study in detail. Additional complications include the very low concentrations of RA that exist in-vivo, the tight binding of RA to CYP26 enzymes and the fact that RA does not ionize well in common LC-MS instruments.
Finally, very little attention has been paid to genetic polymorphisms occurring in the CYP26 enzymes. One study screened ninety-two racially diverse subjects and discovered thirteen single nucleotide polymorphisms (SNPs) in CYP26A1. Two of these SNPs demonstrated significantly reduced (43–89%) metabolism, but due to the lack of above described tools, the exact reason for this reduced activity could not be determined [76]. It is expected that SNPs that alter either the regulation or function of the CYP26 enzymes would impact individual’s health and response to environmental stressors and potentially place the individual at risk for developmental defects, cancer or lowered immunity. It is expected that future studies of genetic polymorphisms in the CYP26 genes will greatly advance our understanding of gene-environment interactions relating to RA homeostasis.
The big question that remains to be answered is why are three different isoforms of CYP26 needed in all species if the three isoforms are functionally identical. Although the evolution of the individual isoforms in not fully characterized, it is unusual for P450 enzymes responsible for metabolizing endogenous compounds to have multiple isoforms within a species. One hypothesis to explain this is that the various CYP26 isoforms have different, yet unknown functions, in addition to clearing RA. For example, they may produce different metabolites that have different biological activity. To test this hypothesis, a detailed biochemical characterization of the three isoforms needs to be conducted including analysis of different substrates. Such information will in turn aid in future design of therapeutic strategies and in understanding biological outcomes of xenobiotic-CYP26 interactions.
Acknowledgments
The authors wish to thank Dr. Alex Zelter for helpful discussions.
Declaration of interest
This work was supported in part by NIH grants RO1-GM081569 and GM07750.
Abbreviations list
- RA
retinoic acid
- at-RA
All-trans-retinoic acid
- ADH
alcohol dehydrogenase
- RDH
retinol dehydrogenase
- REH
retinyl ester hydrolase
- RALDH
retinal dehydrogenase
- NAD
nicotinamide adenine dinucleotide
- NADP
nicotinamide adenine dinucleotide phosphate
- RAR
retinoic acid receptor
- RXR
retinoid X receptor
- CRABP
cellular retinoic acid binding protein
- CYP or P450
cytochrome P450
- RAI
retinoic acid inducible
- mRNA
messenger ribonucleic acid
- RT-PCR
reverse-transcriptase polymerase chain reaction
- OH
hydroxylated (alcohol) metabolite
- oxo
ketone metabolite
- HPLC
high performance liquid chromatograph
- MS
Mass spectrometry
- Cl’int
intrinsic clearance
- Vmax
maximal velocity
- AUC
area under the curve
- RAMBA
retinoic acid metabolism blocking agent
- ER
endoplasmic reticulum
- SNP
single nucleotide polymorphism
- Cl
total body clearance
- fu
fraction unbound
- t½
half life
- V
volume of distribution
- Cmax
maximum concentration achieved
- Tmax
time at which maximum concentration was achieved
- F
bioavailability
- APL
acute promyelocytic leukemia
References
- 1.Tzimas G, Nau H. The role of metabolism and toxicokinetics in retinoid teratogenesis. Curr Pharm Des. 2001;7:803–31. doi: 10.2174/1381612013397708. [DOI] [PubMed] [Google Scholar]
- 2.Blomhoff R, Blomhoff HK. Overview of retinoid metabolism and function. J Neurobiol. 2006;66:606–30. doi: 10.1002/neu.20242. [DOI] [PubMed] [Google Scholar]
- 3.Kane MA, Folias AE, Wang C, et al. Quantitative profiling of endogenous retinoic acid in vivo and in vitro by tandem mass spectrometry. Anal Chem. 2008;80:1702–8. doi: 10.1021/ac702030f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang GW, Russell RM. 13-cis-retinoic acid is an endogenous compound in human serum. J Lipid Res. 1990;31:175–82. [PubMed] [Google Scholar]
- 5.Tzimas G, Collins MD, Burgin H, et al. Embryotoxic doses of vitamin A to rabbits result in low plasma but high embryonic concentrations of all-trans-retinoic acid: risk of vitamin A exposure in humans. J Nutr. 1996;126:2159–71. doi: 10.1093/jn/126.9.2159. [DOI] [PubMed] [Google Scholar]
- 6.Napoli JL. Retinoid binding-proteins redirect retinoid metabolism: biosynthesis and metabolism of retinoic acid. Semin Cell Dev Biol. 1997;8:403–15. doi: 10.1006/scdb.1997.0164. [DOI] [PubMed] [Google Scholar]
- 7.Ross AC. Cellular metabolism and activation of retinoids: roles of cellular retinoid-binding proteins. Faseb J. 1993;7:317–27. doi: 10.1096/fasebj.7.2.8440409. [DOI] [PubMed] [Google Scholar]
- 8.Eckhoff C, Collins MD, Nau H. Human plasma all-trans-, 13-cis- and 13-cis-4-oxoretinoic acid profiles during subchronic vitamin A supplementation: comparison to retinol and retinyl ester plasma levels. J Nutr. 1991;121:1016–25. doi: 10.1093/jn/121.7.1016. [DOI] [PubMed] [Google Scholar]
- 9.Fex G, Felding P. Factors affecting the concentration of free holo retinol-binding protein in human plasma. Eur J Clin Invest. 1984;14:146–9. doi: 10.1111/j.1365-2362.1984.tb02104.x. [DOI] [PubMed] [Google Scholar]
- 10.Eckhoff C, Nau H. Identification and quantitation of all-trans- and 13-cis-retinoic acid and 13-cis-4-oxoretinoic acid in human plasma. J Lipid Res. 1990;31:1445–54. [PubMed] [Google Scholar]
- 11.Kane MA, Chen N, Sparks S, et al. Quantification of endogenous retinoic acid in limited biological samples by LC/MS/MS. Biochem J. 2005;388:363–9. doi: 10.1042/BJ20041867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen H, Namkung MJ, Juchau MR. Biotransformation of all-trans-retinol and all-trans-retinal to all-trans-retinoic acid in rat conceptal homogenates. Biochem Pharmacol. 1995;50:1257–64. doi: 10.1016/0006-2952(95)02005-w. [DOI] [PubMed] [Google Scholar]
- 13.Chen H, Howald WN, Juchau MR. Biosynthesis of all-trans-retinoic acid from all-trans-retinol: catalysis of all-trans-retinol oxidation by human P-450 cytochromes. Drug Metab Dispos. 2000;28:315–22. [PubMed] [Google Scholar]
- 14.Zhang QY, Dunbar D, Kaminsky L. Human cytochrome P-450 metabolism of retinals to retinoic acids. Drug Metab Dispos. 2000;28:292–7. [PubMed] [Google Scholar]
- 15.Marill J, Idres N, Capron CC, et al. Retinoic acid metabolism and mechanism of action: a review. Curr Drug Metab. 2003;4:1–10. doi: 10.2174/1389200033336900. [DOI] [PubMed] [Google Scholar]
- 16.Rothman KJ, Moore LL, Singer MR, et al. Teratogenicity of high vitamin A intake. N Engl J Med. 1995;333:1369–73. doi: 10.1056/NEJM199511233332101. [DOI] [PubMed] [Google Scholar]
- 17.Melhus H, Michaelsson K, Kindmark A, et al. Excessive dietary intake of vitamin A is associated with reduced bone mineral density and increased risk for hip fracture. Ann Intern Med. 1998;129:770–8. doi: 10.7326/0003-4819-129-10-199811150-00003. [DOI] [PubMed] [Google Scholar]
- 18.Ross AC. Vitamin A status: relationship to immunity and the antibody response. Proc Soc Exp Biol Med. 1992;200:303–20. doi: 10.3181/00379727-200-43436a. [DOI] [PubMed] [Google Scholar]
- 19.World_Health_Organization. [[cited 2009 April 4]];Macronutrient deficiencies: vitamin A deficiency. Available from: http://www.who.int/nutrition/topics/vad/en/index.html.
- 20.Jacobs S, Lie DC, DeCicco KL, et al. Retinoic acid is required early during adult neurogenesis in the dentate gyrus. Proc Natl Acad Sci U S A. 2006;103:3902–7. doi: 10.1073/pnas.0511294103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goodman AB. Retinoid receptors, transporters, and metabolizers as therapeutic targets in late onset Alzheimer disease. J Cell Physiol. 2006;209:598–603. doi: 10.1002/jcp.20784. [DOI] [PubMed] [Google Scholar]
- 22.Ding Y, Qiao A, Wang Z, et al. Retinoic acid attenuates beta-amyloid deposition and rescues memory deficits in an Alzheimer’s disease transgenic mouse model. J Neurosci. 2008;28:11622–34. doi: 10.1523/JNEUROSCI.3153-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Girard C, Dereure O, Blatiere V, et al. Vitamin a deficiency phrynoderma associated with chronic giardiasis. Pediatr Dermatol. 2006;23:346–9. doi: 10.1111/j.1525-1470.2006.00261.x. [DOI] [PubMed] [Google Scholar]
- 24.Petkovich M, Brand NJ, Krust A, et al. A human retinoic acid receptor which belongs to the family of nuclear receptors. Nature. 1987;330:444–50. doi: 10.1038/330444a0. [DOI] [PubMed] [Google Scholar]
- 25.Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83:841–50. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- 26.Schug TT, Berry DC, Shaw NS, et al. Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors. Cell. 2007;129:723–33. doi: 10.1016/j.cell.2007.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nowickyj SM, Chithalen JV, Cameron D, et al. Locust retinoid X receptors: 9-Cis-retinoic acid in embryos from a primitive insect. Proc Natl Acad Sci U S A. 2008;105:9540–5. doi: 10.1073/pnas.0712132105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duester G. Retinoic acid synthesis and signaling during early organogenesis. Cell. 2008;134:921–31. doi: 10.1016/j.cell.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Napoli JL. Retinoic acid biosynthesis and metabolism. Faseb J. 1996;10:993–1001. doi: 10.1096/fasebj.10.9.8801182. [DOI] [PubMed] [Google Scholar]
- 30.Budhu AS, Noy N. Direct channeling of retinoic acid between cellular retinoic acid-binding protein II and retinoic acid receptor sensitizes mammary carcinoma cells to retinoic acid-induced growth arrest. Mol Cell Biol. 2002;22:2632–41. doi: 10.1128/MCB.22.8.2632-2641.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fiorella PD, Napoli JL. Expression of cellular retinoic acid binding protein (CRABP) in Escherichia coli. Characterization and evidence that holo-CRABP is a substrate in retinoic acid metabolism. J Biol Chem. 1991;266:16572–9. [PubMed] [Google Scholar]
- 32.Fiorella PD, Napoli JL. Microsomal retinoic acid metabolism. Effects of cellular retinoic acid-binding protein (type I) and C18-hydroxylation as an initial step. J Biol Chem. 1994;269:10538–44. [PubMed] [Google Scholar]
- 33.White JA, Boffa MB, Jones B, et al. A zebrafish retinoic acid receptor expressed in the regenerating caudal fin. Development. 1994;120:1861–72. doi: 10.1242/dev.120.7.1861. [DOI] [PubMed] [Google Scholar]
- 34.White JA, Guo YD, Baetz K, et al. Identification of the retinoic acid-inducible all-trans-retinoic acid 4-hydroxylase. J Biol Chem. 1996;271:29922–7. doi: 10.1074/jbc.271.47.29922. [DOI] [PubMed] [Google Scholar]
- 35.Ray WJ, Bain G, Yao M, et al. CYP26, a novel mammalian cytochrome P450, is induced by retinoic acid and defines a new family. J Biol Chem. 1997;272:18702–8. doi: 10.1074/jbc.272.30.18702. [DOI] [PubMed] [Google Scholar]
- 36.White JA, Beckett-Jones B, Guo YD, et al. cDNA cloning of human retinoic acid-metabolizing enzyme (hP450RAI) identifies a novel family of cytochromes P450. J Biol Chem. 1997;272:18538–41. doi: 10.1074/jbc.272.30.18538. [DOI] [PubMed] [Google Scholar]
- 37.Fujii H, Sato T, Kaneko S, et al. Metabolic inactivation of retinoic acid by a novel P450 differentially expressed in developing mouse embryos. Embo J. 1997;16:4163–73. doi: 10.1093/emboj/16.14.4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y, Zolfaghari R, Ross AC. Cloning of rat cytochrome P450RAI (CYP26) cDNA and regulation of its gene expression by all-trans-retinoic acid in vivo. Arch Biochem Biophys. 2002;401:235–43. doi: 10.1016/S0003-9861(02)00043-7. [DOI] [PubMed] [Google Scholar]
- 39.Swindell EC, Thaller C, Sockanathan S, et al. Complementary domains of retinoic acid production and degradation in the early chick embryo. Dev Biol. 1999;216:282–96. doi: 10.1006/dbio.1999.9487. [DOI] [PubMed] [Google Scholar]
- 40.Kruger KA, Blum JW, Greger DL. Expression of nuclear receptor and target genes in liver and intestine of neonatal calves fed colostrum and vitamin A. J Dairy Sci. 2005;88:3971–81. doi: 10.3168/jds.S0022-0302(05)73083-6. [DOI] [PubMed] [Google Scholar]
- 41.Taimi M, Helvig C, Wisniewski J, et al. A novel human cytochrome P450, CYP26C1, involved in metabolism of 9-cis and all-trans isomers of retinoic acid. J Biol Chem. 2004;279:77–85. doi: 10.1074/jbc.M308337200. [DOI] [PubMed] [Google Scholar]
- 42.White JA, Ramshaw H, Taimi M, et al. Identification of the human cytochrome P450, P450RAI-2, which is predominantly expressed in the adult cerebellum and is responsible for all-trans-retinoic acid metabolism. Proc Natl Acad Sci U S A. 2000;97:6403–8. doi: 10.1073/pnas.120161397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hernandez RE, Putzke AP, Myers JP, et al. Cyp26 enzymes generate the retinoic acid response pattern necessary for hindbrain development. Development. 2007;134:177–87. doi: 10.1242/dev.02706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.MacLean G, Abu-Abed S, Dolle P, et al. Cloning of a novel retinoic-acid metabolizing cytochrome P450, Cyp26B1, and comparative expression analysis with Cyp26A1 during early murine development. Mech Dev. 2001;107:195–201. doi: 10.1016/s0925-4773(01)00463-4. [DOI] [PubMed] [Google Scholar]
- 45.Tahayato A, Dolle P, Petkovich M. Cyp26C1 encodes a novel retinoic acid-metabolizing enzyme expressed in the hindbrain, inner ear, first branchial arch and tooth buds during murine development. Gene Expr Patterns. 2003;3:449–54. doi: 10.1016/s1567-133x(03)00066-8. [DOI] [PubMed] [Google Scholar]
- 46.Zhao Q, Dobbs-McAuliffe B, Linney E. Expression of cyp26b1 during zebrafish early development. Gene Expr Patterns. 2005;5:363–9. doi: 10.1016/j.modgep.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 47.Gu X, Xu F, Song W, et al. A novel cytochrome P450, zebrafish Cyp26D1, is involved in metabolism of all-trans retinoic acid. Mol Endocrinol. 2006;20:1661–72. doi: 10.1210/me.2005-0362. [DOI] [PubMed] [Google Scholar]
- 48.Gu X, Xu F, Wang X, et al. Molecular cloning and expression of a novel CYP26 gene (cyp26d1) during zebrafish early development. Gene Expr Patterns. 2005;5:733–9. doi: 10.1016/j.modgep.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 49.Nelson DR, Koymans L, Kamataki T, et al. P450 superfamily: update on new sequences, gene mapping, accession numbers and nomenclature. Pharmacogenetics. 1996;6:1–42. doi: 10.1097/00008571-199602000-00002. [DOI] [PubMed] [Google Scholar]
- 50.Uehara M, Yashiro K, Mamiya S, et al. CYP26A1 and CYP26C1 cooperatively regulate anterior-posterior patterning of the developing brain and the production of migratory cranial neural crest cells in the mouse. Dev Biol. 2007;302:399–411. doi: 10.1016/j.ydbio.2006.09.045. [DOI] [PubMed] [Google Scholar]
- 51.Gomaa MS, Yee SW, Milbourne CE, et al. Homology model of human retinoic acid metabolising enzyme cytochrome P450 26A1 (CYP26A1): active site architecture and ligand binding. J Enzyme Inhib Med Chem. 2006;21:361–9. doi: 10.1080/14756360600742014. [DOI] [PubMed] [Google Scholar]
- 52.Abu-Abed S, Dolle P, Metzger D, et al. The retinoic acid-metabolizing enzyme, CYP26A1, is essential for normal hindbrain patterning, vertebral identity, and development of posterior structures. Genes Dev. 2001;15:226–40. doi: 10.1101/gad.855001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Niederreither K, Abu-Abed S, Schuhbaur B, et al. Genetic evidence that oxidative derivatives of retinoic acid are not involved in retinoid signaling during mouse development. Nat Genet. 2002;31:84–8. doi: 10.1038/ng876. [DOI] [PubMed] [Google Scholar]
- 54.Yashiro K, Zhao X, Uehara M, et al. Regulation of retinoic acid distribution is required for proximodistal patterning and outgrowth of the developing mouse limb. Dev Cell. 2004;6:411–22. doi: 10.1016/s1534-5807(04)00062-0. [DOI] [PubMed] [Google Scholar]
- 55.Abu-Abed S, MacLean G, Fraulob V, et al. Differential expression of the retinoic acid-metabolizing enzymes CYP26A1 and CYP26B1 during murine organogenesis. Mech Dev. 2002;110:173–7. doi: 10.1016/s0925-4773(01)00572-x. [DOI] [PubMed] [Google Scholar]
- 56.Sakai Y, Meno C, Fujii H, et al. The retinoic acid-inactivating enzyme CYP26 is essential for establishing an uneven distribution of retinoic acid along the anterio-posterior axis within the mouse embryo. Genes Dev. 2001;15:213–25. doi: 10.1101/gad.851501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xi J, Yang Z. Expression of RALDHs (ALDH1As) and CYP26s in human tissues and during the neural differentiation of P19 embryonal carcinoma stem cell. Gene Expr Patterns. 2008;8:438–42. doi: 10.1016/j.gep.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 58.Trofimova-Griffin ME, Brzezinski MR, Juchau MR. Patterns of CYP26 expression in human prenatal cephalic and hepatic tissues indicate an important role during early brain development. Brain Res Dev Brain Res. 2000;120:7–16. doi: 10.1016/s0165-3806(99)00185-6. [DOI] [PubMed] [Google Scholar]
- 59.Loudig O, Babichuk C, White J, et al. Cytochrome P450RAI(CYP26) promoter: a distinct composite retinoic acid response element underlies the complex regulation of retinoic acid metabolism. Mol Endocrinol. 2000;14:1483–97. doi: 10.1210/mend.14.9.0518. [DOI] [PubMed] [Google Scholar]
- 60.Yamamoto Y, Zolfaghari R, Ross AC. Regulation of CYP26 (cytochrome P450RAI) mRNA expression and retinoic acid metabolism by retinoids and dietary vitamin A in liver of mice and rats. Faseb J. 2000;14:2119–27. doi: 10.1096/fj.00-0061com. [DOI] [PubMed] [Google Scholar]
- 61.Lampen A, Meyer S, Nau H. Effects of receptor-selective retinoids on CYP26 gene expression and metabolism of all-trans-retinoic acid in intestinal cells. Drug Metab Dispos. 2001;29:742–7. [PubMed] [Google Scholar]
- 62.Ozpolat B, Mehta K, Lopez-Berestein G. Regulation of a highly specific retinoic acid-4-hydroxylase (CYP26A1) enzyme and all-trans-retinoic acid metabolism in human intestinal, liver, endothelial, and acute promyelocytic leukemia cells. Leuk Lymphoma. 2005;46:1497–506. doi: 10.1080/10428190500174737. [DOI] [PubMed] [Google Scholar]
- 63.Chithalen JV, Luu L, Petkovich M, et al. HPLC-MS/MS analysis of the products generated from all-trans-retinoic acid using recombinant human CYP26A. J Lipid Res. 2002;43:1133–42. doi: 10.1194/jlr.m100343-jlr200. [DOI] [PubMed] [Google Scholar]
- 64.Frolik CA, Roberts AB, Tavela TE, et al. Isolation and identification of 4-hydroxy- and 4-oxoretinoic acid. In vitro metabolites of all-trans-retinoic acid in hamster trachea and liver. Biochemistry. 1979;18:2092–7. doi: 10.1021/bi00577a039. [DOI] [PubMed] [Google Scholar]
- 65.John KV, Lakshmanan MR, Cama HR. Preparation, properties and metabolism of 5,6-monoepoxyretinoic acid. Biochem J. 1967;103:539–43. doi: 10.1042/bj1030539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McCormick AM, Napoli JL, Schnoes HK, et al. Isolation and identification of 5, 6-epoxyretinoic acid: a biologically active metabolite of retinoic acid. Biochemistry. 1978;17:4085–90. doi: 10.1021/bi00612a033. [DOI] [PubMed] [Google Scholar]
- 67.Pijnappel WW, Hendriks HF, Folkers GE, et al. The retinoid ligand 4-oxo-retinoic acid is a highly active modulator of positional specification. Nature. 1993;366:340–4. doi: 10.1038/366340a0. [DOI] [PubMed] [Google Scholar]
- 68.Samokyszyn VM, Gall WE, Zawada G, et al. 4-hydroxyretinoic acid, a novel substrate for human liver microsomal UDP-glucuronosyltransferase(s) and recombinant UGT2B7. J Biol Chem. 2000;275:6908–14. doi: 10.1074/jbc.275.10.6908. [DOI] [PubMed] [Google Scholar]
- 69.Chen H, Fantel AG, Juchau MR. Catalysis of the 4-hydroxylation of retinoic acids by cyp3a7 in human fetal hepatic tissues. Drug Metab Dispos. 2000;28:1051–7. [PubMed] [Google Scholar]
- 70.Marill J, Cresteil T, Lanotte M, et al. Identification of human cytochrome P450s involved in the formation of all-trans-retinoic acid principal metabolites. Mol Pharmacol. 2000;58:1341–8. doi: 10.1124/mol.58.6.1341. [DOI] [PubMed] [Google Scholar]
- 71.McSorley LC, Daly AK. Identification of human cytochrome P450 isoforms that contribute to all-trans-retinoic acid 4-hydroxylation. Biochem Pharmacol. 2000;60:517–26. doi: 10.1016/s0006-2952(00)00356-7. [DOI] [PubMed] [Google Scholar]
- 72.Nadin L, Murray M. Participation of CYP2C8 in retinoic acid 4-hydroxylation in human hepatic microsomes. Biochem Pharmacol. 1999;58:1201–8. doi: 10.1016/s0006-2952(99)00192-6. [DOI] [PubMed] [Google Scholar]
- 73.Package insert for Vesanoid (R) (tretinoin capsules): Roche Pharmaceuticals. Roche Laboratories Inc; 2000. [Google Scholar]
- 74.Cullum ME, Zile MH. Metabolism of all-trans-retinoic acid and all-trans-retinyl acetate. Demonstration of common physiological metabolites in rat small intestinal mucosa and circulation. J Biol Chem. 1985;260:10590–6. [PubMed] [Google Scholar]
- 75.Marill J, Capron CC, Idres N, et al. Human cytochrome P450s involved in the metabolism of 9-cis- and 13-cis-retinoic acids. Biochem Pharmacol. 2002;63:933–43. doi: 10.1016/s0006-2952(01)00925-x. [DOI] [PubMed] [Google Scholar]
- 76.Lee SJ, Perera L, Coulter SJ, et al. The discovery of new coding alleles of human CYP26A1 that are potentially defective in the metabolism of all-trans retinoic acid and their assessment in a recombinant cDNA expression system. Pharmacogenet Genomics. 2007;17:169–80. doi: 10.1097/FPC.0b013e32801152d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lutz JD, Dixit V, Yeung CK, et al. Expression and functional characterization of cytochrome P450 26A1, a retinoic acid hydroxylase. Biochem Pharmacol. 2009;77:258–68. doi: 10.1016/j.bcp.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rostami-Hodjegan A, Tucker GT. Simulation and prediction of in vivo drug metabolism in human populations from in vitro data. Nat Rev Drug Discov. 2007;6:140–8. doi: 10.1038/nrd2173. [DOI] [PubMed] [Google Scholar]
- 79.Huang ME, Ye YC, Chen SR, et al. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood. 1988;72:567–72. doi: 10.1182/blood-2016-11-750182. [DOI] [PubMed] [Google Scholar]
- 80.Jones H, Blanc D, Cunliffe WJ. 13-cis retinoic acid and acne. Lancet. 1980;2:1048–9. doi: 10.1016/s0140-6736(80)92273-4. [DOI] [PubMed] [Google Scholar]
- 81.Altucci L, Leibowitz MD, Ogilvie KM, et al. RAR and RXR modulation in cancer and metabolic disease. Nat Rev Drug Discov. 2007;6:793–810. doi: 10.1038/nrd2397. [DOI] [PubMed] [Google Scholar]
- 82.Center for drug evaluation and research, approval package for application number NDA20-438/S-001, final printed labeling for Accutane (R) (isotretinoin capsules) Roche Pharmaceuticals. Roche Laboratories Inc; 2007. [Google Scholar]
- 83.Ozpolat B, Lopez-Berestein G, Adamson P, et al. Pharmacokinetics of intravenously administered liposomal all-trans-retinoic acid (ATRA) and orally administered ATRA in healthy volunteers. J Pharm Pharm Sci. 2003;6:292–301. [PubMed] [Google Scholar]
- 84.Muindi JR, Frankel SR, Huselton C, et al. Clinical pharmacology of oral all-trans retinoic acid in patients with acute promyelocytic leukemia. Cancer Res. 1992;52:2138–42. [PubMed] [Google Scholar]
- 85.Lazzarino M, Regazzi MB, Corso A. Clinical relevance of all-trans retinoic acid pharmacokinetics and its modulation in acute promyelocytic leukemia. Leuk Lymphoma. 1996;23:539–43. doi: 10.3109/10428199609054862. [DOI] [PubMed] [Google Scholar]
- 86.Sun SY, Lotan R. Retinoids and their receptors in cancer development and chemoprevention. Crit Rev Oncol Hematol. 2002;41:41–55. doi: 10.1016/s1040-8428(01)00144-5. [DOI] [PubMed] [Google Scholar]
- 87.Schwartz EL, Hallam S, Gallagher RE, et al. Inhibition of all-trans-retinoic acid metabolism by fluconazole in vitro and in patients with acute promyelocytic leukemia. Biochem Pharmacol. 1995;50:923–8. doi: 10.1016/0006-2952(95)00213-j. [DOI] [PubMed] [Google Scholar]
- 88.Vanier KL, Mattiussi AJ, Johnston DL. Interaction of all-trans-retinoic acid with fluconazole in acute promyelocytic leukemia. J Pediatr Hematol Oncol. 2003;25:403–4. doi: 10.1097/00043426-200305000-00010. [DOI] [PubMed] [Google Scholar]
- 89.Rigas JR, Francis PA, Muindi JR, et al. Constitutive variability in the pharmacokinetics of the natural retinoid, all-trans-retinoic acid, and its modulation by ketoconazole. J Natl Cancer Inst. 1993;85:1921–6. doi: 10.1093/jnci/85.23.1921. [DOI] [PubMed] [Google Scholar]
- 90.Njar VC. Cytochrome p450 retinoic acid 4-hydroxylase inhibitors: potential agents for cancer therapy. Mini Rev Med Chem. 2002;2:261–9. doi: 10.2174/1389557023406223. [DOI] [PubMed] [Google Scholar]
- 91.Njar VC, Gediya L, Purushottamachar P, et al. Retinoic acid metabolism blocking agents (RAMBAs) for treatment of cancer and dermatological diseases. Bioorg Med Chem. 2006;14:4323–40. doi: 10.1016/j.bmc.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 92.Patel JB, Huynh CK, Handratta VD, et al. Novel retinoic acid metabolism blocking agents endowed with multiple biological activities are efficient growth inhibitors of human breast and prostate cancer cells in vitro and a human breast tumor xenograft in nude mice. J Med Chem. 2004;47:6716–29. doi: 10.1021/jm0401457. [DOI] [PubMed] [Google Scholar]
- 93.Miller VA, Rigas JR, Muindi JR, et al. Modulation of all-trans retinoic acid pharmacokinetics by liarozole. Cancer Chemother Pharmacol. 1994;34:522–6. doi: 10.1007/BF00685665. [DOI] [PubMed] [Google Scholar]
- 94.Stoppie P, Borgers M, Borghgraef P, et al. R115866 inhibits all-trans-retinoic acid metabolism and exerts retinoidal effects in rodents. J Pharmacol Exp Ther. 2000;293:304–12. [PubMed] [Google Scholar]
- 95.Mulvihill MJ, Kan JL, Beck P, et al. Potent and selective [2-imidazol-1-yl-2-(6-alkoxy-naphthalen-2-yl)-1-methyl-ethyl]-dimethyl-ami nes as retinoic acid metabolic blocking agents (RAMBAs) Bioorg Med Chem Lett. 2005;15:1669–73. doi: 10.1016/j.bmcl.2005.01.044. [DOI] [PubMed] [Google Scholar]
- 96.Mulvihill MJ, Kan JL, Cooke A, et al. 3-[6-(2-Dimethylamino-1-imidazol-1-yl-butyl)-naphthalen-2-yloxy]-2,2-dimet hyl-propionic acid as a highly potent and selective retinoic acid metabolic blocking agent. Bioorg Med Chem Lett. 2006;16:2729–33. doi: 10.1016/j.bmcl.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 97.Yee SW, Jarno L, Gomaa MS, et al. Novel tetralone-derived retinoic acid metabolism blocking agents: synthesis and in vitro evaluation with liver microsomal and MCF-7 CYP26A1 cell assays. J Med Chem. 2005;48:7123–31. doi: 10.1021/jm0501681. [DOI] [PubMed] [Google Scholar]
- 98.Van Heusden J, Van Ginckel R, Bruwiere H, et al. Inhibition of all-TRANS-retinoic acid metabolism by R116010 induces antitumour activity. Br J Cancer. 2002;86:605–11. doi: 10.1038/sj.bjc.6600056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fex G, Larsson K, Andersson A, et al. Low serum concentration of all-trans and 13-cis retinoic acids in patients treated with phenytoin, carbamazepine and valproate. Possible relation to teratogenicity. Arch Toxicol. 1995;69:572–4. doi: 10.1007/s002040050215. [DOI] [PubMed] [Google Scholar]
- 100.Nau H, Tzimas G, Mondry M, et al. Antiepileptic drugs alter endogenous retinoid concentrations: a possible mechanism of teratogenesis of anticonvulsant therapy. Life Sci. 1995;57:53–60. doi: 10.1016/0024-3205(95)00242-x. [DOI] [PubMed] [Google Scholar]